REVIEW: Cation–Chloride Cotransporters: Regulation, Physiological Significance, and Role in Pathogenesis of Arterial Hypertension
S. N. Orlov1,2*, S. V. Koltsova1, L. V. Kapilevich2,3, N. O. Dulin4, and S. V. Gusakova3
1Faculty of Biology, Lomonosov Moscow State University, Vorob’evy Gory 1/2, 119991 Moscow, Russia; E-mail: sergeinorlov@yandex.ru2Tomsk State University, pr. Lenina 36, 634050 Tomsk, Russia
3Siberian State Medical University, Moskovskii Trakt 2, 634050 Tomsk, Russia
4University of Chicago, Chicago, 600637 IL, USA
* To whom correspondence should be addressed.
Received July 23, 2014
This review summarizes the data on the functioning of carriers providing electroneutral symport of sodium, potassium, and chloride (Na+,K+,2Cl– cotransport), potassium and chloride (K+,Cl– cotransport), and sodium and chloride (K+,Cl– cotransport) as well as molecular mechanisms of the regulation of these carriers and their physiological significance. We emphasized the involvement of chloride-coupled carriers in the regulation of cell volume and intracellular chloride concentration and novel data on the role of ubiquitous isoform of Na+,K+,2Cl– cotransporter NKCC1 in regulation of vascular smooth muscle contraction and activity of GABAA receptors. Finally, we analyzed the data on activation of NKCC1 in patients with essential hypertension and its role in the long-term maintenance of elevated systemic blood pressure and myogenic response in microcirculatory beds.
KEY WORDS: sodium, potassium, chloride, cotransport, smooth muscle, contraction, myogenic tone, sympathetic nervous system, hypertensionDOI: 10.1134/S0006297914130070
Abbreviations: CCC, cation–chloride cotransporters; CNS, central nervous system; GABA, γ-aminobutyric acid; KCC, K+,Cl– cotransport; NCC, Na+,Cl– cotransport; NKCC, Na+,K+,2Cl– cotransport; OSR1, oxidative stress response kinase; PVN, paraventricular nucleus; SMC, smooth muscle cell; SNS, sympathetic nervous system; SPAK, Ste20-related praline-alanine-rich kinase; WNK, “with no K” lysine kinase.
Cation–chloride cotransporters (CCCs) are members of solute
carriers (SLC) involved in transport of ions across biological
membranes along or against their electrochemical gradient. In the
former case, the energy is provided by the gradient of cotransporting
ions created by Na+,K+-ATPase and other ion
pumps. This group of ion transporters consists of more than 300 genes
of 52 SLC families [1]. CCCs form the SLC12 family,
whose members provide symport of Cl– anions with
Na+ and/or K+ cations. The SLC12 family includes
Na+,Cl– cotransporter (NCC) encoded by the
single gene (SLC12A3),
Na+,K+,2Cl– cotransporters
(NKCC) encoded by SLC12A2 (NKCC1) and SLC12A1 (NKCC2),
and K+,Cl– cotransporters (KCC) encoded by
SLC12A4 (KCC1), SLC12A5 (KCC2), SLC12A6 (KCC3),
and SLC12A7 (KCC4). NKCC is inhibited by bumetanide, furosemide,
and several other structurally similar compounds. Keeping in mind that
the major target of these compounds is ion transport in thick ascending
limb of Henle’s loop, they were termed as high-ceiling diuretics.
NCC is completely blocked by thiazide derivatives. In contrast to NKCC
and NCC, specific inhibitors of KCC remain unknown and their activity
is partially attenuated in the presence of high doses of furosemide [2].
Gene structure, membrane architecture, and pharmacology of CCCs have been subjected to detailed analysis in several comprehensive reviews [2-4]. Considering this, we focus our review on systems involved in the regulation of CCC activity as well as on the role of these carriers in the maintenance of water–salt homeostasis and blood pressure. Recently, it was shown that CCCs are also involved in tumorigenesis [5, 6], the pathogenesis of neuronal disorders [7, 8], and anemia triggered by alterations of erythrocyte volume and hydration [9, 10]. Because of the space limit, these data were out of scope of our review.
REGULATION OF CCC ACTIVITY
Data on the regulation of CCC expression by glucocorticoids and other stimuli affecting transcription via activation of serum- and glucocorticoid-inducible kinase-1 (SGK1) were recently summarized by Lang and Voelkl [11]. Epigenetic mechanisms of regulation of expression of NKCC1 are considered in the section “Physiological Significance”. Here, we briefly summarize the data on the non-genomic mechanism of the regulation of CCC activity.
Second messengers. Data obtained so far show a tissue-specific pattern of CCC regulation by second messengers [11, 12]. Thus, for example, in isolated smooth muscle cells (SMCs) from rat aorta an increment of cAMP content triggered by activation of β-adrenergic receptors and adenylate cyclase led to inhibition of NKCC1, whereas a rise of [Ca2+]i evoked by addition of ionophores increased its activity [13-15]. In SMCs, activation of cGMP-mediated signaling did not affect NKCC1, but it increased activity of KCC [16]. Unlike SMCs, activators of cAMP- and cGMP-mediated signaling as well as chelators of extra- and intracellular calcium had no any impact on NKCC1 activity in renal epithelial cells from canine distal tubule (MDCK cells) [17-19], whereas in secreting epithelium of shark rectal gland [20] and human airways [21] cAMP augmented NKCC1 activity. It is known that diacyl glycerol production triggered by agonists of GTP-binding protein-coupled receptors results in activation of protein kinase C. It was shown that activation of this enzyme by phorbol esters does not influence NKCC1 in SMCs [13], but completely blocks this ion carrier in MDCK cells [17-19]. Viewed collectively, these data allowed researchers to propose that cAMP, cGMP, Ca2+, diacyl glycerol, and other canonical second messengers affect CCCs via their interaction with tissue-specific intermediates of intracellular signaling cascades rather than with ion carriers per se [4]. The tissue-specific intermediates of CCC include protein kinases and phosphoprotein phosphatases considered in the next section.
Protein kinases and phosphatases. The first indication on the regulation of ion transport by CCC phosphorylation was obtained in the laboratory of B. Forbush. Their experiments demonstrated that bumetanide-sensitive K+ fluxes in shark rectal glands triggered by hyperosmotic shrinkage and activation of adenylate cyclase correlate with phosphorylation of serine and threonine residues in NKCC1 [20]. Later on, they proposed that reciprocal regulation of CCCs by volume changes (cell shrinkage activates NKCC but inhibits KCC) and intracellular chloride concentration (elevation of [Cl–]i inhibits NKCC but activates KCC) is mediated by reciprocal actions of these stimuli on the phosphorylation of CCCs by unknown kinases and phosphatases [22-24] (Fig. 1).
Fig. 1. Regulation of cation–chloride cotransporters by cell volume and concentration of intracellular chloride mediated by reciprocal phosphorylation of the carriers.
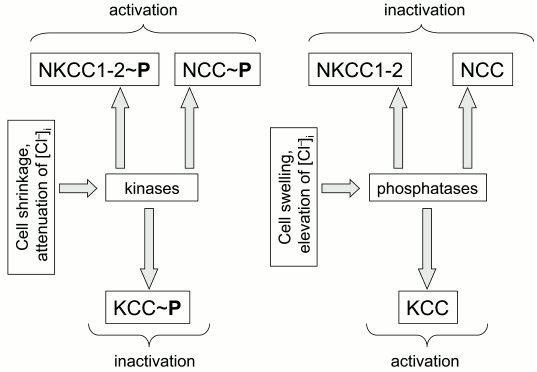
All cloned CCCs contain consensuses that might be phosphorylated by protein kinase C, protein kinase A, and casein kinase II. However, to the best of our knowledge, there is not any report showing that CCC phosphorylation by these canonical protein kinases correlates with the activity of these carriers [2, 25]. In contrast, studies performed in dozens of laboratories have demonstrated a key role in regulation of CCC activity of serine–threonine kinases of the WNK family. These “with no K” lysine kinases are lacking a lysine reside that is present in the ATP-binding site of all other serine-threonine kinases [26, 27]. Side-by-side with WNK, the activity of CCC also correlates with phosphorylation by sterile-20 (Ste20)-related praline-alanine-rich kinase (SPAK) and oxidative stress response kinase (OSR1) [28].
In the human genome, four WNKs (WNK1-4) have been located on chromosomes 12, 9, X, and 17, respectively [25]. The first evidence for their involvement in regulation of ion transport were obtained in the study of the physiological consequences of WNK1 and WNK4 mutations in patients with pseudohypoaldosteronism type II (PHAII) – a monogenic form of inherited hypertension accompanied by hyperkalemia and metabolic acidosis [29]. Because thiazide derivatives known as potent NCC inhibitors are widely used for the treatment of this disorder, it has been proposed that both WNK1 and WNK4 are involved in the regulation of NCC activity. Indeed, it was shown that a conserved WNK4 sequence consisting of catalytic site and autoinhibitory domain contains mutations detected in patients with PHAII [29].
Later on, it was demonstrated that WNK3 increases activity of NCC, NKCC1, and NKCC2 in oocytes subjected to hypotonic swelling via phosphorylation of two threonine residues located within their N-termini [30, 31]. Also, it was reported that coexpression of WNK3 leads to complete inactivation of KCC1-4, which was abolished by inhibitors of protein phosphatases PP1 and PP2B – calyculin A and cyclosporin A, respectively [32] (Fig. 2). These data suggested that WNKs are primary sensors of cell volume and [Cl–]i changes involved in reciprocal regulation of NKCC and KCC activity (Fig. 1).
Fig. 2. Scheme showing crosstalk of serine–threonine protein kinases and phosphatases in regulation of the activity of cation–chloride cotransporters.
