REVIEW: Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Health and Disease
N. V. Vorobjeva1* and B. V. Pinegin2
1Biology Faculty, Lomonosov Moscow State University, Lenin Hills 1/12, 119991 Moscow, Russia; E-mail: nvvorobjeva@mail.ru2Institute of Immunology, Federal Medical-Biological Agency, Kashirskoe Shosse 24/2, 115478 Moscow, Russia
* To whom correspondence should be addressed.
Received July 5, 2014
Recent insights into the specific type of cell death characteristic of neutrophils, called NETosis, are summarized. NETosis is a process of generation of Neutrophil Extracellular Traps (NETs), whose main components are DNA, granular antimicrobial peptides, and nuclear and cytoplasmic proteins. The structure of NETs determines their bactericidal, fungicidal, antiprotozoal, and antiviral properties. Therefore, NETs production by neutrophils is an essential immune response to infection. In addition to the antimicrobial function, NETosis is involved in many inflammatory and autoimmune disorders and participates in the regulation of noninfectious processes. The molecular mechanisms of NET formation, bactericidal effect, and involvement in some noninfectious, autoimmune, and inflammatory processes are discussed in detail in this review.
KEY WORDS: neutrophils, neutrophil extracellular traps, NETosis, autoimmune diseases, inflammationDOI: 10.1134/S0006297914120025
Abbreviations: APC, antigen-presenting cell; CIC, circulating immune complex; IFN, interferon; LPS, lipopolysaccharide; MHC, major histocompatibility complex; pDC, plasmacytoid dendritic cell; TNF, tumor necrosis factor.
In 2004, Zychlinsky and coauthors [1] discovered
that neutrophils were able to kill pathogens outside the cells by
releasing so-called Neutrophil Extracellular Traps (NETs). Since
neutrophils lose viability in the process of trap formation, in 2007
Steinberg and Grinstein [2] denoted this form of
neutrophil cell death as “NETosis”. It has been shown that
the bactericidal action of NETs is associated with their unique
composition: DNA, granular components, histones, and some cytoplasmic
proteins. It should be noted that trap formation has also been shown
for eosinophils [3], mast cells [4], chicken heterophils [5], and
macrophages/monocytes [6]. In addition to humans
and mice, nucleic acid trap formation has been described in other
animals, e.g. cattle, horses, fish, cats, rabbits, and invertebrates.
The release of nucleic acids from oenocytoid cells of the wax moth
Galleria mellonella is an important protective mechanism against
pathogens in insects [7]. The formation of
extracellular DNA filaments has also been shown for plants, and this
mechanism is supposed to play a key role in protection of root tips
from fungal infections [8, 9].
Since chromatin is released into extracellular space not only by
neutrophils but also by other types of cells, and due to the prevalence
of this phenomenon in vertebrates, invertebrates, and plants, the
broader term for this mechanism is ETosis (from English: Extracellular
Traps (ETs)). This review considers the mechanism of trap formation by
neutrophils, i.e. NETosis.
NET morphology. NETs have a unique ultrastructure. Their framework is formed by chromatin filaments of ~15-17-nm diameter [1] consisting of modified nucleosomes [10]. This framework is dotted with globular structures about 50-nm in diameter, being the proteins of granules and other cell compartments [1]. Surprisingly, NETs may not only have the morphology of elongated thin filaments, but can also be cloud-like structures that occupy 10-15-fold greater area compared to the initial cell size. It is supposed that such NET morphology occurs in vivo under conditions of sufficient intercellular space, e.g. in lung alveoli.
Mechanism of NET formation. NETs are formed because of a unique form of cell death, when the initial loss of all intracellular membranes is followed by disintegration of the cytoplasmic membrane. To date, very little is known about the mechanisms of NETosis. This is due primarily to the fact that neutrophils are short-lived, terminally differentiated leukocytes, which are unable to divide, and genetic manipulations with them are difficult. In addition, there are almost no neutrophil cell lines fully representing the physiology of primary blood cells.
