Mouse Lymphomyeloid Cells Can Function with Significantly Decreased Expression Levels of Cytochrome c
E. S. Shilov1*, I. V. Kislyakov1, E. A. Gorshkova1, R. V. Zvartsev2, M. S. Drutskaya2, I. A. Mufazalov2, V. P. Skulachev2, and S. A. Nedospasov3*
1Lomonosov Moscow State University, Biological Faculty, 119991 Moscow, Russia; E-mail: shilov_evgeny@inbox.ru2Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia
3Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 32 Vavilova st., 119991 Moscow, Russia; E-mail: sergei.nedospasov@gmail.com
* To whom correspondence should be addressed.
Received July 23, 2014
Cytochrome c is an indispensable electron carrier in the mitochondrial respiratory chain and also an important mediator of the internal pathway triggering apoptosis. Mice with a complete deficiency of the Cycs gene encoding the somatic cytochrome c die during the embryogenesis. Using the technology of LoxP-cre-dependent tissue-specific recombination, we obtained some mouse strains with significantly reduced expression of cytochrome c in certain cell types (“conditional genetic knockdown”). This knockdown was achieved by abrogation of the normal splicing of the Cycs locus pre-mRNA due to an additional acceptor site inside the stop-cassette neor. Previously, we observed embryonic lethality in homozygous mice with the same knockdown of cytochrome c in all cells of the organism. In the present work we studied two novel mouse strains with conditional knockdown of the Cycs gene in T lymphocytes and macrophages. Somewhat surprisingly, the mice of these two strains under normal conditions were not phenotypically different from the wild-type mice, either on the whole organism level or on the level of activity of individual target cells. Thus, the amount of cytochrome c in lymphomyeloid cells does not affect their development and normal functioning.
KEY WORDS: cytochrome c, genetic knockdown, T lymphocytes, macrophages, cre recombinase, splicingDOI: 10.1134/S0006297914120177
Cytochrome c is a vitally important component of the oxidative phosphorylation system playing an important role in protection against reactive oxygen species, and it also triggers apoptosome generation on initiating the internal pathway of apoptosis regulation [1]. Many animals have this protein in some isoforms, e.g. rodents have two genes (Cycs and Cyct) which encode, respectively, the somatic and testicular isoforms of cytochrome c [2]. Knockout of testicular cytochrome c results in an early atrophy of testicles but does not cause sterility of males [3]. On the other hand, the complete deletion of the Cycs gene encoding the major somatic variant of cytochrome c is lethal at the E8-E10 stage of embryogenesis [4]. The absence of cytochrome c in cells does not abrogate the mouse fibroblasts cultivation, but pyruvate and uridine need to be added into the medium, similarly to the case of ρ0 cells that have damage of mitochondrial DNA [5]. However, cells deprived of cytochrome c have defects in assembly of the respiratory chain full-size complexes I and IV, a decrease in cardiolipin fraction among membrane lipids of mitochondria [6], and also disorders in induction of the cell response to hypoxia because of inability to accumulate stabilized prohypoxic transcriptional factors HIF-1 and HIF-2 [7]. However, it is known that the budding yeast S. cerevisiae with different mutations affecting functions of cytochrome c isoform 1, which constitutes ~95% of the total amount of this protein, can support the functioning of the respiratory chain by 5% present as isoform 2. And the ability of yeast to grow on medium with glycerol or ethanol as the only metabolic substrate allowed researchers to discriminate mutants by gene cyc1 encoding this major isoform from double mutants or mutants in other unique genes encoding the respiratory chain proteins [8].
Thus, it is reasonable to suppose that cytochrome c could be present in the respiratory chain of mitochondria in a significantly higher amount that it was for the least sufficient of its functioning. In the present work we studied this problem in vivo and in vitro using unique mice in which the amount of cytochrome c in the cells could be regulated by LoxP-Cre technology.
MATERIALS AND METHODS
Obtaining mice with conditional deficiency of cytochrome c in T cells and macrophages. Mice genetically based on the C57BL/6 strain were used. The genetic construct cfKW was created as described earlier [9, 10]. To obtain mice with macrophages deficient in cytochrome c, the wild-type allele of the Cycs gene was deleted using the cre recombinase under control of the macrophagal lysozyme LysM promoter, and for T cells the Cre gene was used under control of the Cd4 gene promoter. Mice from the same litter with other genotypes that had the normal amount of this protein in all cells were used as control.
Mice were genotyped by PCR based on DNA taken from their tail tips (age > P25). The protocol of tissue lysis and DNA isolation was identical to the protocol of DNA isolation from macrophages and is described in the corresponding section. To determine the genotype of the Cycs locus, we used primers cytex2 – 5′-AGAGATGCAGAGGTTAAGTG-3′ and cytl2 – 5′-TGACCTTGCCTTCTTCGG-3′ (the wild-type locus gives a 520-bp PCR product, and the cfKW locus gives a 609-bp PCR product). The presence in the mice of the recombinase LysM-Cre gene was tested using the primers Cre8 – 5′-CCCAGAAATGCCAGATTACG-3′ and Mlys2 – 5′-CTTGGGCTGCCAGAATTTCTC-3′. To determine the locus Cd4 genotype, competitive PCR was used with three primers:
CD4cre1 – 5′-ATCAAGGTCCTGAGGAAGAG-3′,
CD4cre2 – 5′-ACCTCATCACTCGTTGCATC-3′,
CD4cre3 – 5′-CTAGGAGTTGTGCTGCACAG-3′.
For all the above-mentioned PCRs, the same optimized program was used: 94°C, 3 min; then 35 cycles – 94°C, 40 s; 60°C, 45 s; 72°C, 60 s; terminating elongation – 72°C, 5 min. The PCR products were analyzed by electrophoresis in 1% agarose gel and visualized by staining with ethidium bromide.