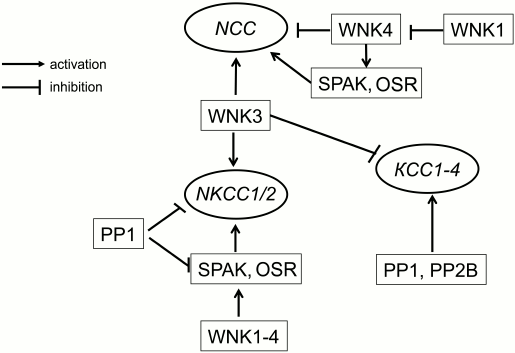
SPAK and OSR1 belong to the family of mammalian kinases that are homologous to yeast Ste20/Sts kinase having 90% identical amino acid in the catalytic domain [33]. Experiments with permeabilized oocytes failed to find any impact of SPAK on the activity of NKCC1, whereas expression of catalytically inactive SPAK inhibited NKCC1 and activated KCC2 [34, 35]. SPAK-mediated signaling was also revealed in the study of activation by WNK3 of NKCC2 [36]. Later, it was demonstrated that SPAK interacts with the N-terminal fragment of NKCC1 as well as with regulatory domain of WNKs [27, 34, 37]. In another set of experiments, it was shown that NKCC activity of sensory neurons is decreased by two-fold in homozygous SPAK knockout (SPAK–/–) mice as compared to the wild-type. These results suggested that along with direct action on CCCs, these carriers can be regulated by WNKs via their phosphorylation by SPAK and OSR1 [28, 38] (Fig. 2).
More recently, it was shown that in contrast to the direct inhibitory action of WNK4 on NCC activity seen in patients with PHAII, activation of this carrier by angiotensin II is caused by WNK4-mediated phosphorylation of SPAK/OSR1 [39, 40]. To explain this phenomenon, it should be noted that transcription of the WNK1-4 genes resulted in accumulation of a dozen splice-variants involved in different tissue-specific cellular responses including unknown ones. It should also be emphasized that along with WNK, SPAK, and OSR1, CCC activity might be affected by distinct unidentified protein kinases. Indeed, it has been reported that increased NKCC1 activity detected in cells treated with the activator of adenylate cyclase forskolin and transfected with SPAK is accompanied by phosphorylation of distinct threonine residues [41, 42].
PHYSIOLOGICAL SIGNIFICANCE
Mechanisms of the implication of CCCs in salt reabsorption and secretion by epithelial cells are subjected to detailed analysis in several comprehensive reviews [2-4]. Keeping this in mind, we devote this section to functions of CCCs detected in non-epithelial cells and having general biological significance.
Functional responses mediated by alteration of intracellular Cl– concentration. In all types of cells studied so far, CCCs generate both inwardly- and outwardly-directed ion movements, and the direction of net flux depends on the stoichiometry of the carrier and transmembrane gradients of cations created by Na+,K+-ATPase. Thus, the stoichiometry 1 : 1 predicts that the value of ion flux is in direct proportion to concentration of cotransporting ions. Because [Na+]o >> [Na+]i, [K+]i >> [K+]o, and [Cl–]o > [Cl–]i, net fluxes generated by NCC and KCC exhibit inward and outward directions, respectively. More complex behavior was found for ion fluxes mediated by NKCC. In the overwhelming number of cells, [Cl–]o2 >>> [Cl–]i2, and the net flux mediated by NKCC functioning with stoichiometry 1 Na+ : 1 K+ : 2 Cl– has inward direction.
The data considered above suggest that CCCs contribute to regulation of [Cl–]i, whereas their contribution to the adjustment of intracellular concentration of monovalent cation is negligible because of the highly active Na+,K+-pump. Indeed, it was shown that inhibition of NCC and NKCC results in attenuation of [Cl–]i, whereas suppression of KCC activity increases this parameter [43]. Importantly, the alteration of CCC activity can lead to adjustment of [Cl–]i above or below the values corresponding to Nernst equilibrium potential. This means that in cells abundant with anion channels, CCCs contribute to maintenance of electrical potential, thus having an impact on the whole spectrum of cellular functions controlled by potential-sensitive proteins localized within the plasma membrane. This conclusion is supported by data considered in the next section.
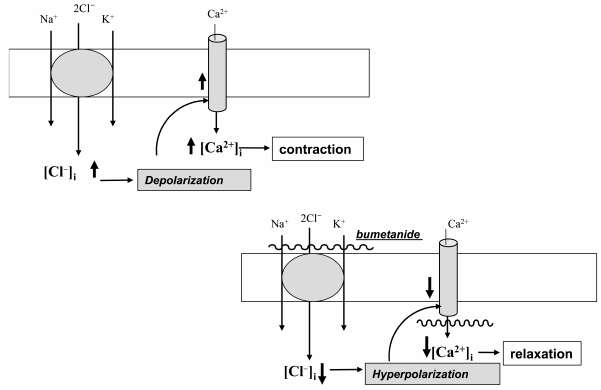
SMC contraction and myogenic response. In contrast to the major role of plasma membrane permeability for K+ (PK) in the maintenance of electrical resistance and resting potential (Em) in skeletal and cardiac muscle, the values of PK and PCl in SMCs are about the same [44]. This feature suggests that in SMCs CCCs may be involved in regulation of the [Cl–]i/[Cl–]o ratio and therefore Em and excitation–contraction coupling. Indeed, the NKCC inhibitors furosemide and bumetanide decreased [Cl–]i [45, 46] and led to hyperpolarization of rat vascular SMCs [45]. These data suggest that decreased baseline tonus seen in SMCs treated with Henle’s loop diuretics [47-49] as well as attenuation by these compounds contraction of smooth muscle strips evoked by modest increment of [K+]o [46], electrical stimulation [50], additions of histamine [51], angiotensin II [52], thromboxane A2 [53, 54], oxytocin [55, 56], agonists of α-adrenergic [46, 57-59] and purinergic receptors [60] is caused by Cl–i-dependent hyperpolarization and suppression of the activity of voltage-gated L-type Ca2+ channels (Fig. 3).
Fig. 3. Mechanism of the involvement of ubiquitous isoforms of Na+,K+,2Cl– cotransport (NKCC1) in the regulation of smooth muscle contraction. NKCC1 mediates increase in [Cl–]i, depolarization of SMC whose sarcolemma is abundant in anion channels, opening of voltage-gated Ca2+ channels, elevation of [Ca2+]i, and contraction. Bumetanide and other inhibitors of NKCC1 decrease [Cl–]i that, in turn, leads to hyperpolarization, the closure of voltage-gated Ca2+ channels, and relaxation of SMCs.
Myogenic tonus (response) is unique property of small (<100-200 µm) blood vessels to decrease rather than increase their inner diameter in response to elevated intraluminal pressure. Both kinetic and the attitude of myogenic response is different in blood vessels of different origin. Importantly, myogenic response in blood vessels from the brain, skeletal muscle, and renal afferent arteriole provides constant blood supply of these tissues independently of the changes in the systemic blood pressure [61-63].
It was reported that bumetanide decreases the myogenic tonus of mesenteric arteries [64] and completely abolished myogenic response of renal afferent arteriole [54]. We demonstrated that the inhibitory action of bumetanide but not the L-type Ca2+ channel blocker nicardipine on myogenic response as well as on contraction triggered by α-adrenergic stimulation is absent in mesenteric arteries from NKCC1–/– mice [64]. Because NKCC2 is not expressed in SMCs, these data demonstrated for the first time that bumetanide and other loop diuretics inhibit contraction and myogenic response of vascular SMCs via their interaction with NKCC1, i.e. a ubiquitous isoform of Na+,K+,2Cl– cotransporter.
Synaptic transmission. Neuron–neuron interactions are controlled by neurotransmitters via regulation of transduction of electrical signals. Excitatory and inhibitory neurotransmitters lead to depolarization and hyperpolarization of postsynaptic membrane, respectively. Thus, for example, ionotropic glutamate receptors and acetylcholine receptors cause depolarization of postsynaptic membrane via an increment of ion current mediated by ion channels permeable for Na+ and Ca2+. In contrast, hyperpolarization results from the increment of the permeability of K+ channels triggered by activation of metabotropic acetylcholine receptors. Unlike the above-listed neurotransmitters, gamma-aminobutyric acid (GABA) increases permeability for Cl– and other low molecular weight anions via its interaction with ionotropic GABAA receptors. The direction of net flux mediated by these receptors is determined by transmembrane chloride gradient and electrical potential of the postsynaptic membrane. In case RT/F·ln([Cl–]o/[Cl–]in) < Em, the net chloride flux will be directed into the cells, which leads to hyperpolarization and attenuation of neuronal activity. An elevation of [Cl–]in changes the direction of net chloride flux, and in this case activation of GABAA leads to elevation of neuronal activity (Fig. 4).
Fig. 4. Mechanisms of the implication of NKCC1 and KCC2 in the function of GABAA receptors. The differences in the symbol size reflect the differences in the activity of ion carriers involved in [Cl–]i regulation. The equation shows conditions when net anion flux through GABAA receptors is absent.
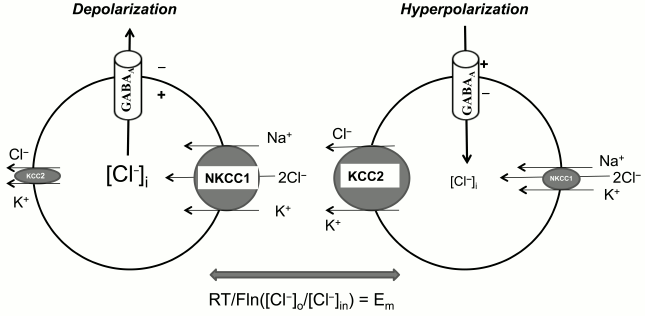
Keeping these data in mind, it might be assumed that the ratio of activity of NKCC1 and KCC2, providing inwardly and outwardly directed Cl– fluxes, respectively, plays a key role in the function of GABAA receptors. Indeed, the attenuation of NKCC1 activity on the background of elevated KCC2 is the major mechanism of the alteration of the functional properties of GABAA receptors in central neuronal system (CNS) of mammals during ontogenesis: GABAA receptors function as activatory receptors in the prenatal stage but become inhibitory in few days after birth (for review see [7, 8, 28]). It was demonstrated that the content of WNK3 RNA in brain neurons is correlated with KCC2 expression, achieving its maximal values 21 days after birth [30]. These data suggest that expression of this kinase determines the drastic age-dependent changes in the functioning of GABAA receptors.