Nevertheless, neutrophils are known to undergo major morphological modifications in the process of NETosis (figure). Several minutes after activation, the cells lie flat, being tightly attached to the substrate. During the next hour the nucleus loses its lobules, chromatin is decondensed, and the inner and outer leaflets of the nuclear membrane are separated from each other. Disintegration of the granules occurs simultaneously. Within another hour, the nuclear membrane breaks up into separate vesicles, while the nucleoplasm and cytoplasm merge into a homogenous mass. Finally, the cells become rounded and seem to be contracted until the cytoplasmic membrane is broken; then the cell contents are excreted to the exterior and form bundles of thin filaments, i.e. NETs.
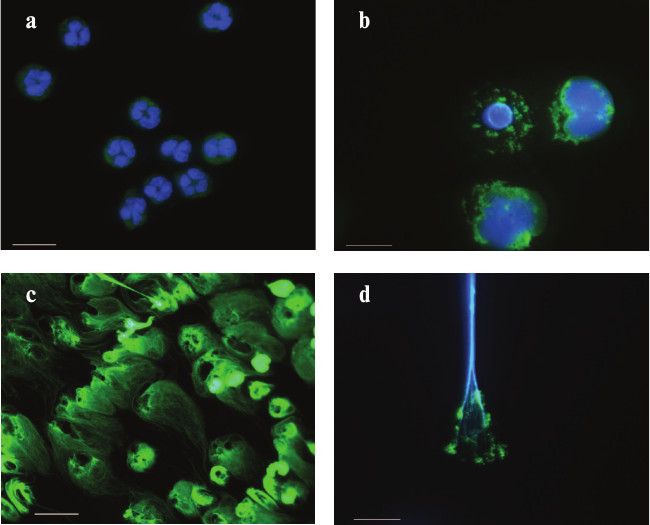
NETosis induced by phorbol myristate acetate. Immunofluorescence microscopy. Neutrophils were isolated from peripheral blood of a healthy donor. The cells were adhered to a coverglass, stimulated with low doses of PMA, fixed in 4% paraformaldehyde, and, where necessary, permeabilized with saponin. The preparations were stained with monoclonal FITC-labeled antibodies to the marker of azurophilic granules – myeloperoxidase (green fluorescence (a, b, d)). Chromatin was stained with specific dyes: DAPI (a, b, d) and SYBR green (c). The photographs were made with a Leica DM LB microscope (Leica Microsystems, Germany). Magnification: 100× (a, b, d) and 40× (c). Scale: 10 μm (a, b, d) and 25 μm (c). The photographs show: a) nonstimulated human neutrophils; b) neutrophils stimulated with PMA for 2 h. In the upper left cell, the nucleus has lost its segmental structure, while chromatin has been partially decondensed. The cell on the right is at a later stage of NETosis: the nuclear membrane is destroyed, and the contents of all compartments are mixed, while the cytoplasmic membrane is still intact; c) neutrophils stimulated with PMA for 3 h. Chromatin fibrils protrude far beyond the cells; d) neutrophilic trap. Numerous chromatin fibrils (blue fluorescence) with adhered MPO molecules (green fluorescence). The photographs were taken by N. V. Vorobjeva
Trap composition. Surprisingly, even after the contents of all cell compartments have been mixed, no more than 30 different proteins are detected in NET composition (Table 1) [10]. Most of these proteins are of granular origin; only a few types of proteins get into NETs from the nucleus (histones) and very few come from the cytoplasm [10]. It has been shown in some works that NETs are formed by living cells – eosinophils and neutrophils – by release of mitochondrial DNA into the extracellular space [3, 11]. However, it should be noted that there are extremely few mitochondria in neutrophils [12]; therefore, there was 100,000 times less mitochondrial DNA found in these experiments compared to the nuclear DNA.
Table 1. NET-associated proteins [10]

Recently, a unique mechanism of NET formation was described: the cytoplasmic membrane remained intact, and the cells preserved viability [13]. In these experiments, neutrophils were stimulated by Staphylococcus aureus for 5-60 min (NETosis usually develops during 3-4 h), followed by the release of vesicles with the included decondensed chromatin and bactericidal proteins of the granules. All these components were mixed, and they formed traps in the extracellular space.