Isolation and polarization of bone marrow macrophages. Mice (age 6-8 weeks) were euthanized, prepared, the femoral bones were taken, and the bone marrow was isolated by washing from the bone diaphyses into a tube with a sterile serum-free DMEM. The bone marrow cells were counted in a Goryaev hemocytometer and placed into 100-mm Petri dishes (7·106 in 7 ml of cultural medium). The medium composition stimulated differentiation of the bone marrow cells into macrophages and corresponded to the standard protocol described earlier [11]. On the seventh day of cultivation, the cells were removed from the dishes with a sterile solution of 0.05% EDTA in PBS and replaced for polarization into 35-mm Petri dishes at 106 cells in 2 ml of culture medium per dish. The macrophages were polarized using recombinant mouse cytokines IFN-γ and IL-4 (Peprotech, USA) and also lipopolysaccharide (LPS) from E. coli (Sigma, USA). Each clone was M1-polarized with γ-interferon (50 ng/ml) and LPS (50 ng/ml). Polarization with γ-interferon at the same concentration without LPS and M2 polarization with IL-4 (20 ng/ml) were also performed, and non-polarized cells were used as control. The next day the cells were stained for the presence of specific markers, and DNA and RNA were isolated.
Western-blotting. Macrophages lysed in Laemmli buffer were used (2·106 cells per sample). Lysates of macrophages were subjected to SDS-PAGE in a Mini-Protean system (Bio-Rad, USA) and then to wet transfer onto an Amersham Hybond-P methanol-activated PVDF-membrane (GE Healthcare, GB), blocked with a bovine serum albumin solution (Sigma) in PBS, and treated with primary monoclonal antibodies against cytochrome c (BD Pharmingen, USA) and polyclonal rabbit antibodies to histone H2B (Abcam, GB). For the detection, secondary antibodies conjugated with horseradish peroxidase and an ECL West Dura kit (Pierce, USA) were used, and the signal was recorded with ChemiDoc luminescent imager (Bio-Rad).
Isolation of DNA. To quantitatively evaluate functioning of the LysMcre/loxP-system and determine the efficiency of excising the wild-type Cycs allele, DNA was isolated from (2-5)·106 bone marrow macrophages. The isolation was performed according to the standard protocol of the Jackson Laboratory for DNA isolation from mouse cells using proteinase K (Amresco, USA).
RNA was isolated from bone marrow-derived macrophages (BMDM) using Chomczynski method [12] with TRI Reagent (Sigma) according to the manufacturer’s protocol.
Reverse transcription reaction was performed with random nine-nucleotide primers using a Reverse Transcriptase SS III kit (Invitrogen, USA) according to the manufacturer’s protocol. For every specimen, 400 ng of total RNA was used that was pretreated with DNase I (Thermo Scientific, USA) for eliminating residual genomic DNA.
Real-time PCR. The relative expression of the alleles and their ratio were determined using the earlier described ΔΔCt approach [13]. Expression was determined with the mouse β-actin gene as a reference, and the ratio of WT and K72W was determined using as reference allele the allele K72W, which was not excised. Expression of the wild-type allele was determined using primers Cyc-1exF: 5′-CGTCTGTCTTCGAGTCCG-3′ and CycLys-R: 5′-TTCCAGGGATGTACTTTTTG-3′. For determination of the expression of allele K72W, primers Cyc-1exF and CycTrp-R: 5′-TTCCAGGGATGTACTTCCAT-3′ were used. Expression of the testicular form of cytochrome c was determined with primers CycT-F: 5′-CAGGGCACGGCTGCTGTGAT-3′ and CycT-R: 5′-CCACCGTGTGGCACTGAGCA-3′. The β-actin expression was determined using primers actin-longF: 5´-CAGGGTGTGATGGTGGGAATG-3′ and actin-longR: 5´-CCAGAGGCATACAGGGACAGC-3′. To determine the ratio of the WT and K72W alleles, for the WT allele the primers Cyc-2intF: 5′-AGAACAAAGGTAACGGGG-3′ and the above-mentioned CycLys-R were used, and the above-mentioned primers Cyc-2intF and CycTrp-R were used for the K72W allele.
PCR was performed on a CFX96 OneTouch device (Bio-Rad) using kits for real-time PCR with EvaGreen intercalator (Syntol, Russia). For all pairs of primers, the amplification was performed according to the following program: 94°C, 3 min; then 40 cycles – 94°C, 20 s; 60°C, 30 s; 72°C, 30 s; the terminating elongation — 72°C, 5 min; then for the PCR-products a melting curve from 50 to 95°C was designed.
Flow cytofluorimetry: staining of specific surface markers, intracellular staining of cytochrome c, testing phagocytosis, and cell sorting. Before staining, Fc-receptors on the cells were blocked with specific antibodies against them (anti-Fcγ, clone 2.4G2; diluted 1 : 400). The fractions of differentiated macrophages in the population were assessed by staining of the macrophagal marker F4/80 (clone BM8, conjugate with FITC) or of Mac-1 (clone M1/70, conjugate with PE). For staining the polarization markers, antibodies against CD206 and MHCII conjugated with fluorochromes allophycocyanin (APC) (clone C068C2) and phycoerythrin (PE) (clone M5/114) (Biolegend, USA), respectively, were used. Antibodies against CD11c (clone N418, conjugate with APC) were also used. All antibodies used were diluted 1 : 400, the staining of the surface markers was performed using standard methods, and the incubation time with antibodies was 30 min. The number of dead cells with damaged integrity of the plasma membrane was assessed by staining with propidium iodide (Sigma). The hydrogen peroxide level was measured by staining with H2DCF-DA (2,7-dichlorofluorescin-diacetate) (Sigma) that was initially hydrolyzed to H2DCF in the cells and then oxidized to fluorochrome DCF (2,7-dichlorofluorescein).