Cell volume regulation. Animal cells maintain their volume with an accuracy of 1-2% by means of systems providing inwardly and outwardly directed fluxes of monovalent ions and organic osmolytes termed as regulatory volume increase (RVI) and regulatory volume decrease (RVD), respectively [65, 66]. In early experiments with mammalian erythrocytes, it was shown that cell shrinkage and swelling result in activation of NKCC and KCC [67-69]. Later, we demonstrated that in vascular SMCs subjected to hyperosmotic shrinkage, RVI is caused by activation of NKCC [15]. It should be emphasized that under isoosmotic conditions the inhibitor of this carrier bumetanide did not significantly affect the volume of human lung fibroblasts [70]. This means that in several cell types in the absence of external stimuli NKCC has a minor impact on the generation of net osmolyte fluxes as compared with Na+,K+-ATPase and other ion transport systems. Indeed, recently we reported that addition of bumetanide resulted in ~2-fold elevation of human lung fibroblast volume only after inhibition of the Na+,K+-ATPase by ouabain [70]. This observation indicates that dissipation of the transmembrane gradient of monovalent cations evoked by Na+,K+-ATPase inhibition results in generation of inwardly-directed ion flux mediated by NKCC1, the only isoforms of NKCC expressed in these cells. The functional consequences of disturbances of the cell volume regulatory machinery are considered elsewhere [65, 71, 72]. The pathophysiological implications of cell volume disturbances are demonstrated under analysis of astrocyte swelling evoked by brain hypoxia (see section “Role in Pathogenesis of Arterial Hypertension”).
ROLE IN PATHOGENESIS OF ARTERIAL HYPERTENSION
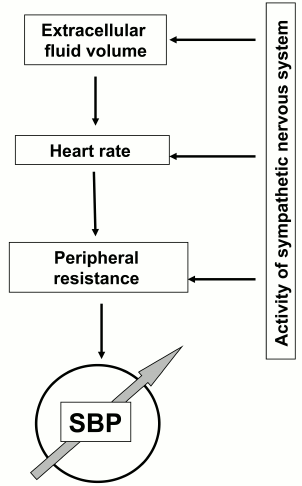
Elevation of systemic blood pressure has been documented in 25% of adults and is a major risk factor for complications including stroke, heart failure, and renal disease resulting in premature death [73]. In 2001, the USA alone spent 54 billion dollars for the treatment of hypertension [74]. The general thermodynamic model predicts that elevation of systemic blood pressure can be a consequence of the increase in peripheral resistance of blood flow, heart rate, and the extracellular volume (Fig. 5). In turn, the above-listed parameters are under the control of dozen of hormones and neurotransmitters and sympathetic nerve system (SNS) affecting heart, blood vessels, and renal function [75]. The involvement of these systems in the pathogenesis of hypertension is supported by a set of the major pharmacological tools used for the normalization of blood pressure. The list of antihypertensive drugs include diuretics decreasing salt reabsorption in the kidney via inhibition of NKCC2 (furosemide) and NCC (thiazides), vasodilators including inhibitors of angiotensin-converting enzyme (ramipril), antagonists of angiotensin II (lozartan) and α-adrenergic (doxazosin) receptors, Ca2+ channel blockers (amlodipin), as well as antagonists of β-adrenergic receptors (atenol) that decrease activity of the SNS [73].
Fig. 5. Major systems involved in elevation of systemic blood pressure (SBP).
In several forms of systematic hypertension, servomechanisms underlying long-term elevation of blood pressure are well-documented. This is the case of hypertension caused by adrenal tumors and renal insufficiency as well as the set of monogenous hypertension caused by single gene mutations. However, these forms of disease found in less than 5% of patients with elevated blood pressure are combined by the common name of secondary hypertension. In the rest of patients with primary or essential hypertension, the mechanisms of blood pressure elevation remain unknown [76]. In this section, we summarize data on the involvement of CCCs in the pathogenesis of hypertension.
Monogenous forms of secondary hypertension and hypotension. Monogenous forms of hypertension and hypotension identified so far are caused by mutations of genes involved in regulation of extracellular fluid volume by renal epithelial cells [77, 78]. This observation is consistent with the data showing a key role of the kidney in long-term maintenance of elevated blood pressure demonstrated by Artur Guyton [79]. Among monogenous forms of hypertension and hypotension, three types of mutations result in altered function of CCCs. Thus, loss-of-function mutations of NKCC2 and NCC detected in patients with Barter type I and Gitelman syndrome, respectively, lead to attenuated salt reabsorption in the thick ascending limb of Henle’s loop and distal nephron that, in turn, decreases extracellular fluid volume and systemic blood pressure [80, 81]. In both diseases inherited in accordance with classical Mendelian genetics, hypotension is accompanied by hypokalemia and alkaloidosis, i.e. universal markers of decrease reabsorption of salt in the distal nephron. In contrast, in patients with PHAII, also known as Gordon’s syndrome, hypertension is caused by mutations within WNK1 and WNK4 resulting in activation of NCC, increased sodium reabsorption, and hyperkalemia [29]. Data on the involvement of WNKs in CCC regulation are considered in the section “Regulation of CCC Activity”.
Primary hypertension. Unlike monogenous forms of secondary hypertension, elevation of blood pressure in primary hypertension is a consequence of a complex combination of inherited traits and several environmental factors including limited physical activity, obesity, smoking, and excess consumption of salt and alcohol. Inherited traits are probably caused by altered function of 4-5 genes whose combination can be different even within the same human population [82]. This feature emphasizes the mosaic origin of the pathogenesis of essential hypertension as first noted by Pickering [83].
In the middle of the 1970s, we initiated a search for inherited factors of primary hypertension by investigation of the activity of plasma membrane ion transporters in spontaneously hypertensive rats (SHR) and Milan hypertensive strain (MHS) known as the most adequate experimental models of human essential hypertension. We found that elevated permeability of the erythrocyte membrane for monovalent cations shown in early studies [84, 85] is caused by augmented activity of NKCC (for review see [86-90]). Later, the involvement of this carrier in the pathogenesis of primary hypertension was supported by the following data. (i) In erythrocytes of the first generation of hybrids obtained by crossing of MHS and Milan normotensive strain MNS (F1 MHS × MNS) and subjected to X-ray irradiation, NKCC activity was increased after transplantation of bone marrow from MHS but not from MNS [91]. These results indicate that increased activity of NKCC is an inherited property of erythrocytes from MHS rather than a consequence of long-term elevation of blood pressure. (ii) In erythrocytes of the second generation hybrids obtained by crossing of SHR and normotensive Kyoto–Wistar rats (F2 SHR × WKY) as well as in F2 MHS × MNS hybrids, NKCC activity positively correlated with blood pressure [91, 92]. (iii) Several researchers demonstrated decreased blood pressure in NKCC1–/– knockout mice [93-95]. This finding, however, was not confirmed by Kim et al. [96]. The reasons underlying this discrepancy remain unknown. (iv) Administration of bumetanide, i.e. a potent inhibitor of Na+,K+,2Cl– cotransport, decreased blood pressure in wild-type but not in NKCC1–/– mice [58].
Because NKCC1 is the only isoform of Na+,K+,2Cl– cotransporters identified in erythrocytes and SMCs, the data considered above indicate that at least in these experimental models of human primary hypertension augmented activity of this carrier contributes to activation of servomechanisms of long-term elevation of systemic blood pressure. These findings also raise the question of the mechanisms of this phenomenon. Data considered in the section “Physiological Significance” indicated that these mechanisms may involve NKCC1-mediated regulation of [Cl–]in that, in turn, affects SMC contraction and SNS activity. Indeed, it was shown that inhibitory action of bumetanide on the contraction of mesenteric arteries evoked by activation of α-adrenergic receptor is increased in SHR compared to normotensive controls [97, 98]. Due to methodological problems, data on the activity of NKCCs in freshly isolated SMCs in primary hypertension are limited to a few publications (for review see [86-90]). It was shown, however, that the content of NKCC1 mRNA and immunoreactive protein is increased in aorta and heart from SHR [97]. The possible mechanism of this phenomenon is considered below.
As mentioned above, along with abnormalities of the ion-transport system in kidney and blood vessels, alterations in the CNS results in elevation of SNS tone that, in turn, leads to elevation of systemic blood pressure via its impact on the cardiovascular system and kidneys (Fig. 6) [99-102]. This hypothesis is consistent with numerous reports on activation of the SNS in patients with essential hypertension [99] and SHR [103], as well as with data on the major role in SNS activation of the hypothalamic paraventricular nuclei (PVN) [104], whose hyperreactivity in primary hypertension is well-documented [105, 106].
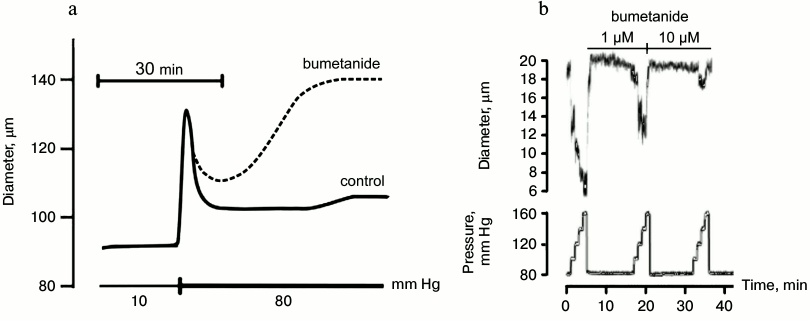
Fig. 6. Effects of bumetanide on myogenic response of mouse mesenteric arteries (a) and afferent arteriole of rat kidney (b) [54, 64].
It is known that excitability of presynaptic neurons in the PVN is activated by excitatory glutamatergic neurons and suppressed by inhibitory GABAergic neurons, respectively [107]. It was also shown that activity of GABAergic neurons is decreased in the PVN of SHR [107, 108]. As mentioned above, the relationship between inhibitory and activatory actions of GABAA receptors is determined by intracellular chloride concentration, which is under the control of the ratio of NKCC1 and KCC2 activities (Fig. 4). Investigations performed at the University of Texas demonstrated that the electrical potential, at which ion current mediated by GABAA receptors (EGABA) is reversible, shifted to positive values in the PVN of SHR by 15 mV compared to the normotensive control, which corresponds to 2-fold elevation of [Cl–]i [109]. This difference as well as decreased inhibitory activity of GABAergic neurons in SHR was abolished by addition of low doses of bumetanide but not furosemide. These results suggested that augmented [Cl–]i in SHR neurons is caused by activation of NKCC1 rather than inhibition of KCC2. This conclusion is an agreement with elevation of NKCC1 mRNA and immunoreactive protein in the PVN of SHR without any changes in KCC2 content [109].