NETosis inducers include many physiological stimuli, e.g. bacteria, fungi, protozoa, and viruses (Table 2). NETosis can be induced by reactive oxygen species (ROS) such as hydrogen peroxide [14]; antibodies [15] and antigen–antibody complexes [16, 17]; microbial components, lipopolysaccharide (LPS) [18, 19], M1 protein from Streptococcus pyogenes [20], and the lipophosphoglycans from Leishmania amazonensis [21]. Traps can also be induced by TLR4-activated platelets [22].
Table 2. Microorganisms and chemical factors
inducing NET formation [14]

It has been shown that cell–substrate adhesion, when MAC-1 integrin receptors are activated, is necessary for the induction of NETosis [23]. Neutrophils can also form traps in suspension, but much less intensively. It seems that this situation triggers a mechanism of blocking (or the absence of stimulation) of NETosis, which protects an organism from possible formation of clots in blood vessels by NETs.
Molecular mechanisms of NET formation. As reported previously, NET formation is a gradual process with several successive steps: 1) ROS generation; 2) transport of neutrophil elastase (NE), and, later, myeloperoxidase (MPO) from the granules to the nucleus; 3) histone modification, and, finally, 4) disruption of cytoplasmic membrane and release of chromatin. It would be worth dwelling on these steps in more detail, because together they compose a unique mechanism of cell death, i.e. NETosis.
Thus, it has been shown that ROS are necessary for NETosis. In neutrophils, ROS are formed during the so-called “respiratory burst” with the involvement of the NADPH oxidase complex [24]. This multicomponent enzyme complex is assembled during activation in membranes of specific granules and on the cytoplasmic membrane of neutrophils and performs electron transfer from NADPH localized in the cytoplasm across the membrane to molecular oxygen. Then the oxygen undergoes one-electron reduction leading to the formation of superoxide anion radical (O2.-). This, in turn, undergoes redox transformation, spontaneously or with the involvement of superoxide dismutase, with the formation of hydrogen peroxide. These primary ROS (O2.- and H2O2) can undergo further transformation resulting in the production of more active metabolites, e.g. hydroxyl radical (OH˙) and hypochlorous acid (HOCl). Hypochlorous acid has the strongest microbicidal effect and is formed with the involvement of myeloperoxidase, the enzyme of azurophilic granules.
The involvement of ROS in NET formation has been proved not only pharmacologically but also when studying the neutrophils of patients with chronic granulomatous disease (CGD). This pathology is determined by mutations in NADPH oxidase subunits resulting in the assembly of a nonfunctional or lowly functional enzyme complex, which is unable to synthesize ROS. People with such mutations suffer from recurrent infections life-long [25, 26], and their neutrophils do not form neutrophil traps [14]. However, it has been shown that the addition of H2O2 to the neutrophils of CGD patients restores the ability to release NETs [14]. Thus, in the information transfer pathway, ROS are between the enzyme complex and the subsequent mediators.
The molecules involved in signal transduction from the receptors to NADPH oxidase were revealed by rigorous inhibition analysis carried out in Zychlinsky’s laboratory [27]. The matter is that the most effective and frequently used NETosis activator is phorbol-12-myristate-13-acetate (PMA), the immediate stimulator of protein kinase C. It has been shown that the activation of NETosis by PMA is accompanied by the induction of the Raf/MEK/ERK signaling pathway [27], as well as the Rac2 (a small GTPase of the Rho-family)-mediated pathway [19].