For intracellular staining of cytochrome c, macrophages were fixed with 2% solution of p-formaldehyde, then permeabilized with 0.5% Triton X-100 and stained with specific antibodies according to the manufacturer’s protocol. Cytochrome c was stained with specific monoclonal mouse antibody clone 6H2.B4 (BD Pharmingen) diluted 1 : 200 and secondary monoclonal anti-IgG1 rat antibodies of clone M1-14D12 (eBioscience, USA) conjugated with PE, diluted 1 : 200.
To determine the phagocytic activity of macrophages, DCF fluorochrome-labeled E. coli were used. To do this, overnight culture of the bacteria was grown in liquid LB medium, and then 1 ml aliquot was taken and supplemented with 10 nmol DCF-DA, which was converted in the cells into the fluorescent derivative DCF by bacterial esterases. After incubation with DCF-DA for 30 min at 37°C, the bacterial suspension was centrifuged at 3000g for 10 min, and the medium with the excess fluorochrome was removed. Then the bacterial cells were washed in PBS, centrifuged once more, added to the macrophage suspension in DMEM medium (100 µl of the overnight culture per 106 macrophages), and incubated for 1 h at 37°C. Then the macrophages were precipitated by centrifugation (300g for 3 min) and washed in PBS from the unbound bacteria. The phagocytic activity was determined by DCF fluorescence in the macrophagal gate. The measurements were performed using a Guava 8HT capillary flow cytofluorimeter (Merck Millipore, Germany).
Lymphocytes were sorted with a BD FacsAria cell sorter, and populations of CD4+ and CD8+ T cells were isolated using antibodies against CD3 (clone 145-2C11, Alexa750), CD4 (clone GK 1.5, FITC), and CD8 (clone 53-6.72, PE).
Induction of apoptosis and analysis of cell death. Apoptosis of macrophages was induced with the nonspecific inhibitor of protein kinases staurosporine (Sigma) at the concentration of 500 nM and also with the reversible inhibitor of glycolysis 2-deoxyglucose (20 mM), which could be phosphorylated by hexokinase but is not subject to hexose phosphate isomerization and accumulated in glycolytic cells. The macrophages were incubated with both substances separately or together for 6 h, then the cells were removed from the culture plastic with cold PBS on ice and stained with annexin V conjugated with APC (BD Pharmingen) and propidium iodide according to the manufacturer’s protocol. The staining was analyzed using a BD FacsCanto II flow cytofluorimeter.
Measurement of respiratory activity of cells. The respiratory activity of the macrophage suspension was measured using a Clark polarographic electrode with an Oxygraph device (Hansatech, South Korea) using (2-3)·106 cells (the cell number was pre-calculated in a Goryaev hemocytometer) in 0.3 ml of DMEM medium. After recording the curve of normal oxygen consumption by the cells from the medium during 2-3 min, an uncoupler of oxidative phosphorylation, the protonophore CCCP (carbonyl cyanide m-chlorophenylhydrazone (Sigma)) in ethanol, was added into the working chamber to the final concentration of 6.7 µM. Then the oxygen consumption was calculated in pmol for 106 cells per minute in the normal state and in the presence of the protonophore.
Statistical analyses. The ratio of genotypes was tested using the χ2 test at the level of p = 0.05. Significance of the ratio of cell populations obtained by flow cytometry was tested using the Mann–Whitney test. In other cases, the data were statistically assessed with Student’s t-test.
RESULTS
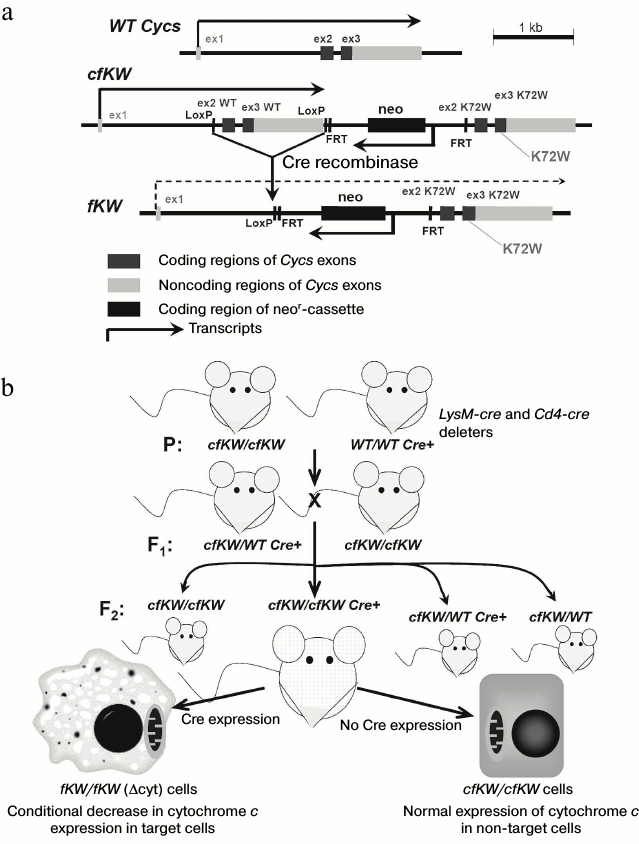
Characteristics of model with decreased expression of cytochrome c. Based on a sequence of the mouse somatic gene Cycs, mice with genotype cfKW were previously obtained. In these mice the second and third exons of the Cycs wild type allele were flanked with LoxP-sites, in the 3′-direction from them there was a neor-cassette, and in the 3′-direction from it there were the second and third exons of the “knock-in” allele of Cycs with the K72W substitution. The detailed scheme of the Cycs locus is presented in Fig. 1a. We described the cfKW mice obtaining in previous paper [10]. Due to Cre recombinase activity, these mice could be in vivo deprived of the encoding part of the wild-type Cycs allele to obtain a genotype denoted as fKW (in homozygous state denoted by us as Δcyt). In this genotype, the first and second exons of Cycs are separated by neor gene which is inserted into the first intron of Cycs and acts as a stop-cassette in this case. We used this approach to obtain on the base of the cfKW/cfKW mouse strain some independent strains: with full deletion of the wild-type Cycs allele (because of embryonic lethality this strain is maintained in the heterozygous state), and also mouse strains with conditional deletions in T cells and macrophages. For this purpose, auxiliary deleter mice were used with genotypes CMV-Cre, Cd4-Cre, and LysM-Cre, respectively [14]. Mice with the genotypes cfKW/fKW and WT/fKW had normal viability, and their phenotype was undistinguishable from that of the wild-type and cfKW/cfKW mice, but we failed to obtain newborn fKW/fKW mice because this genotype was associated with embryonal lethality. Autopsy of pregnant females revealed that the fKW/fKW embryos stop in their development at the E9.5-E11.5 period, and they reduce and die [14], thus demonstrating full knockout phenotype of the Cycs gene [4]. Nevertheless, the Δcyt-genotype that we obtained was not a full genetic knockout because expression of the somatic cytochrome c was retained in the Δcyt-cells, though the Cycs transcript level was about an order of magnitude lower than that in normal cells, and the transcript itself was mainly represented by the “knock-in” form with the K72W substitution [14]. Thus, the Δcyt-genotype is a combination of genetic knock-in and knockdown. The scheme for obtaining mice with the conditional Δcyt-genotype is presented in Fig. 1b. It should be noted that the K72W substitution itself did not affect the respiratory function of cytochrome c but made difficult the induction of apoptosis in primary cell lines with a mixed genetic background resulting from crossbreeding of 129 and B6 mice [10]. However, this mutation was not phenotypically manifested on the genetically pure background of the B6 strain.