Mechanisms of elevation of NKCC1 activity in primary hypertension remains poorly investigated, which probably reflects the polygenic and mosaic origin of this disease as well as diverse mechanisms of regulation of the activity and expression of CCCs. Thus, for example, elevation of [Ca2+]i activates whereas cAMP inhibits this carrier in SMCs [13, 57, 110]. Numerous investigations have documented abnormal activity of both signaling systems in primary hypertension [111, 112]. A key role of WNK, SPAK, and OSR1 kinases in regulation of several CCCs including NKCC1, NKCC2, and NCC were considered in the section “Regulation of CCC Activity” and their implication in the regulation of blood pressure in monogenous hypertension [113, 114] and in genetically engineered animals [115] has been documented. Bergaya and coworkers reported that both phosphorylation of NKCC1 and increment of blood pressure evoked by activation of α-adrenergic receptors are decreased in Wnk+/– mice [116]. The bumetanide-sensitive component of vessel contraction was also attenuated in SPAK knockout mice [117]. It should be noted, however, that in contrast to monogenous hypertension, there is no evidence for mutations of genes encoding CCCs or the WNK/SPAK/OSR1 regulatory pathway in primary hypertension.
Recent investigations suggest that CCC-mediated abnormalities of ion transport have epigenetic origin. Indeed, it was shown that the content of NKCC1 mRNA and protein content is increased in aorta, heart, and PVN neurons from rats with spontaneous hypertension [97, 109]. In case of SHR aorta and heart, increased expression of the carrier is accompanied by attenuated methylation of the NKCC1 gene promoter [97]. Importantly, methylation of the NKCC1 promoter exhibited age-dependent increase in normotensive rats, without any changes in SHR [98]. It was also shown that activity of DNA methyltransferase 3B (DNTB3B) is three-fold higher in 18-week-old normotensive rats as compared to age-matched SHR. These results assume that in this experimental model of primary hypertension hypomethylation of NKCC1 promoter is caused by decreased activity of DNTB3B that, in turn, leads to augmented NKCC1 expression, increment of [Cl–]i, depolarization and contraction of SMCs, increased vascular tonus resistance, and blood pressure elevation.
The role of epigenetic factors in augmented expression of NKCC1 in neurons of the PVN of SHR, controlling activity of the SNS, remains unknown. It was shown, however, that in these cells from SHR NKCC1 is highly glycosylated [109]. The authors assume that this phenomenon contributes to increased content of membrane-bound protein, i.e. the fraction of the carrier providing Na+,K+,Cl– cotransport.
Complications caused by elevation of systemic blood pressure. The major cause of the premature death in patients with essential hypertension is damage of the target organs such as brain vessels and kidney caused by elevation of local blood pressure [118]. In the brain, the long-term elevation of systemic blood pressure increases the probability of irreversible damage of the blood flow that results in stroke, whereas in kidney it leads to structural changes in the nephron, changes of salt–water homeostasis, and proteinuria [73].
Because Rbf ~ 1/d4 (where Rbf is the blood flow resistance and d is the inner diameter of a vessel) [119], the role of myogenic tonus as a nature-created tool for protection of target organs from elevation of systemic arterial pressure has been subjected to investigation by numerous research groups [120]. It was shown that chronic suppression of myogenic response in patients with essential hypertension as a consequence of hypertrophy of a vessel wall leads to attenuation of its sensitivity to changes in intraluminal pressure. As a results of these changes, an increment of systemic blood pressure is transferred to microcirculation beds incorporated in the brain, heart, retina, and kidney that causes irreversible changes in the structure and function in these and other target organs of hypertension [121, 122]. Keeping this in mind, the action of antihypertensive drugs on myogenic response should be subjected to detailed investigations.
Nifedipine, amlodipine, diltiazem, and other inhibitors of voltage-gated L-type Ca2+ channels evoke prolonged attenuation of systemic blood pressure, taking the second position on the antihypertensive drug market. It is generally assumed that the blood–brain barrier protects brain vessels from their action. However, such kind of protection is absent in the kidney and other target organs. Considering this, it should be noted that during the last two decades an extended number of clinical studies showed the development of renal insufficiency and heart failure in patients with essential hypertension subjected to prolonged treatment with Ca2+ blockers [122-126]. Both in vivo and in vitro studies demonstrated that development of renal insufficiency is due to suppression by Ca2+ blockers of myogenic response in renal afferent arteriole. Indeed, in contrast to mesenteric arteries possessing slowly developing myogenic response resulting in partial normalization of inner diameter after elevation of intraluminal pressure by 70 mm Hg (Fig. 6a), myogenic response of renal afferent arteriole develops during the first 10-100 msec after elevation of intraluminal pressure by 20 mm Hg and leads to attenuation of inner diameter by 20-30% (Fig. 6b). Unlike SMCs of afferent arteriole abundant with L-type Ca2+ channels, their content in renal efferent arteriole is negligible [127]. Considering this, it can be assumed that in spite of attenuation of systemic blood pressure, inhibition of myogenic response by Ca2+ antagonists increases blood pressure in the local microcirculatory bed of the kidney. Indeed, it was shown that these drugs increase rather that decrease the glomerular filtration rate (for review, see [128-130]).
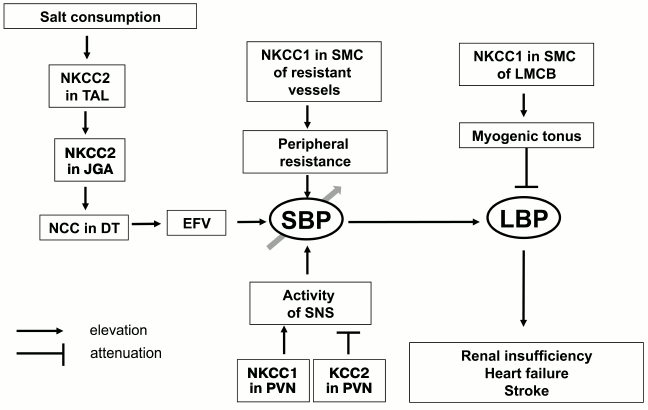
For investigation of the role of myogenic response in kidney function, Loutzenhiser and coworkers employed isolated perfused kidney. This experimental approach allowed them to study renal microcirculation in the absence of its modulation by the juxtaglomerular apparatus [54]. With this model, it was shown that bumetanide completely suppresses myogenic response of afferent arteriole in the rat kidney (Fig. 6b). These results considered together with the absence of myogenic response in Nkcc1–/– mice [64] suggested that augmented activity of NKCC1 documented in the study of erythrocytes from SHR, MHS, and patients with essential hypertension protects the kidney from damage by prolonged elevation of systemic blood pressure, whereas chronic administration of furosemide and other NKCC inhibitors accelerates renal insufficiency and proteinuria [89, 90, 131]. In other words, high activity of NKCC1 in SMCs of the afferent arteriole maintains constant renal blood flow even after elevation of systemic blood pressure caused by activation of this carrier in mesenteric arteries and other vessels, contributing to regulation of peripheral resistance (Fig. 7). This hypothesis is consistent with four-fold increase of renal complications in Blacks with hypertension possessing up to three-fold attenuation of NKCC activity in erythrocytes compared with age-matched hypertensive Caucasians [131, 132]. The relative contribution of Ca2+ channels and NKCC1 in the myogenic response of coronary and CNS microcirculatory beds remains unexplored.
Fig. 7. Scheme showing implication of cation–chloride cotransporters in the pathogenesis of essential hypertension and its cardiovascular and renal complications. TAL, thick ascending limb of Henle; JGA, juxtaglomerular apparatus; DT, distal tubule; EFV, extracellular fluid volume; SNS, sympathetic nervous system; PVN, paraventricular nuclei; LMCB, local microcirculatory bed; SBP, systemic blood pressure; LBP, local blood pressure. For other abbreviations, see text.
Ischemia is the major consequence of even brief disturbance of blood circulation in the brain vessels that leads to irreversible damage of neuronal function. It was shown that attenuation of oxygen partial pressure results in astrocyte swelling that, in turn, leads to release of glutamate and other neurotransmitters triggering massive Ca2+ influx into neurons and their death [133, 134]. It was shown that bumetanide suppresses astrocyte swelling, neurotransmitter release [135], and the death of neurons in the hippocampus subjected hypoxia and hypoglycemia [136]. Moreover, both [K+]o-induced swelling and release of neurotransmitters evoked by ischemia were sharply decreased in astrocytes from NKCC1–/– mice [137], thus indicating activation of NKCC1 as a mechanism of astrocyte swelling. We were first to report that NKCC activity in SMCs decreases on elevation of [HCO3–]o [138]. Thus, activation of NKCC1 may in hypoxic conditions be caused by acidosis-mediated attenuation of [HCO3–] in cerebrospinal fluid. Hypoxia is also accompanied by sharp attenuation of intracellular ATP content and inhibition of the Na+,K+-ATPase [139]. As shown in our recent studies, inhibition of the Na+,K+-ATPase changes the direction of NKCC1-mediated net ion fluxes and evokes cell swelling [70]. Additional experiments should be performed for evaluation of the relative impact of Na+,K+-pump and NKCC1 in regulation of astrocyte volume under ischemic conditions.
CONCLUSION
The data considered above lead us to several conclusions. First, the transport of monovalent ions across epithelial cells as well regulation of cell volume and intracellular chloride concentration are major functions of CCCs. In renal epithelial cells, reabsorption of salt and osmotically-obliged water is provided by NKCC2 and NCC, whereas cell volume is under tissue-specific control of all seven identified CCCs. In smooth muscle cells, NKCC1 has a major impact on [Cl–]i, whereas in neurons this parameter is under the control of NKCC1 and KCC2.
Second, regulation of CCC activity by diverse stimuli, including cell volume changes, is provided by serine–threonine kinases WNK, SPAK, and OSR1 and phosphoprotein phosphatases PP1 and PP2B. There is no evidence for direct impact on CCC phosphorylation of cAMP-, cGMP-, diacylglycerol-, and Ca2+-sensitive protein kinase.
Third, in several rare monogenous disorders, blood pressure changes are caused by mutations of NKCC2 and NCC that lead to altered reabsorption of salt and osmotically-obliged water in renal epithelial cells. In primary hypertension, NKCC1 activity is increased in vascular SMCs and neurons of the PVN, which leads to elevation of peripheral resistance in the systematic circulation and activation of SNS, respectively. In both cases, these abnormalities are caused by elevation of [Cl–]i and plasma membrane depolarization.