Another feature of NETosis is the loss of chromatin segregation into eu- and heterochromatin [14]. This process involves neutrophil elastase and myeloperoxidase, the enzymes of azurophilic granules. Both enzymes move from the granules into the nucleus at the earliest stages of NETosis by a still unknown way. Neutrophil elastase is the first to be transported into the nucleus, where it catalyzes the cleavage of the linker histone H1 and modifies the core histones [28]. It has been shown that elastase is extremely important for trap formation, because mice deficient in this enzyme were incapable of producing NETs [28]. MPO migrates into the nucleus later, and its function is associated with intensification of chromatin decondensation, probably due to the synthesis of hypochlorous acid [28]. It should be noted that patients with mutations in the MPO gene cannot form valid NETs [29], probably because of insufficient amount of hypochlorous acid [30].
In addition to partial cleavage of histones by elastase and MPO, another modification intensifies chromatin decondensation. Peptidylarginine deiminase 4 (PAD4) induced in the neutrophil after proper activation catalyzes deimination of arginine residues to citrulline in three out of four core histones, which results in their weaker binding to DNA. Some works have shown that histones as part of NETs, as well as in decondensed chromatin, are citrullinated [18, 23, 31]. The role of this process in NETosis was demonstrated pharmacologically for cell lines forming a limited number of NETs. In addition, PAD4 knockout mice were unable to citrullinate histone H3, and, therefore, did not form NETs [32, 33].
Recently, it has been shown that autophagy is necessary for NETosis, and this mechanism follows NADPH oxidase activation in the information transfer pathway [34]. It has been shown that the stimulation of neutrophils by PMA leads to the formation of giant vacuoles similar to autophagosomes. However, the involvement of autophagy in NETosis has still not been sufficiently studied.
Neeli et al. [18, 23] first demonstrated that the cytoskeleton plays an important role in the regulation of NETosis of human neutrophils. Previously, it was shown that the tubulin cytoskeleton determined the direction of movement of the granules during exocytosis and phagocytosis of neutrophils. In their work, Neeli et al. [18] showed that depolymerization of the tubulin cytoskeleton by Nocodazole stopped the nuclear membrane disruption during the induction of NETosis by lipopolysaccharide (LPS), and, therefore, suppressed NET formation. In addition, in the same work they showed the involvement of the actin cytoskeleton in NETosis, since the addition of cytochalasin D to neutrophils also resulted in the inhibition of chromatin release [18]. The data obtained in these works could be interpreted as follows. To date, it is still unclear how elastase and MPO get into the nucleus for chromatin decondensation. It is quite probable (the authors’ hypothesis) that it is exactly the cytoskeleton that ensures the transport of azurophilic granules toward the nucleus, followed by enzyme transfer from one compartment to another by a still unknown pathway. In addition, Neeli et al. [23] assumed that MAC-1 integrins activated during NETosis could initiate the proper rearrangement of the cytoskeleton.
The regulatory role of the cytoskeleton in cell contraction at the terminal stage of NETosis and release of chromatin outside the cell has been hypothesized. However, this hypothesis needs to be supported by clear evidence.
Inhibitory pathways. At present, rather many facts point to the negative effect of NETosis on a host organism (this problem will be elucidated in the respective section). It has been shown that NETs can trigger serious local inflammatory processes and autoimmune diseases. In this context, mechanisms capable of negative regulation of NETosis are very relevant. It has been shown recently that the serpin B1 protein (an inhibitor of serine proteases such as elastase, cathepsin G, and proteinase-3) is transported from the cytoplasm to the nucleus at early stages of NETosis, where it blocks chromatin decondensation [35].
Another factor capable of inhibiting NETosis has been revealed: MUNC13-4, which is a member of the MUNC13 protein family [36]. This previously described protein participates in ROS generation, exocytosis of azurophilic granules, and maturation of phagolysosomes in neutrophils.
In addition, serum endonuclease DNase 1 is able to catalyze the degradation of chromatin released from neutrophils and to restrict NETosis [37]. Thus, the regulatory pathways inhibiting trap formation can limit the damage caused by excessive activation of neutrophils.
Antimicrobial activity of NETs. As known, the main function of neutrophils is the elimination of pathogens. According to Zychlinsky, NETs have evolved for repression of infections by entrapment and focalization of pathogens through inactivation of virulence factors and destruction of pathogens [1].