Fig. 1. a) Scheme of Cycs locus organization in mice with different genotypes used in the present work; b) scheme of breeding offspring with conditional decrease in cytochrome c expression. The Cre recombinase expression is controlled by a specific promoter that allows us to remove the cytochrome c gene in some cell types such as macrophages (see figure) or T cells. Upon removal of the wild-type allele, expression of the K72W allele begins; however, it is significantly lower than the basal level of expression.
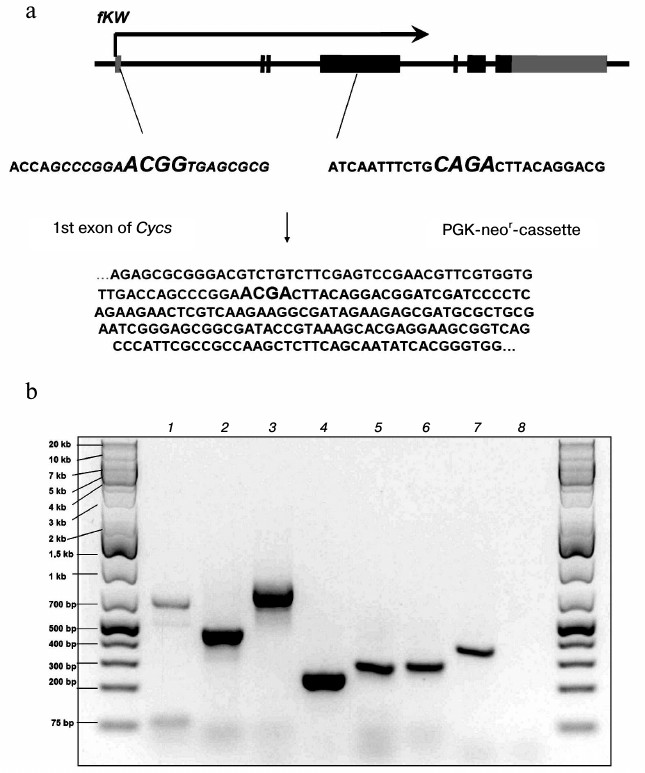
The mechanism responsible for a decrease in K72W allele expression due to the presence of the neor-cassette between the first and second exons in the fKW genotype is based on disorders in splicing of the Cycs gene primary transcript. We found that in fKW/fKW cells, chimeric transcripts were produced which consisted of the Cycs gene first exon and a part of the neor-cassette. Such mRNAs did not contain the reading frame of cytochrome c and were results of alternative splicing of the Cycs primary transcript. The splicing alternativeness in this case was realized due to a noncanonical acceptor site presented in the neor-cassette sequence, which in the fKW genotype was located significantly nearer to the donor site of the first exon than the canonical acceptor site of the second exon splicing. It should be noted that the neor-cassette was oriented opposite to the Cycs gene orientation; therefore, chimeric RNAs were non-functional not only for the gene of cytochrome c, but also for the gene of aminoglycoside phosphotransferase. A part of the chimeric Cycs-neor mRNA sequence was cloned as cDNA and then sequenced, which confirmed its structure (Fig. 2a). In the Δcyt-cells, the primary transcript processing with production of chimeric mRNA was more intensive than production of the K72W allele mRNA, which led to predominance of aberrant transcripts compared to those correctly spliced ones (Fig. 2b). In addition to a physical shortage of the correctly spliced mRNA, another cause for the decrease in total Cycs expression in the Δcyt-cells could be the RNA interference mechanism associated with the mutual complementarity of the chimeric mRNA and the neor-cassette mRNA, which together could produce double-stranded RNA.
Fig. 2. a) Scheme of formation of chimeric Cycs-neor mRNA and a fragment of its sequence. Boundaries of the donor and acceptor sites of the splicing are shown in italics; b) some fragments of transcripts detected in Δcyt-cells by PCR on cDNA: 1) first intron of Cycs; 2, 3, 7) different PCR products based on the chimeric Cycs-neor mRNA; 4) fragment of neor mRNA; 5) mRNA of the K72W allele; 6) mRNA of the WT allele; 8) the absence of a fragment corresponding to mRNA of the Cyct gene encoding the testicular cytochrome c.