Fourth, furosemide and other loop diuretics decreases systemic blood pressure via inhibition of NKCC2 in the thick ascending limb of Henle’s loop and NKCC1 in SMCs of resistant vessels. However, the same compounds suppress the myogenic response of SMCs in the microcirculatory beds of kidney and brain, thus increasing the risk of renal and cerebral complications.
In spite of progress in the understanding of the molecular mechanisms of the implication of CCC in the regulation of cellular functions in physiological and pathophysiological conditions, several key questions remain unanswered. (i) What is a molecular origin of cell volume sensor(s) transducing signal to volume-sensitive WNKs? (ii) Is the functional significance of SPAK/OSR1/WNK signaling limited to regulation of CCCs? What is the mechanism of SPAK/OSR1-independent regulation of CCCs by WNKs? (iii) In cultured SMCs as well as in isolated blood vessels, NKCC1 is activated by phenylephrine, angiotensin II, and other vasoconstrictors and inhibited by vasodilators whose action is mediated by cAMP [13, 57]. Does NKCC1 contribute to regulation of vascular tone by these compounds? (iv) Do rennin, angiotensin, aldosterone, and other hormones involved in blood pressure regulation affect SPAK/OSR1/WNK signaling? (v) A key role of NKCC1 in regulation of myogenic response of SMCs in renal afferent arteriole is well-documented. What is the relative contribution of NKCC1 in regulation of myogenic response in SMCs of the microcirculatory beds of the brain and other target-organs of hypertension? (vi) Experiments in vitro demonstrated that loop diuretics can be used as a pharmacological tool augmenting inhibitory function of GABAA receptors. Do these compounds penetrate through the blood–brain barrier? In other words, can we use these drugs as well as modulators of SPAK/OSR1/WNK-mediated signaling for regulation of SNS activity?
These questions are important for better understanding of the crosstalk of biochemical signaling systems and systems involved in regulation of ion homeostasis and cell volume. We firmly believe that they are also important for the development of novel antihypertensive drugs lacking side effects caused by inhibition of CCCs in epithelial cells and myogenic tonus in microcirculatory beds. Indeed, currently used high-ceiling diuretics exhibit the same affinity for NKCC1 and NKCC2. Because the apparent affinity for furosemide and bumetanide is in proportion to carrier activity [140], inhibition of highly active NKCC2 and diuretic action of this compounds is much greater than their vasodilatory effects. It is also important to mention another side effect of these drugs: their prolonged administration resulted in the development of deafness due to inhibition of NKCC1 in epithelial cells of the inner ear [141, 142]. Thus, the development of tissue-specific inhibitors of SPAK and other regulators of CCCs should be considered as potential targets for novel antihypertensive drugs.
This work was supported by grants from the Canadian Institutes for Health Research (MOP-81392, MOP-81392), the Heart and Stroke Foundation of Canada, the Kidney Foundation of Canada, the Russian Foundation for Fundamental Research (09-0073/04, 14-04-31705), Federal Program 2009-2013 of the Russian Federation for Research Staff and Innovations.
REFERENCES
1.Hediger, M. A., Romero, M. F., Peng, J.-B., Rolfs,
A., Takanaga, H., and Bruford, E. A. (2004) The ABCs of solute
carriers: physiological, pathophysiological and therapeutic
implications of human membrane transport protein, Pfluger Arch.
Europ. J. Physiol., 447, 465-468.
2.Gamba, G. (2005) Molecular physiology and
pathophysiology of electroneutral cation-chloride cotransporters,
Physiol. Rev., 85, 423-493.
3.Orlov, S. N., and Mongin, A. A. (2007) Salt sensing
mechanisms in blood pressure regulation and hypertension, Am. J.
Physiol. Heart Circ. Physiol., 293, H2039-H2053.
4.Markadieu, N., and Delpire, E. (2014) Physiology
and pathophysiology of SLC12A1/2 transporters, Pfluger Arch. Europ.
J. Physiol., 466, 91-105.
5.Garzon-Mudvi, T., Schiapparelli, P., ap Rhys, C.,
Guerrero-Cazares, H., Smith, C., Kim, D.-H., Kone, L., Farber, H., Lee,
D. Y., An, S. S., Levchenko, A., and Quinones-Hinojosa, A. (2012)
Regulation of brain tumor dispersal by NKCC1 through a novel role in
focal adhesion regulation, PLoS Biol., 10, e1001320.
6.Chen, Y.-F., Chou, C.-Y., Ellory, J. C., and Shen,
M.-R. (2010) The emerging role of KCl cotransport in tumor biology,
Am. J. Transl. Res., 2, 345-355.
7.Kahle, K. T., Staley, K. J., Nahed, B. V., Gamba,
G., Hebert, S. C., Lifton, R. P., and Mount, D. B. (2008) Roles of the
cation-chloride cotransporters in neurological disease,
Nature Clin. Pract. Neurol., 4, 490-503.
8.Loscher, W., Puskarjov, M., and Kaila, K. (2013)
Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for
novel antiepileptic and antiepileptogenic treatments,
Neuropharmacology, 69, 62-74.
9.Rust, M. B., Alper, S. L., Rudhard, Y., Shmukler,
B. E., Vicente, R., Brugnara, C., Trudel, M., Jentsch, T. J., and
Hubner, C. A. (2007) Disruption of erythroid K-Cl cotransporters alters
erythrocyte volume and partially rescues erythrocyte dehydration in SAD
mice, J. Clin. Investig., 117, 1708-1717.
10.Rinehart, J., Gulcicek, E. E., Joiner, C. H.,
Lifton, R. P., and Gallagher, P. G. (2010) Determinants of erythrocyte
hydration, Curr. Opin. Haematol., 17, 191-197.
11.Lang, F., and Voelkl, J. (2013) Therapeutic
potential of serum and glucocorticoid inducible kinase inhibition,
Expert Opin. Investig. Drugs, 22, 701-714.
12.Gagnon, F., Hamet, P., and Orlov, S. N. (1999)
Na+,K+ pump and Na+-coupled ion
carriers in isolated mammalian kidney epithelial cells: regulation by
protein kinase C, Canad. J. Physiol. Pharmacol., 77,
305-319.
13.Orlov, S. N., Resink, T. J., Bernhardt, J., and
Buhler, F. R. (1992) Na+-K+ pump and
Na+-K+ cotransport in cultured vascular smooth
muscle cells from spontaneously hypertensive rats: baseline activity
and regulation, J. Hypertens., 10, 733-740.
14.Smith, J. B., and Smith, L. (1987)
Na+/K+/Cl– cotransport in
cultured vascular smooth muscle cells: stimulation by angiotensin II
and calcium ionophores, inhibition by cyclic AMP and calmodulin
antagonists, J. Membr. Biol., 99, 51-63.
15.Orlov, S. N., Tremblay, J., and Hamet, P. (1996)
Cell volume in vascular smooth muscle is regulated by
bumetanide-sensitive ion transport, Am. J. Physiol., 270,
C1388-C1397.
16.Adragna, N., White, R. E., Orlov, S. N., and
Lauf, P. K. (2000) K-Cl cotransport in vascular smooth muscle and
erythrocytes: possible implication in vasodilation, Am. J.
Physiol., 278, C381-C390.
17.Gagnon, F., Orlov, S. N., Tremblay, J., and
Hamet, P. (1998) Complete inhibition of
Na+,K+,Cl– cotransport in
Madin–Darby canine kidney cells by PMA-sensitive protein kinase
C, Biochim. Biophys. Acta, 1369, 233-239.
18.Gagnon, F., Dulin, N. O., Tremblay, J., Hamet,
P., and Orlov, S. N. (1999) ATP-induced inhibition of
Na+,K+,Cl– cotransport in
Madin–Darby canine kidney cells: lack of involvement of known
purinoceptor-coupled signaling pathways, J. Membr. Biol.,
167, 193-204.
19.Orlov, S. N., Dulin, N. O., Gagnon, F., Gekle,
M., Douglas, J. G., Schwartz, J. H., and Hamet, P. (1999) Purinergic
regulation of Na+,K+,Cl–
cotransport and MAP kinases is limited to C11-MDCK cells
resembling intercalated cells from collecting ducts, J. Membr.
Biol., 172, 225-234.
20.Lytle, C., and Forbush III, B. (1992) The Na-K-Cl
cotransport protein of shark rectal gland. II. Regulation by direct
phosphorylation, J. Biol. Chem., 267, 25438-25443.
21.Grubb, B. R., Pace, A. J., Lee, E., Koller, B.
H., and Boucher, R. C. (2001) Alterations in airway ion transport in
NKCC1-deficient mice, Am. J. Physiol. Cell Physiol., 281,
C615-C623.
22.Lytle, C. (1997) Activation of avian erythrocyte
Na-K-Cl cotransport by cell shrinkage, cAMP, fluoride, and calyculin A
involves phosphorylation at common sites, J. Biol. Chem.,
272, 15069-15077.
23.Lytle, C. (1998) A volume-sensitive protein
kinase regulates the Na-K-Cl cotransporter in duck red blood cells,
Am. J. Physiol., 274, C1002-C1010.
24.Lytle, C., and McManus, T. (2002) Coordinate
modulation of Na-K-2Cl cotransport and K-Cl cotransport by cell volume
and chloride, Am. J. Physiol. Cell Physiol., 283,
C1422-C1431.
25.Kahle, K. T., Rinehart, J., Ring, A., Gimenez,
I., Gamba, G., Hebert, G., and Lifton, R. P. (2006) WNK protein kinase
modulate cellular Cl-flux altering the phosphorylation state of the
Na-K-Cl and K-Cl cotransporters, Physiology, 21,
326-335.
26.Dowd, B. F., and Forbush, B. (2003) PASK
(proline-alanine-rich STE20-related kinase), a regulatory kinase of the
Na-K-Cl cotransporter (NKCK1), J. Biol. Chem., 278,
27347-27353.
27.Piechotta, K., Lu, J., and Delpire, E. (2002)
Cation chloride cotransporters interact with the stress-related kinases
Ste20-related proline-alanine-rich kinase (SPAK) and oxidative response
1 (OSR1), J. Biol. Chem., 277, 50812-50819.
28.Delpire, E., and Austin, T. M. (2010) Kinase
regulation of Na+-K+-2Cl–
cotransport in primary neurons, J. Physiol. (L),
588, 3365-3373.