The entrapment of microorganisms limits their dissemination from the initial site of infection. It is supposed that pathogens are entrapped due to electrostatic interactions between negatively charged chromatin fibrils and positively charged bacterial surface [38]. Indeed, it has been shown that the pathogens that have capsules or can change their surface charge are not entrapped [39]. Another way to avoid entrapping is the synthesis of nucleases and their attachment to the surface of the pathogen [40]. The group A streptococcus (GAS) Streptococcus pyogenes [41], Pneumococcus species, and Staphylococcus aureus [42] synthesize endonucleases that release them from NETs and allow their penetration into deeper organs [43].
In addition, NETs can inactivate microbial proteins (so-called virulence factors) that can alter the functions of host cells. Elastase as a trap component was shown to specifically catalyze the splitting of the virulence factors of Shigella flexneri, Salmonella typhimurium, and Yersinia enterocolitica [1, 44]. NETs were also shown to contain Cathepsin G and Proteinase 3, which are similar to elastase and can split virulence factors of pathogens of other classes [45].
There is evidence that NETs contain proteins that are able to kill or inhibit pathogens. They include enzymes such as lysozyme and proteases, antimicrobial peptides (the BPI protein and defensins), ion chelators (calgranulin), and, interestingly, histones. The antimicrobial activity of NETs seems to be determined by the joint action of these components, as well as by their high local concentration on chromatin fibrils.
Some components of NETs proved to be capable of independent action. For example, Parker et al. [46] demonstrated that the MPO activity was sufficient to kill Staphylococcus aureus. Another protein found in the traps, calgranulin, was shown to have fungicidal activity [10, 47]. This protein chelates zinc needed for fungal growth and turned out to effectively inhibit the growth of Candida albicans and Aspergillus species. Finally, the important role of histones in killing pathogens has been demonstrated. Experiments with the addition of antibodies against histones showed inhibition of the killing of different classes of microorganisms determined by NET formation [1]. These facts confirm the supposition that histones have the strongest bactericidal activity.
In addition to the activities against bacteria, fungi, and parasites, NETs have antiviral effect. Recently, Saito et al. showed [48] that NETs are induced by the human immunodeficiency virus 1 (HIV-1), apparently via endosomal TLR7- and TLR8-receptors sensitive to viral RNA. Interestingly, the entrapped virions were completely inactivated, and the process of their inhibition was blocked by the addition of DNase leading to degradation of DNA fibrils [48].
Thus, it has been shown in the in vitro system that NETs inhibit Gram-positive and Gram-negative bacteria, fungi, viruses, and parasites. However, data on the effects of NETs in in vivo systems are not so numerous.
It is known that newborn children are rather susceptible to infections; it has been shown that NET formation in their bodies is reduced [49-51]. It is important that the patients with mutations in the genes coding for NADPH oxidase, MPO, elastase, i.e. deficient in the main active mediators of NETosis, suffer from recurrent infections. However, since all of these components are also involved in neutrophil effector functions such as phagocytosis and degranulation, elucidation of their specific contributions to each of these functions is still needed.
A case reported by Bianchi et al. [26] is a striking example of the role of NETs in infection repression. This article describes a patient with CGD suffering from severe Aspergillosis, whose neutrophils could not kill the hyphae of some Aspergillus strains because of inability of its cells to form NETs. However, after gene therapy, the NADPH oxidase function and the ability to form NETs were restored, and the patient recovered.
Involvement of NETs in inflammatory processes. Many facts published in the past decade demonstrate a negative effect of neutrophil traps. Several inflammatory processes have been described with NETosis playing a key role as a negative regulator. These processes include lung diseases such as acute respiratory distress syndrome, acute lung injury, and cystic fibrosis. In thrombosis, NETosis plays a dual role: both positive and negative. Below we consider some of the above pathologies where NETosis is involved.