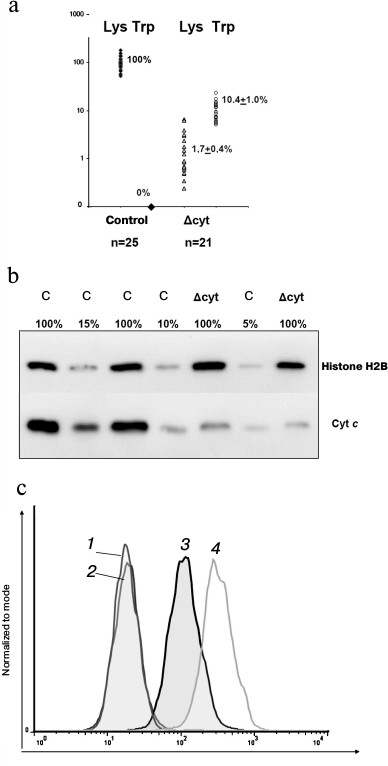
Expression of the K72W and WT alleles of the Cycs gene revealed that the Δcyt-cells and normal macrophages differed in the amount of total cytochrome c on the level of both mRNA (Fig. 3a) and protein (Fig. 3, b and c). We earlier found a similar decrease in expression in thymocytes from the mouse strain with T cell knockdown (published in [14]) but not in splenocytes, represented by the total fraction of all leukocytes isolated from the spleen, because the difference in the amount of cytochrome c in the T cells of the spleen was masked by its normal amount in the other leukocytes. In the control WT/WT and cfKW/cfKW Cre-mice, expression of the tryptophan allele in the studied leukocytes either could not be detected by RT-PCR or was at least three orders of magnitude weaker than the lysine allele expression in some of the cfKW/cfKW mice. In populations of Δcyt-macrophages and T cells, the lysine allele expression was partially preserved due to the presence of a small fraction of deletion-free cells (due to the actual efficiency of Cre recombinase in the range of 94-97%). Note that the decrease in the wild-type allele expression was greater than the degree of its excision. This finding seems to indicate potential allelic exclusion in wt Cycs and K72W alleles. The allele K72W expression in Δcyt-cells was comparable with the wild-type allele expression or slightly higher. Thus, the total expression of cytochrome c on the mRNA level in Δcyt-cells varied in individual mice from 5 to 20% of its normal amount. And no compensatory expression of the testicular cytochrome was observed either in control cells or in cytochrome c-deficient cells. Detection of the intracellular cytochrome c amount by flow cytofluorimetry (Fig. 3c) revealed a decreased level of this protein in cultures of bone marrow macrophages from cfKW/cfKW LysM-cre mice as compared with the control mice, and this is also confirmed by Western blotting (Fig. 3b). Based on the quantitative data on mRNA and protein expression levels, we conclude that in our model the total amount of cytochrome c is decreased in the target cell population, e.g. in macrophages, to ~10% of its initial level. We earlier showed a similar decrease in cytochrome c expression for Δcyt T cells [14].
Fig. 3. Expression of cytochrome c in the Δcyt and control bone marrow macrophages. a) Expression of different forms of cytochrome c determined on the mRNA level by allele-specific real time PCR. The results were normalized to the average level of expression in macrophages with intact Cycs locus. b) Total expression of cytochrome c determined on the protein level by Western blotting. Lysate of the control macrophages was titrated into 100, 15, 10, and 5% of the standard number (2·106 cells), and lysates of two primary cell lines of Δcyt macrophages were taken in standard quantities. The load and normalization were controlled using antibodies against histone H2B. c) Intracellular staining of bone marrow macrophages for cytochrome c and its determination by flow cytofluorimetry. Curves: 1, 2) respectively, the control and Δcyt macrophages stained with the secondary antibodies without addition of the primary antibodies; 3) Δcyt macrophages upon complete staining; 4) stained control cfKW/cfKW macrophages.
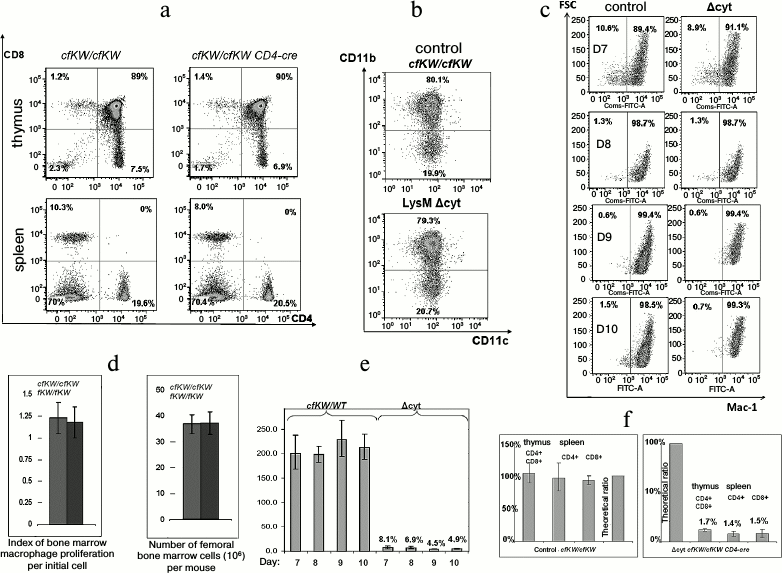
Decrease in amount of cytochrome c does not influence development of lymphomyeloid cells. Notwithstanding the embryonal lethality at the full knockdown of cytochrome c obtained by us, mice with macrophagal knockdown and mice with T cell knockdown did not differ from the control mice either in the T cellular compartment of the thymus (Fig. 4a) or in the myeloid compartment of the bone marrow (Fig. 4b). Moreover, no difference was found during in vitro cultivation of bone marrow macrophages between the normal and Δcyt cells in the level of proliferation or degree of differentiation (Fig. 4, c and d). The efficiency of excision of the LoxP-flanked wild-type allele by Cre recombinase was analyzed in the Δcyt T cellular mouse strain. This parameter was determined earlier for Δcyt macrophages and Δcyt thymocytes by approaches of allele-specific real-time PCR and Southern blotting and was found to be ~90-98%, which corresponded to the expected efficiency of Cre recombinase [14]. And during the cultivation of bone marrow macrophages, the excision degree of the wild-type allele was smoothly decreasing in correlation with the degree of cell differentiation (Fig. 4e). Having this in mind, we performed fluorescently activated cell sorting of CD4-positive T cells from the thymus and spleen of cfKW/cfKW Cd4-cre mice. Considering a possible positive or negative selection of cytochrome c-deficient cells during their maturation in the thymus and later in the peripheral immune system, we could expect changes in the fraction of the Δcyt T cells and correspondingly in the ratio of the wild-type and K72W alleles. However, this ratio corresponded to the theoretical efficiency of Cd4-cre recombinase and it was the same for the central and peripheral T cells (Fig. 4f). This fact shows the absence of any in vivo selection of cytochrome-deficient T lymphocytes compared to the normal lymphocytes. Thus, the decrease in the amount of cytochrome c influenced the cells significantly less than chemical modifications of this protein or some amino acid replacements in it (for review, see [15] by Kulikov et al.).