29.Wilson, F. H., Disse-Nicodeme, S., Choate, K. A.,
Ishikawa, K., Nelson-Williams, C., Desitter, I., Gunel, M., Milford, D.
V., Lipkin, G. W., Achard, J. M., Feely, M. P., Dussil, B., Berland,
Y., Unwin, R. J., Mayan, H., Simon, D. B., Farfel, Z., Jeunemaitre, X.,
and Lifton, R. P. (2001) Human hypertension caused by mutations in WNK
kinases, Science, 293, 1107-1112.
30.Kahle, K. T., Rinehart, J., de los Heros, P.,
Louvi, A., Meade, P., Vazquez, N., Hebert, S. C., Gamba, G., Gimenez,
I., and Lifton, R. P. (2005) WNK3 modulates transport of Cl–
in and out of cells: implications for control of cell volume and
neuronal excitability, Proc. Natl. Acad. Sci. USA, 102,
16783-16788.
31.Rinehart, J., Kahle, K. T., de los Heros, P.,
Vazquez, N., Meade, P., Wilson, F. H., Hebert, S. C., Gimenez, I.,
Gamba, G., and Lifton, R. P. (2005) WNK3 kinase is a positive regulator
of NKCC2 and NCC, renal cation-Cl– cotransporters
required for normal blood pressure homeostasis, Proc. Natl. Acad.
Sci. USA, 102, 16777-16782.
32.De los Heros, P., Kahle, K. T., Rinehart, J.,
Bobadilla, N. A., Vazquez, N., San Cristobal, P., Mount, D. B., Lifton,
R. P., Hebert, S. C., and Gamba, G. (2006) WNK3 bypasses the tonicity
requirement for K-Cl cotransporter activation via phosphatase-dependent
pathway, Proc. Natl. Acad. Sci. USA, 103, 1976-1981.
33.Delpire, E. (2009) The mammalian family of
sterile 2p-like protein kinases, Pfluger Arch. Europ. J.
Physiol., 458, 953-967.
34.Piechotta, K., Garbarini, N. J., England, R., and
Delpire, E. (2003) Characterization of the interaction of the stress
kinase SPAK with the Na+-K+-2Cl–
cotransporter in the nervous system: evidence for a scaffolding
role of the kinase, J. Biol. Chem., 278, 52848-52856.
35.Gagnon, K. B., England, R., and Delpire, E.
(2006) Volume sensitivity of cation-Cl– cotransporters
is modulated by the interaction of two kinases: SPAK and WNK4, Am.
J. Physiol. Cell Physiol., 290, C134-C142.
36.Ponce-Coria, J., San-Cristobal, P., Kahle, K. T.,
Vazquez, N., Pacheco-Alvarez, D., de los Heros, P., Juarez, P., Munoz,
E., Michel, G., Bobadilla, N. A., Gimenez, I., Lifton, R. P., Hebert,
S. C., and Gamba, G. (2008) Regulation of NKCC2 by a chloride-sensing
mechanism involving the WNK3 and SPAK kinases, Proc. Natl. Acad.
Sci. USA, 105, 8458-8463.
37.Delpire, E., and Gagnon, K. B. (2007) Genome-wide
analysis of SPAK/OSR1 binding motifs, Physiol. Genom.,
28, 223-231.
38.Richardson, C., and Alessi, D. (2008) The
regulation of salt transport and blood pressure by the WNK-SPAK/OSR1
signaling pathway, J. Cell Sci., 121, 3293-3304.
39.Castaneda-Bueno, M., Cervantes-Perez, L. G.,
Vazquez, N., Uribe, N., Kantesaria, S., Morla, L., Bobadilla, N. A.,
Alessi, D. R., and Gamba, G. (2012) Alteration of the renal
Na+/Cl– cotransporter by angiotensin II is
a WNK4-dependent process, Proc. Natl. Acad. Sci. USA,
109, 7929-7934.
40.Castaneda-Bueno, M., and Gamba, G. (2012)
Mechanisms of sodium-chloride cotransporter modulation by angiotensin
II, Curr. Opin. Nephrol. Hypertens., 21, 516-522.
41.Darman, R. B., and Forbush, B. (2002) A
regulatory locus of phosphorylation in the N-terminus of the Na-K-Cl
cotransporter, NKCC1, J. Biol. Chem., 277,
37542-37550.
42.Vitari, A. C., Thastrup, J., Rafigi, F. H., Deak,
M., Morrice, N. A., Karlsson, H. K., and Alessi, D. R. (2006)
Functional interactions of the SPAK/OSR1 kinases with their upstream
activator WNK1 and downstream substrate NKCC1, Biochem. J.,
397, 223-231.
43.Alvarez-Leefmans, F. J. (2001) Intracellular
chloride regulation, in Cell Physiology Source Book. A Molecular
Approach (Sperelakis, N., ed.) Academic, San Diego, pp.
301-318.
44.Chipperfield, A. R., and Harper, A. A. (2001)
Chloride in smooth muscle, Progr. Biophys. Mol. Biol.,
74, 175-221.
45.Davis, J. P. L., Chipperfield, A. R., and Harper,
A. A. (1993) Accumulation of intracellular chloride by (Na-K-Cl)
cotransport in rat arterial smooth muscle is enhanced in
deoxycorticosterone acetate (DOCA)/salt hypertension, J. Mol. Cell.
Cardiol., 25, 233-237.
46.Anfinogenova, Y. J., Baskakov, M. B., Kovalev, I.
V., Kilin, A. A., Dulin, N. O., and Orlov, S. N. (2004)
Cell-volume-dependent vascular smooth muscle contraction: role of
Na+,K+,2Cl– cotransport,
intracellular Cl– and L-type Ca2+ channels,
Pflug. Arch., 449, 42-55.
47.Barthelmebs, M., Stephan, D., Fontaine, C.,
Grima, M., and Imbs, J. L. (1994) Vascular effects of loop diuretics:
an in vivo and in vitro study in the rat,
Naunyn-Schmiedebergs Arch. Pharmacol., 349, 209-216.
48.Lavallee, S. L., Iwamoto, L. M., Claybaugh, J.
R., Dressel, M. V., Sato, A. K., and Nakamura, K. T. (1997)
Furosemide-induced airway relaxation in guinea pigs: relation to
Na-K-2Cl cotransport function, Am. J. Physiol., 273,
L211-L216.
49.Tian, R., Aalkjaer, C., and Andreasen, F. (1990)
Mechanisms behind the relaxing effect of furosemide on the isolated
rabbit ear artery, Pharmacol. Toxicol., 67, 406-410.
50.Kovalev, I. V., Baskakov, M. B., Anfinogenova, Y.
J., Borodin, Y. L., Kilin, A. A., Minochenko, I. L., Popov, A. G.,
Kapilevich, L. V., Medvedev, M. A., and Orlov, S. N. (2003) Effect of
Na+,K+,2Cl– cotransport
inhibitor bumetanide on electrical and contractile activity of smooth
muscle cells in guinea pig ureter, Byul. Eksp. Biol. Med.,
136, 145-149.
51.Kovalev, I. V., Baskakov, M. B., Medvedev, M. A.,
Minochenko, I. L., Kilin, A. A., Anfinogenova, Y. J., Borodin, I. V.,
Gusakova, S. V., Popov, A. G., Kapilevich, L. V., and Orlov, S. N.
(2008) Na+,K+,2Cl–cotransport
and chloride permeability of the cell membrane in mezaton and histamine
regulation of electrical and contractile activity in smooth muscle
cells from the guinea pig ureter, Ross. Fiziol. Zh., 93,
306-317.
52.Stanke, F., Devillier, P., Breant, D., Chavanon,
O., Sessa, C., Bricca, G., and Bessard, G. (1998) Furosemide inhibits
angiotensin II-induced contraction on human vascular smooth muscle,
Brit. J. Clin. Pharmacol., 46, 571-575.
53.Stanke-Labesque, F., Craciwski, J. L., Bedouch,
P., Chavanon, O., Magne, J. L., Bessard, G., and Devillier, P. (2000)
Furosemide inhibits thromboxane A2-induced contraction in isolated
human internal artery and saphenous vein, J. Cardiovasc.
Pharmacol., 35, 531-537.
54.Wang, X., Breaks, J., Loutzenhiser, K., and
Loutzenhiser, R. (2007) Effects of inhibition of the
Na+/K+/2Cl– cotransporter on
myogenic and angiotensin II responses of the rat afferent arteriole,
Am. J. Physiol. Renal Physiol., 292, F999-F1006.
55.Mozhayeva, M. G., and Bagrov, Y. Y. (1995) The
inhibitory effects of furosemide on Ca2+ influx pathways
associated with oxytocin-induced contractions of rat myometrium,
Gen. Physiol. Biophys., 14, 427-436.
56.Mozhayeva, M. G., Bagrov, Y. Y., Ostretsova, I.
B., and Gillespie, J. I. (1994) The effect of furosemide on
oxytocin-induced contractions of the rat myometrium, Exp.
Physiol., 79, 661-667.
57.Akar, F., Skinner, E., Klein, J. D., Jena, M.,
Paul, R. J., and O’Neill, W. C. (1999) Vasoconstrictors and
nitrovasodilators reciprocally regulate the
Na+-K+-2Cl– cotransporter in rat
aorta, Am. J. Physiol., 276, C1383-C1390.
58.Garg, P., Martin, C., Elms, S. C., Gordon, F. J.,
Wall, S. M., Garland, C. J., Sutliff, R. L., and O’Neill, W. C.
(2007) Effect of the Na-K-2Cl cotransporter NKCC1 on systematic blood
pressure and smooth muscle tone, Am. J. Physiol. Heart Circ.
Physiol., 292, H2100-H2105.
59.Palacios, J., Espinoza, F., Munita, C.,
Cifuentes, F., and Michea, L. (2006)
Na+-K+-2Cl– cotransporter is
implicated in gender differences in the response of the rat aorta to
phenylephrine, Brit. J. Pharmacol., 148, 964-972.
60.Koltsova, S. V., Maximov, G. V., Kotelevtsev, S.
V., Lavoie, J. L., Tremblay, J., Grygorczyk, R., Hamet, P., and Orlov,
S. N. (2009) Myogenic tome in mouse mesenteric arteries: evidence for
P2Y receptor-mediated, Na+,K+,2Cl–
cotransport-dependent signaling, Purinergic Signaling,
5, 343-349.
61.Davis, M. J., and Hill, M. A. (1999) Signaling
mechanisms underlying the vascular myogenic response, Physiol.