Cystic fibrosis (mucoviscidosis). Cystic fibrosis is a severe hereditary disease caused by a mutation in the CFTR (cystic fibrosis transmembrane conductance regulator) gene and characterized by effects on exocrine glands and severe disorders of the respiratory system and gastrointestinal tract. These gene mutations result in disturbance of normal Cl– transport across the layer of epithelial cells, and, consequently, cause dehydration, condensation, and impeded drainage of mucins. In the case of pulmonary cystic fibrosis, enhanced mucous viscosity facilitates colonization of the organ by bacteria such as Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa.
Another factor responsible for high mucous viscosity is the great number of DNA molecules detected in the sputum of patients with mucoviscidosis [52], which correlates with the high concentration of neutrophils and the presence of neutrophil traps in the lungs [53]. It was shown that the presence of NETs in this case had no influence at all on the number of pathogens but, in fact, even increased their titers in mucus.
It is interesting that patients with mucoviscidosis undergo recombinant DNase therapy to reduce mucous viscosity. However, it has been shown that recombinant DNase is able to catalyze the cleavage of free DNA but not DNA/protein complexes typical for NETs. The potential positive role of elastase in this process is discussed in the context of this problem. According to Papayannopoulos et al. [54], elastase must facilitate dissolution of secreted fluids by degrading histones and providing access to chromatin for DNase. Otherwise, free elastase and other proteolytic components of NETs can damage tissues of the lungs and thereby intensify the immune response. It is probable that patients with mucoviscidosis need a complex therapy including both recombinant DNase and protease inhibitors.
Coagulation (blood clotting). Coagulation is a process of blood clot formation inside blood vessels that reduces blood loss in trauma and contributes to limitation of microbial infection spread in an organism. It has been shown that neutrophil traps are involved as a positive factor in clot formation during coagulation. However, their excessive formation may result in excessive blood clotting, which can cut off blood flow to vital organs and cause severe ischemia.
Neutrophils are known to adhere to the vascular endothelium during the formation of both arterial and venous blood clots. In the places of tight adhesion, they release chromatin fibrils (traps) to form stoppers that stimulate subsequent thrombosis [55]. Elastase and cathepsin G are adsorbed on chromatin fibrils and being serine proteases degrade coagulation inhibitors. In support of this mechanism, it was shown that arterial thrombosis in mice with the deficiency of both enzymes was decreased due to reduced blood clotting and fibrin polymerization. The same effect was observed in mice administered antibodies to NET components [56].
We should note another surprising fact associated with the action of NETs. When mice were infected with Escherichia coli, the bacteria were better isolated in hepatic microvessels of the animals with functional NETosis compared to those administered with the anti-DNA antibodies blocking NETosis [56]. These experiments provide direct evidence for the fact that coagulation with the involvement of NETosis reduces the spread of pathogens in a host organism.
Thus, based on the facts presented we conclude that neutrophils and their traps effectively interact with coagulation factors and vascular endothelium, while the effects of NETs such as bactericidity and procoagulation can be synergically involved in homeostatic maintenance in infectious diseases, e.g. sepsis.
Periodontitis. Another pathology where NETs play a key role is periodontitis. Periodontitis is an inflammatory disease of periodontium caused by microorganisms colonizing the gingival cavity. Periodontitis is most often associated with Porphyromonas gingivalis [57]; this bacterium enters the gingival cavity and attracts neutrophils, which then release chromatin traps [58] to prevent the spread of the pathogen.
Involvement of NETs in autoimmune diseases. Like many other physiological processes in an organism, NET formation plays not only positive but also negative role. The negative effect of NETs is related to their participation in the development of autoimmune diseases based on self-sustained adaptive immune response to self-antigens, which results in cell and tissue damage [59]. The initial basis for the induction of adaptive response is chronic activation of cells of the innate immune system and synthesis of antiinflammatory cytokines by these cells. One of the main cytokines activating the innate immune cells is type I interferons (IFNs). Their synthesis in an organism is strictly regulated. Type I IFNs deficiency leads to enhanced susceptibility to infectious diseases and some types of cancer, while its excess results in the development of autoimmune disorder [16, 17].