Fig. 4. Viability of cytochrome-deficient cells is comparable with the viability of control cells. a) Staining of cells from thymus and spleen for CD4 and CD8 in control (cfKW/cfKW) and Δcyt (cfKW/cfKW Cd4-cre) mice. Here and further, mice with cfKW/cfKW genotype from the same litter as Δcyt mice were taken as the control (in the case of no special indication). b) Staining of bone marrow cells of the control and Δcyt mice for CD11b and CD11c. c) Maturation of bone marrow macrophages of the control and Δcyt mice from the 7th to 10th day after the bone marrow isolation, staining for CD11b, and the direct light scattering. d) Number of bone marrow cells and yield of mature macrophages per bone marrow cell. e) Changes in the quantity of the wild-type residual allele during the maturation of bone marrow macrophages. Cells with the cfKW/WT genotype with ratio of Lys and Trp alleles equal to 2 : 1 were taken as the control. f) Ratio of alleles in the sorted T lymphocytes of a mouse with genotype cfKW/cfKW Cd4-cre (to the right) and of control mouse (to the left). The first column of each diagram corresponds to thymus DP-cells, the second column to spleen CD4+ cells, the third column to spleen CD8+ cells, and the fourth column shows a theoretical ratio of the alleles in the absence of recombinase activity.
To study further the cytochrome c-depending viability aspects, we used bone marrow macrophages because they could be obtained in an amount sufficient for several parallel experimental procedures, could be synchronized with the moment of excision of the wild-type Cycs allele, and were metabolically more active than T cells. We determined oxygen consumption by normal and cytochrome-deficient macrophages and also analyzed the fraction of apoptotic cells under condition of apoptosis induction by staurosporine or the glycolysis inhibitor 2-deoxyglucose and also by their combination. Oxygen consumption was not limited by mitochondrial cytochrome c because the total oxygen consumption by both types of cells under normal conditions and on uncoupled oxidation—phosphorylation with a protonophore was virtually the same and was at rest 0.64 ± 0.10 nmol O2/106 control cells and 0.68 ± 0.08 nmol O2/106 cytochrome-deficient macrophages, whereas in the presence of uncoupler it was 1.36 ± 0.21 nmol O2/106 control cells and 1.29 ± 0.12 nmol O2/106 cytochrome-deficient macrophages (Fig. 5a). The efficiency of the respiratory control of the electron transfer chain in the cytochrome-deficient macrophages was also similar to that in the control cells. The addition to bone marrow macrophages of 2-deoxyglucose and staurosporine in all cases stimulated cell death, though cytochrome-deficient macrophages were more resistant to apoptosis in response to deoxyglucose and especially to deoxyglucose combined with staurosporine, whereas staurosporine itself acted similarly on the cells of all genotypes (Fig. 5b). At first glance, this result seems to be paradoxical but it must be taken into account that staurosporine is now thought to act on cells without any association with Apaf-1 and cytochrome c [16]. Nevertheless, we cannot unambiguously believe the decreased amount of cytochrome c in our genetic model to be the major cause of an increase in the resistance of bone marrow macrophages to apoptosis because we deal with a combination of genetic knockdown and knock-in, and cytochrome c with K72W substitution could increase this resistance [10].
Fig. 5. Respiration and cell death of bone marrow macrophages. a) Average oxygen consumption by control and Δcyt macrophages (pmol O2/min per 106 cells) in the absence and presence of an oxidation—phosphorylation uncoupler (6.7 µM CCCP). b) Staining with PI/AnnexinV-APC of control and Δcyt macrophages after induction of cell death by 6-h incubation with 20 mM 2-deoxyglucose (2DG) or 500 nM staurosporine (STS) or their combination at the same concentrations.
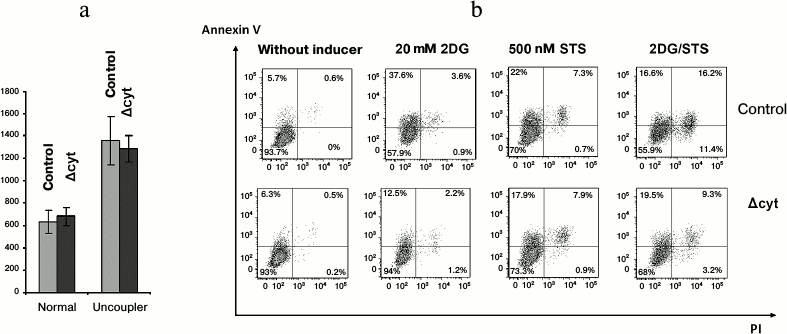
Thus, it occurred unexpectedly that a decrease in the amount of cytochrome c in lymphomyeloid cells did not decrease their viability as compared to normal cells and did not affect parameters of their vital activity under normal conditions, except for a decrease in the induction of the internal pathway of apoptosis.