Rev., 79, 387-423.
62.Hill, M. A., Davis, M. J., Meininger, G. A.,
Potocnik, S. J., and Murphy, T. V. (2006) Arteriolar myogenic signaling
mechanisms: implications for local vascular functions, Clin.
Hemorheol. Microcirc., 34, 67-79.
63.Schubert, R., and Mulvany, M. J. (1999) The
myogenic response: established facts and attractive hypothesis,
Clin. Sci., 96, 313-326.
64.Koltsova, S. V., Kotelevtsev, S. V., Tremblay,
J., Hamet, P., and Orlov, S. N. (2009) Excitation-contraction coupling
in resistant mesenteric arteries: evidence for NKCC1-mediated pathway,
Biochem. Biophys. Res. Commun., 379, 1080-1083.
65.Lang, F., Busch, G., Ritter, M., Volkl, H.,
Waldegger, S., Gulbins, E., and Haussinger, D. (1998) Functional
significance of cell volume regulatory mechanisms, Physiol.
Rev., 78, 247-306.
66.Mongin, A. A., and Orlov, S. N. (2001) Mechanisms
of cell volume regulation and possible nature of the cell volume
sensor, Pathophysiology, 8, 77-88.
67.Orlov, S. N., Pokudin, N. I., Kotelevtsev, Yu.
V., and Gulak, P. V. (1989) Volume-dependent regulation of ion
transport and membrane phosphorylation in human and rat erythrocytes,
J. Membr. Biol., 107, 105-117.
68.Adragna, N., Di Fulvio, M., and Lauf, P. K.
(2004) Regulation of K-Cl cotransport: from function to genes, J.
Membr. Biol., 201, 109-137.
69.Orlov, S. N. (1994) Ion transport across
erythrocyte membrane: mechanisms and volume-dependent regulation,
Sov. Sci. Rev. F. Physiol. Gen. Biol., 8, 1-48.
70.Koltsova, S. V., Akimova, O. A., Orlov, S. N.,
and Dulin, N. O. (2013) Both Na+/K+ pump and
Na+,K+,2Cl– cotransport
contribute to cell volume control in human lung fibroblasts, Byul.
Sib. Med., 12, 42.
71.Hoffmann, E. K., Lambert, I. H., and Pedersen, S.
F. (2009) Physiology of cell volume regulation in vertebrates,
Physiol. Rev., 89, 193-277.
72.Orlov, S. N., Platonova, A. A., Hamet, P., and
Grygorczyk, R. (2013) Cell volume and monovalent ion transporters:
their role in the triggering and progression of the cell death
machinery, Am. J. Physiol. Cell Physiol., 305,
C361-C372.
73.O’Shaughnessy, K. M., and Karet, F. E.
(2006) Salt handling in hypertension, Ann. Rev. Nutr.,
26, 343-365.
74.Balu, S., and Thomas, J. (2006) Incremental
expenditure of treating hypertension in the United States, Am. J.
Hypertens., 19, 810-816.
75.Guyton, A. C. (1980) Arterial Pressure and
Hypertension, WB Saunders Co, Philadelphia.
76.Postnov, Y. V., and Orlov, S. N. (1987)
Primary Hypertension as a Cell Membrane Pathology [in Russian],
Meditsina, Moscow.
77.Lifton, R. P., Gharavi, A. G., and Geller, D. S.
(2001) Molecular mechanisms of human hypertension, Cell,
104, 545-556.
78.Lifton, R. P. (2005) Genetic dissection of human
blood pressure variation: common pathways from rare phenotypes,
Harvey Lectures, 100, 71-101.
79.Guyton, A. C. (1991) Blood pressure control
– special role of the kidney and body fluids, Science,
252, 1813-1816.
80.Simon, D. B., Karet, F. E., Hamdan, J. M., Di
Pietro, A., Sanjad, S. A., and Lifton, R. P. (1996) Bartter’s
syndrome, hypokalemic alkalosis with hypercalciuria, is caused by
mutation of Na-K-2Cl cotransporter NKCC2, Nature Genet.,
13, 183-188.
81.Simon, D. B., Nelson-Williams, C., Bia, J.,
Ellison, D., Karet, F. E., Molina, A. M., Vaara, I., Iwata, F.,
Cushner, M., Koolen, M., Gainza, F. J., Gitelman, H. J., and Lifton, R.
P. (1996) Gitelman’s variant of Bartter’s syndrome,
inherited hypokalemic alkalosis, is caused by mutations in the
thiazide-sensitive Na-Cl cotransporter, Nature Genet.,
12, 24-30.
82.Hamet, P., Pausova, Z., Adarichev, V.,
Adaricheva, K., and Tremblay, J. (1998) Hypertension: genes and
environment, J. Hypertens., 16, 397-418.
83.Pickering, G. W. (1964) Systematic arterial
pressure, in Circulation of the Blood. Men and Ideas (Fishman,
A. P., et al., eds.) London, pp. 487-541.
84.Jones, A. W. (1973) Altered ion transport in
vascular smooth muscle from spontaneously hypertensive rats. Influence
of aldosterone, norepinephrine and angiotensin, Circ. Res.,
33, 563-572.
85.Postnov, Yu. V., Orlov, S. N., Shevchenko, A. S.,
and Adler, A. M. (1977) Altered sodium permeability, calcium binding
and Na-K-ATPase activity in the red blood cell membrane in essential
hypertension, Pflug. Arch. Europ. J. Physiol., 371,
263-269.
86.Postnov, Yu. V., and Orlov, S. N. (1985) Ion
transport across plasma membrane in primary hypertension, Physiol.
Rev., 65, 904-945.
87.Orlov, S. N., Adragna, N., Adarichev, V. A., and
Hamet, P. (1999) Genetic and biochemical determinants of abnormal
monovalent ion transport in primary hypertension, Am. J.
Physiol., 276, C511-C536.
88.Garay, R. P., and Alda, O. (2007) What can we
learn from erythrocyte Na-K-Cl cotransporter NKCC1 in human
hypertension, Pathophysiology, 14, 167-170.
89.Orlov, S. N., Tremblay, J., and Hamet, P. (2010)
NKCC1 and hypertension: a novel therapeutic target involved in
regulation of vascular tone and renal function, Curr. Opin. Nephrol.
Hypertens., 19, 163-168.
90.Orlov, S. N., Koltsova, S. V., Tremblay, J.,
Baskakov, M. B., and Hamet, P. (2012) NKCC1 and hypertension: role in
the regulation of vascular smooth muscle contractions and myogenic
tone, Ann. Med., 44, S111-S118.
91.Bianchi, G., Ferrari, P., Trizio, P., Ferrandi,
M., Torielli, L., Barber, B. R., and Polli, E. (1985) Red blood cell
abnormalities and spontaneous hypertension in rats. A genetically
determined link, Hypertension, 7, 319-325.
92.Kotelevtsev, Yu. V., Orlov, S. N., Pokudin, N.
I., Agnaev, V. M., and Postnov, Yu. V. (1987) Genetic analysis of
inheritance of Na+,K+ cotransport, calcium level
in erythrocytes and blood pressure in F2 hybrids of spontaneously
hypertensive and normotensive rats, Byul. Eksp. Biol. Med.,
103, 456-458.
93.Flagella, M., Clarke, L. L., Miller, M. L.,
Erway, L. C., Giannella, R. A., Andriga, A., Gawenis, L. R., Kramer,
J., Duffy, J. J., Doetschman, T., Lorenz, J. N., Yamoah, E. N.,
Cardell, E. L., and Shull, G. E. (1999) Mice lacking the basolateral
Na-K-2Cl cotransporter have impaired epithelial chloride secretion and
are profoundly deaf, J. Biol. Chem., 274,
26946-26955.
94.Meyer, J. W., Flagella, M., Sutliff, R. L.,
Lorenz, J. N., Nieman, M. L., Weber, G. S., Paul, R. J., and Shull, G.
E. (2002) Decreased blood pressure and vascular smooth muscle tone in
mice lacking basolateral Na+-K+-2Cl–
cotransporter, Am. J. Physiol., 283,
H1846-H1855.
95.Wall, S. M., Knepper, M. A., Hassel, K. A.,
Fischer, M. P., Shodeinde, A., Shin, W., Pham, T. D., Meyer, J. W.,
Lorenz, J. N., Beierwaltes, W. H., Dietz, J. R., Shull, G. E., and Kim,
Y.-H. (2006) Hypotension in NKCC1 null mice: role of the kidney, Am.
J. Physiol. Renal Physiol., 290, F409-F416.
96.Kim, S. M., Eisner, C., Faulhaber-Walter, R.,
Mizel, D., Wall, S. M., Briggs, J. P., and Schnermann, J. (2008) Salt
sensitivity of blood pressure in NKCC1-deficient mice, Am. J.
Physiol. Renal Physiol., 295, F1230-F1238.
97.Lee, H.-A., Baek, I., Seok, Y. M., Yang, E., Cho,
H.-M., Lee, D.-Y., Hong, S. H., and Kim, I. K. (2010) Promoter
hypomethylation upregulates
Na+-K+-2Cl– cotransporter 1 in
spontaneously hypertensive rats, Biochem. Biophys. Res. Commun.,
396, 252-257.
98.Cho, H.-M., Lee, H.-A., Kim, H. Y., Han, H. S.,
and Kim, I. K. (2011) Expression of
Na+,K+-2Cl– cotransporter is
epigenetically regulated during postnatal development of hypertension,
Am. J. Hypertens., 12, 1286-1293.
99.Mancia, G., Grassi, G., Giannattasio, C., and
Seravalle, G. (1999) Sympathetic activation in the pathogenesis of
hypertension and progression of organ damage, Hypertension,
34, 724-728.
100.Schlaich, M. P., Lambert, E., Kaye, D. M.,
Krozowski, Z., Campbell, D. J., Lambert, G., Hastings, J., Aggarwal,
A., and Esler, M. D. (2004) Sympathetic augmentation in hypertension:
role of nerve firing, norepinephrine reuptake, and angiotensin
neuromodulation, Hypertension, 43, 169-175.
101.Huang, B. S., Amin, M. S., and Leenen, F. H. H.
(2006) The central role of the brain in salt-sensitive hypertension,
Curr. Opin. Cardiol., 21, 295-394.