Type I IFNs accelerate the maturation of antigen-presenting cells (APC), macrophages, and dendritic cells and enhances the expression of class I and II MHC and co-stimulatory molecules. The effects of type I IFNs on the B-lymphocytes are particularly strong. Together with IL-6, they induce the maturation of plasmoblasts into mature plasma cells and stimulate the synthesis of all IgG subclasses and the synthesis of autoantibodies by B1 cells.
Type I IFNs are synthesized by nearly all nucleated cells of an organism, but most intensively by plasmacytoid dendritic cells (pDC). The main inducers of their synthesis are nucleic acids (NAs). Under the conditions of normal functioning, there are always small amounts of nucleic acids outside cells. They probably “leak” from apoptotic cells untimely removed by macrophages through phagocytosis. Extracellular occurrence of NAs can also be a result of NETosis. However, the healthy organism has mechanisms that prevent the receptors of the innate immune cells from contacting NAs. First, the NA-recognizing domains of these receptors are localized on the inner surface of endosomes of innate immune cells, and, therefore, are inaccessible to NAs. In addition, in the presence of a great arsenal of endonucleases in the extracellular medium, NAs are almost instantly cleaved when released from cells.
The secondary granules of neutrophils contain the protein cathelicidin hCAP-18, which is cleaved during cell activation with the formation of cationic peptide LL-37. This peptide uses electrostatic bonds to form complexes with nearly all kinds of nucleic acids, including auto-DNA and auto-RNA with marked immunostimulating properties, in particular, the ability to induce the synthesis of type I IFNs [60-66].
The LL-37 peptide causes the aggregation of NAs and protects them from the influence of DNA/RNA nucleases, contributes to endocytosis and penetration of NAs into the endosomal compartment of dendritic cells [61], and is a component of neutrophil extracellular traps [16, 17]. It has been shown that NET fragments obtained from neutrophils are able to induce the synthesis of IFN-α in the culture of plasmacytoid dendritic cells (pDC) [17]. Hence, there are grounds to believe that NETosis plays an important role in the development of autoimmune diseases. In the presence of nuclease inhibitors or with reduced functional activity of nucleases (DNase 1) in people with the respective predisposition, NETs are a source of autoantigens and cause the development of an autoimmune response to DNA and its protein components [37]. NET formation has been shown in systemic lupus erythematosus (SLE), ANCA vasculitis, type 2 diabetes, atherosclerosis, rheumatoid arthritis, psoriasis, and gout.
The role of NETs in SLE has been studied more thoroughly. It is known that SLE is an immune complicated pathology. In the 1970s, it was shown that the circulatory immune complexes (CIC) of patients with SLE consisted of DNA/RNA/protein and IgG antibodies specific to these components. The CIC isolated recently from the sera of patients with SLE consist of DNA and IgG only. Such CIC always contained the LL-37 peptide. The addition of CIC containing the LL-37 peptide, the pDC culture was accompanied by a marked synthesis of IFN-α. The removal of LL-37 from CIC abolished their ability of stimulate IFN-α [17].
The source of LL-37 peptide in CIC of SLE patients is neutrophils. In SLE patients, these cells are essentially different from the neutrophils of healthy people. These neutrophils are characterized by enhanced apoptosis, necrosis, and NETosis. However, phagocytosis of apoptotic neutrophils in such patients is substantially reduced. The degradation of apoptotic material is reduced too. Higher density of LL-37 molecules on the surface of neutrophils can be observed. Another difference of SLE patients from healthy donors is the higher level of low-density granulocytes (LDG) [67]. This unique subpopulation of neutrophils is characterized by immature phenotype (CD14–CD15+CD16+CD10+), enhanced synthesis of IFN-α and TNF, and enhanced expression of LL-37, myeloperoxidase, elastase, and cathepsin G. LDG differ from usual neutrophils in their evident disposition to NETosis [67, 68].
In addition to antibodies to various proteins and NAs, the serum of SLE patients was shown to contain antibodies against LL-37 peptide, α-defensins (HNP), and antimicrobial proteins of the primary granules of neutrophils [69]. Anti-LL-37 and anti-HNP antibodies are strong inducers of NETosis in SLE patients [17].