In addition to testing cell respiration and apoptosis induction as functions associated with cytochrome c, we analyzed such specific cellular functions inherent in macrophages as expression of the major histocompatibility complex II (MHC II) (Fig. 6, a and b), generation of reactive oxygen species (Fig. 6c), polarization under the influence of exogenous cytokines (Fig. 6d), and phagocytosis (Fig. 6e). Macrophages with cytochrome c deficiency were not significantly different from the control cells.
Fig. 6. Analysis of specific functions of macrophages. a) Staining of unpolarized (M0) and polarized in M1 and M2 directions macrophages for MHC II (Δcyt macrophages are to the right, control macrophages are to the left). b) Average value of the fraction of MHC II-high macrophages (here and further dark columns are Δcyt macrophages, light columns are control macrophages). c) Staining of macrophages for intracellular reactive oxygen species (ROS) using H2DCF-DA. Curves: 1, 2) unstained macrophages; 3, 4) stained macrophages characterizing the intracellular ROS level; 5, 6) positive control, i.e. hydrogen peroxide-treated cells with fully oxidized H2DCF-DA (in each pair Δcyt cells are presented by odd curves, and control cells by even curves). d) Fraction of CD206-positive macrophages among polarized cells and in non-polarized control (M0). e) Fraction of macrophages phagocytizing DCF-labeled bacteria.
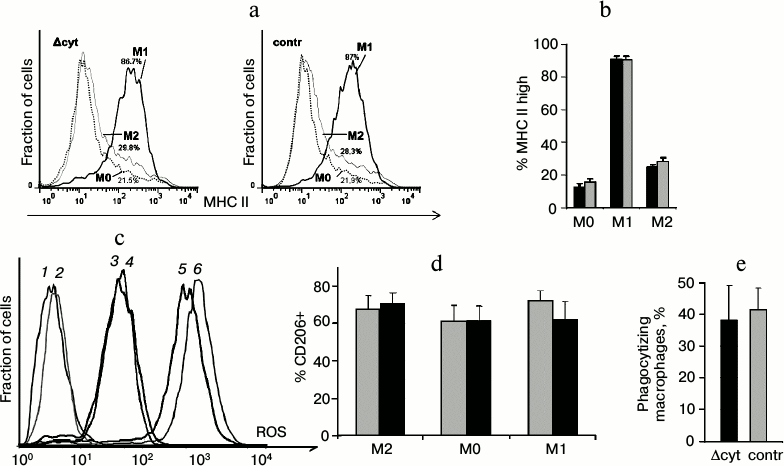
Although in our experimental system we have obtained nearly an order decrease in the intracellular level of cytochrome c, we have not observed any influence on the vital activity and functions of macrophages except for increased resistance to apoptosis due to inhibition of glycolysis. Because of the specific design of the recombinant locus in our model, we cannot neglect an effect of the amino acid substitution K72W in cytochrome c in addition to the decrease in its total amount. On the other hand, just the same decrease in the amount of cytochrome c in all cell types during embryogenesis leads to lethal phenotype, and this suggests that cells with another physiological limits of cytochrome c should exist. The approximately tenfold decreased level of cytochrome c in lymphomyeloid cells seems to be sufficient to provide normal functioning of the respiratory chain of mammals. Considering that the absence of cytochrome c in mitochondria affected the assembly of not only the full-size cytochrome oxidase complex, but also the assembly of NADH-dehydrogenase complex [6], which was not directly connected with cytochrome c, it may be supposed that cytochrome c should be a component closely associated with a respiratory complex – a respirasome. The respirasome physical structure with appropriate resolution shows that respiratory complexes I, III, and IV are associated in the consequent order [17]. The association between cytochrome c and the respirasome can potentially be realized, in particular, due to interaction between a cytochrome c molecule and a fatty acid residue within cardiolipin [18] or somewhat otherwise including interactions of amino acid cationic residues of cytochrome c and anionic heads of phospholipids [19]. Thus, we suppose that at least in some type cells the major part of the electron flow between complexes III and IV occurs not due to their stochastic transfer by cytochrome c molecules freely swimming in the intermembrane space between independent respiratory complexes, but within boundaries of the same respirasome that is served by a relatively small and possibly stoichiometric amount of cardiolipin-associated cytochrome c. From this standpoint, the remaining pool of mitochondrial cytochrome c can play an auxiliary role and be responsible for more versatile regulation of the respiratory chain functions, similarly to the regulatory function of the respirasome itself [20]. Therefore, the loss of cytochrome c from this pool will not affect oxidative phosphorylation in the majority of cell types and will not affect the phenotype. However, the respiratory chain architecture can be different in different types of cells, which is indicated by mitochondria-associated diseases that as a rule damage metabolically active cells (photoreceptors, neurons, muscle cells). Searches for “vulnerable” type cells that determine lethal phenotype in homozygous mice with the fKW/fKW genotype should be continued in the future.
The authors are grateful to D. M. Potashnikova, D. V. Kuprash, V. G. Gogvadze, A. V. Kulikov, A. Yu. Sazykin, and Yu. V. Shebzukhov for valuable advice and methodical help during this work.
This work was supported by the Russian Foundation for Basic Research (project 14-04-31403) and the Research Institute of Mitoengineering, Lomonosov Moscow State University.
REFERENCES
1.Skulachev, V. P. (1998) Cytochrome c in the
apoptotic and antioxidant cascades, FEBS Lett., 423,
275-280.