102.Leenen, F. H. H. (2010) The central role of the
brain aldosterone–“ouabain” pathway in salt-sensitive
hypertension, Biochim. Biophys. Acta, 1802,
1132-1139.
103.Judy, W. V., Watanabe, A. M., Henry, P. D.,
Besch, H. R., Murphy, W. R., and Hockel, G. M. (1976) Sympathetic nerve
activity: role in regulation of blood pressure in the spontaneously
hypertensive rats, Circ. Res., 38, 21-29.
104.Pyner, S., and Coote, J. H. (2000)
Identification of branching paraventricular neurons of the hypothalamus
that project to the rostroventrolateral medulla and spinal cord,
Neuroscience, 100, 549-556.
105.Allen, A. M. (2002) Inhibition of the
hypothalamic paraventricular nucleus in spontaneously hypertensive rats
dramatically reduces sympathetic vasomotor tone, Hypertension,
39, 275-280.
106.Li, D. P., and Pan, H. L. (2007) Glutamatergic
inputs in the hypothalamic paraventricular nucleus maintain sympathetic
vasomotor tone in hypertension, Hypertension, 49,
916-925.
107.Li, D. P., and Pan, H. L. (2007) Role of
GABAA and GABAB receptors in paraventricular
nucleus in control sympathetic vasomotor tone in hypertension, J.
Pharmacol. Exp. Ther., 320, 615-626.
108.Li, D. P., and Pan, H. L. (2006) Plasticity of
GABAergic control of hypothalamic presympathetic neurons in
hypertension, Am. J. Physiol. Heart Circ. Physiol., 290,
H1110-H1119.
109.Ye, Z.-Y., Li, D.-P., Byun, H. S., Li, L., and
Pan, H.-L. (2012) NKCC1 upregulation disrupts chloride homeostasis in
the hypothalamus and increases neuronal-sympathetic drive in
hypertension, J. Neurosci., 32, 8560-8568.
110.Jiang, G., Cobbs, S., Klein, J. D., and
O’Neill, W. C. (2003) Aldosterone regulates the Na-K-Cl
cotransporter in vascular smooth muscle, Hypertension,
41, 1131-1135.
111.Orlov, S. N., Li, J.-M., Tremblay, J., and
Hamet, P. (1995) Genes of intracellular calcium metabolism and blood
pressure control in primary hypertension, Semin. Nephrol.,
15, 569-592.
112.Hamet, P., Orlov, S. N., and Tremblay, J.
(1995) Intracellular signaling mechanisms in hypertension, in
Hypertension: Pathophysiology, Diagnosis, and Treatment (Laragh,
J. H., et al., eds.) Raven Press, New York, pp. 575-608.
113.Kahle, K. T., Rinehart, J., Giebisch, G.,
Gamba, G., Hebert, S. C., and Lifton, R. P. (2008) A novel protein
kinase signaling pathway essential for blood pressure regulation in
humans, Trends Endocrinol. Metab., 19, 91-95.
114.Susa, K., Kita, S., Iwamoto, T., Yang, S.-S.,
Lin, S.-H., Ohta, A., Sohara, E., Rai, T., Sasaki, S., Alessi, D. R.,
and Uchida, S. (2012) Effect of heterozygous deletion of WNK1 on the
WNK-OSR1/SPAK-NCC/NKCC1/NKCC2 signal cascade in the kidney and blood
vessels, Clin. Exp. Nephrol., 16, 530-538.
115.Rafigi, F. H., Zuber, A. M., Glover, M.,
Richardson, C., Fleming, S., Jovanovic, A., O’Shaughnessy, K. M.,
and Alessi, D. R. (2010) Role of the WNK-activated SPAK kinase in
regulating blood pressure, EMBO Mol. Med., 2, 63-75.
116.Bergaya, S., Faure, S., Baudrie, V., Rio, M.,
Escoubet, B., Bonnin, P., Henrion, D., Loirand, G., Achard, J. M.,
Jeunemaitre, X., and Hadchouel, J. (2011) WNK1 regulates
vasoconstriction and blood pressure response to a1-adrenergic
stimulation in mice, Hypertension, 58, 439-445.
117.Yang, S.-S., Lo, Y.-F., Wu, C.-C., Lin, S.-W.,
Yeh, C.-J., Chu, P., Sytwu, H.-K., Uchida, S., Sasaki, S., and Lin,
S.-H. (2010) SPAK-knockout mice manifest Gitelman syndrome and impaired
vasoconstriction, J. Am. Soc. Nephrol., 21,
1868-1877.
118.Janardhan, V., and Qureshi, A. I. (2004)
Mechanisms of ischemic brain injury, Curr. Cardiol. Rep.,
6, 117-123.
119.Folkow, B. (2010) Cardiovascular
“remodeling” in rat and human: time axis, extent, and in
vivo relevance, Physiology, 25, 264-265.
120.Loutzenhiser, R., Griffin, K., Williamson, G.,
and Bidani, A. (2006) Renal autoregulation: new perspectives regarding
the protective and regulatory roles of the underlying mechanisms,
Am. J. Physiol. Regul. Integr. Compar. Physiol., 290,
R1153-R1167.
121.Liu, Y., and Gutterman, D. D. (2009) Vascular
control in humans: focus on the coronary microcirculation, Basic
Res. Cardiol., 104, 211-227.
122.Bidani, A., Griffin, K. A., Williamson, G.,
Wang, X., and Loutzenhiser, R. (2009) Protective importance of the
myogenic response in the renal circulation, Hypertension,
54, 393-398.
123.Nathan, S., Pepine, C. J., and Bakris, G. L.
(2005) Calcium antagonists: effects on cardio-renal risk in
hypertensive patients, Hypertension, 46, 637-642.
124.Griffin, K. A., Picken, M. M., Bakris, G. L.,
and Bidani, A. K. (1999) Class differences in the effects of calcium
channel blockers in the rat remnant kidney model, Kidney Int.,
55, 1849-1860.
125.Bakris, G. L., Toto, R. D., McCullough, P. A.,
Rocha, R., Purkayastha, D., and Davis, P. (2008) Effect of different
ACE inhibitor combinations on albuminuria: results of the GUARD study,
Kidney Int., 73, 1303-1309.
126.Shibata, M. C., Leon, H., Chatterley, T.,
Dorgan, M., and Vandermeer, B. (2010) Do calcium channel blockers
increase the diagnosis of heart failure with hypertension? Am. J.
Cardiol., 106, 228-235.
127.Hansen, P. B., Jensen, B. L., Andreasen, D.,
and Scott, O. (2001) Differential expression of T- and L-type
voltage-dependent calcium channels in renal resistance vessels,
Circ. Res., 89, 630-638.
128.Loutzenhiser, R., and Epstein, M. (1990) The
renal hemodynamic effects of calcium antagonists, in Calcium
Antagonists and the Kidney (Epstein, M., et al., eds.) Hanley &
Belfus, Inc., Philadelphia, pp. 33-74.
129.Hayashi, K., Homma, K., Wakino, S., Tokuyama,
H., Sugano, N., Saruta, T., and Itoh, H. (2010) T-Type channel blockage
as a determinant of kidney protection, Keio J. Med., 59,
84-95.
130.Inscho, E. W., Cook, A. K., Imig, J. D., Vial,
C., and Evans, R. J. (2003) Physiological role for P2X1 receptors in
renal microvascular autoregulatory behavior, J. Clin. Investig.,
112, 1895-1905.
131.Orlov, S. N. (2005) Decreased
Na+,K+,Cl– cotransport and salt
retention in Blacks: a provocative hypothesis, J. Hypertens.,
23, 1929-1930.
132.Orlov, S. N., Gossard, F., Pausova, Z.,
Akimova, O. A., Tremblay, J., Grim, C. E., Kotchen, J. M., Kotchen, T.
A., Gaudet, D., Cowley, A., and Hamet, P. (2010) Decreased NKCC1
activity in erythrocytes from African-Americans with hypertension and
dyslipidemia, Am. J. Hypertens., 23, 321-326.
133.Khodorov, B. (2004) Glutamate-induced
deregulation of calcium homeostasis and mitochondrial dysfunction in
mammalian central neurons, Progr. Biophys. Mol. Biol.,
86, 279-351.
134.Mongin, A. A. (2007) Disruption of ionic and
cell volume homeostasis in cerebral ischemia: the perfect storm,
Pathophysiology, 14, 183-193.
135.Su, G., Kintner, D. B., and Sun, D. (2002)
Contribution of Na+,K+,Cl–
cotransporter to high [K+]o-induced
swelling and EAA release is astrocytes, Am. J. Physiol. Cell
Physiol., 282, C1136-C1146.
136.Busse, S., Breder, J., Dinkel, K., Reymann, K.
G., and Schroder, U. H. (2005) Inhibitors of
cation-chloride-cotransporters affect hypoxic/hypoglycemic injury in
hyppocampal slices, Brain Res., 194, 116-121.
137.Su, G., Kintner, D. B., Flagella, M., Shull, G.
E., and Sun, D. (2002) Astrocytes from
Na+,K+,Cl– cotransporter-null
mice exhibit absence of swelling and decrease in EAA release, Am. J.
Physiol. Cell Physiol., 282, C1147-C1160.
138.Koltsova, S. V., Luneva, O. G., Lavoie, J. L.,
Tremblay, J., Maksimov, G. V., Hamet, P., and Orlov, S. N. (2009)
HCO3-dependent impact of
Na+,K+,2Cl– cotransport in
vascular smooth muscle excitation-contraction coupling, Cell.
Physiol. Biochem., 23, 407-414.
139.Williams, R. S., and Benjamin, I. J. (2000)
Protective responses in the ischemic myocardium, J. Clin.
Investig., 106, 813-818.
140.Hannaert, P., Alvarez-Guerra, M., Pirot, D.,
Nazaret, C., and Garay, R. P. (2002) Rat NKCC2/NKCC1 cotransport
selectivity for loop diuretic drugs, Naunyn-Schmiedebergs Arch.
Pharmacol., 365, 193-199.
141.Delpire, E., Lu, J., England, R., Dull, C., and
Thorne, T. (1999) Deafness and imbalance associated with inactivation
of the secretory Na-K-2Cl cotransporter, Nature Genet.,
22, 192-195.
142.Lang, F., Vallon, V., Knipper, M., and
Wangemann, P. (2007) Functional significance of channels and
transporters expressed in the inner ear and kidney, Am. J. Physiol.
Cell Physiol., 293, C1187-C1208.