2.Hake, L. E., Kuemmerle, N., Hecht, N. B., Kozak, C.
A., Zou, H., Li, Y., Liu, X., and Wang, X. (1994) The genes encoding
the somatic and testis-specific isotypes of the mouse cytochrome
c genes map to paralogous regions of chromosomes 6 and 2,
Genomics, 20, 503-505.
3.Narisawa, S., Hecht, N. B., Goldberg, E.,
Boatright, K. M., Reed, J. C., and Millan, J. L. (2002) Testis-specific
cytochrome c-null mice produce functional sperm but undergo
early testicular atrophy, Mol. Cell Biol., 22,
5554-5562.
4.Li, K., Li, Y., Shelton, J. M., Richardson, J. A.,
Spencer, E., Chen, Z. J., Wang, X., and Williams, R. S. (2000)
Cytochrome c deficiency causes embryonic lethality and
attenuates stress-induced apoptosis, Cell, 101,
389-399.
5.King, M. P., and Attardi, G. (1989) Human cells
lacking mtDNA: repopulation with exogenous mitochondria by
complementation, Science, 246, 500-503.
6.Vempati, U. D., Han, X., and Moraes, C. T. (2009)
Lack of cytochrome c in mouse fibroblasts disrupts
assembly/stability of respiratory complexes I and IV, J. Biol.
Chem., 284, 4383-4391.
7.Mansfield, K. D., Guzy, R. D., Pan, Y., Young, R.
M., Cash, T. P., Schumacker, P. T., and Simon, M. C. (2005)
Mitochondrial dysfunction resulting from loss of cytochrome c
impairs cellular oxygen sensing and hypoxic HIF-alpha activation,
Cell. Metab., 6, 393-399.
8.Sherman, F., Stewart, J. W., Jackson, M., Gilmore,
R. A., and Parker, J. H. (1974) Mutants of yeast defective in
iso-1-cytochrome c, Genetics, 77, 255-284.
9.Mufazalov, I. A., Kruglov, A. A., Efimov, G. A.,
Drutskaya, M. S., Penkov, D. N., Kuprash, D. V., and Nedospasov, S. A.
(2011) Effects of mutation in the cytochrome c gene and of
inhibitors of reactive oxygen species on the accelerated involution of
the thymus in mice heterogeneous by the TNF/LT, Ros. Immunolog.
Zh., 2, 112-120.
10.Mufazalov, I. A., Penkov, D. N., Chernyak, B. V.,
Pletyushkina, O. Yu., Vysokikh, M. Yu., Chertkova, R. V., Kirpichnikov,
M. P., Dolgikh, D. A., Kuprash, D. V., Skulachev, V. P., and
Nedospasov, S. A. (2009) Embryonal mouse fibroblasts with the K72W
mutation in the gene of somatic cytochrome c: obtaining and
characteristics, Mol. Biol. (Moscow), 43, 648-656.
11.Boltz-Nitulescu, G., Wiltschke, C., Holzinger,
C., Fellinger, A., Scheiner, O., Gessl, A., and Forster, O. (1987)
Differentiation of rat bone marrow cells into macrophages under the
influence of mouse L929 cell supernatant, J. Leukoc. Biol.,
41, 83-91.
12.Chomczynski, P., and Sacchi, N. (1987)
Single-step method of RNA isolation by acid guanidinum
thiocyanate–phenol–chloroform extraction, Anal.
Biochem., 162, 156-159.
13.Livak, K. J., and Schmittgen, T. D. (2001)
Analysis of relative gene expression data using real-time quantitative
PCR and the 2ΔΔCT method, Methods,
25, 402-408.
14.Shilov, E. S., Mufazalov, I. A., Shebzukhov, Yu.
V., Zvartsev, R. V., Drutskaya, M. S., and Nedospasov, S. A. (2013)
Conditional genetic knockdown of the mouse somatic cytochrome c
in lymphomyelid cells, Ros. Immunol. Zh., 4, 361-371.
15.Kulikov, A. V., Shilov, E. S., Mufazalov, I. A.,
Gogvadze, V., Nedospasov, S. A., and Zhivotovsky, B. (2012) Cytochrome
c: the Achilles’ heel in apoptosis, Cell. Mol. Life
Sci., 69, 1787-1797.
16.Imao, T., and Nagata, S. (2013) Apaf-1- and
caspase-8-independent apoptosis, Cell Death Differ., 20,
343-352.
17.Dudkina, N. V., Kudryashev, M., Stahlberg, H.,
and Boekema, E. J. (2011) Interaction of complexes I, III, and IV
within the bovine respirasome by single particle cryoelectron
tomography, Proc. Natl. Acad. Sci. USA, 108,
15196-15200.
18.Tuominen, E. K., Wallace, C. J., and Kinnunen, P.
K. (2002) Phospholipid–cytochrome c interaction: evidence
for the extended lipid anchorage, J. Biol. Chem., 277,
8822-8826.
19.Sinibaldi, F., Howes, B. D., Piro, M. C.,
Polticelli, F., Bombelli, C., Ferri. T., Coletta, M., Smulevich, G.,
and Santucci, R. (2010) Extended cardiolipin anchorage to cytochrome
c: a model for protein–mitochondrial membrane binding,
J. Biol. Inorg. Chem., 15, 689-700.
20.Lapuente-Brun, E., Moreno-Loshuertos, R.,
Acin-Perez, R., Latorre-Pellicer, A., Colas, C., Balsa, E.,
Perales-Clemente, E., Quiros, P., Calvo, E., Rodriguez-Hernandez, M.
A., Navas, P., Cruz, R., Carracedo, A., Lopez-Otin, C., Perez-Martos,
A., Fernandez-Silva, P., Fernandez-Vizarra, E., and Enriquez, J. A.
(2013) Supercomplex assembly determines electron flux in the
mitochondrial electron transport chain, Science, 340,
1567-1570.