Thus, partial elucidation of the mechanisms of SLE development and involvement of NETosis in this process make it possible to elaborate a strategy for treating such patients. It should be mentioned that about 30% of SLE patients are unable to degrade NETs because of low activity of serum DNase 1 (gene mutation), the expression of DNase 1 inhibitors, or higher titers of autoantibodies that protect NETs from degradation by DNase 1 [37, 70]. It has been shown [70] that the above groups of SLE patients have higher titers of autoantibodies and suffer from severe concomitant disease – lupus nephritis. Thus, the strategies aimed at elimination of NETs and their components are now most promising for therapy of SLE patients.
NETosis, or formation of neutrophil extracellular traps, is an important strategic response of the immune system to various pathogens. The trap component chromatin captures pathogens, thereby limiting their spread within the host organism, while bactericidal proteins of the nucleus and granules effectively reduce their virulence or eliminate them completely.
In addition to the antimicrobial function, NETs are involved in regulation of a number of noninfectious processes and are an aetiological factor of many inflammatory and autoimmune diseases.
In spite of the fact that exactly 10 years have passed since the first description of NETosis by A. Zychlinsky, there are still many blank spots in the biology of this phenomenon. It is unclear, for example, what mechanisms elastase and MPO use to enter the nucleus from azurophilic granules at the initial stages of NETosis, what the role of autophagy is in this process, and how the cell chooses a particular program aimed at protecting the host from pathogens: phagocytosis, degranulation, or NETosis.
Nevertheless, we assume that the current interest of researchers in NETosis will lead to answers of many questions concerning this unique process in the near future.
The authors are sincerely grateful to Prof. S. A. Nedospasov, Corresponding Member of the Russian Academy of Sciences, for organizing this issue of Biochemistry (Moscow) dedicated to the memory of Prof. A. A. Yarilin and for his most valuable recommendations for preparing this manuscript.
We are grateful to E. S. Shilov for reading the text and making suggestions for its improvement.
REFERENCES
1.Brinkmann, V., Reichard, U., Goosmann, C., Fauler,
B., Uhlemann, Y., Weiss, D. S., Weinrauch, Y., and Zychlinsky, A.
(2004) Neutrophil extracellular traps kill bacteria, Science,
303, 1532-1535.
2.Steinberg, B. E., and Grinstein, S. (2007)
Unconventional roles of the NADPH oxidase: signaling, ion homeostasis,
and cell death, Sci. STKE, 379, pe11.
3.Yousefi, S., Gold, J. A., Andina, N., Lee, J. J.,
Kelly, A. M., Kozlowski, E., Schmid, I., Straumann, A., Reichenbach,
J., Gleich, G. J., and Simon, H.-U. (2008) Catapult-like release of
mitochondrial DNA by eosinophils contributes to antibacterial defense,
Nature Med., 14, 949-953.
4.Von Kockritz-Blickwede, M., Goldmann, O., Thulin,
P., Heinemann, K., Norrby-Teglund, A., Rohde, M., and Medina, E. (2008)
Phagocytosis-independent antimicrobial activity of mast cells by means
of extracellular trap formation, Blood, 111,
3070-3080.
5.Chuammitri, P., Ostojic, J., Andreasen, C. B.,
Redmond, S. B., Lamont, S. J., and Palic, D. (2009) Chicken heterophil
extracellular traps (HETs): novel defense mechanism of chicken
heterophils, Vet. Immunol. Immunopathol., 129,
126-131.
6.Chow, O. A., von Kockritz-Blickwede, M., Bright, A.
T., Hensler, M. E., Zinkernagel, A. S., Cogen, A. L., Gallo, R. L.,
Monestier, M., Wang, Y., Glass, C. K., and Nizet, V. (2010) Statins
enhance formation of phagocyte extracellular traps, Cell Host
Microbe, 8, 445-454.
7.Altincicek, B., Stotzel, S., Wygrecka, M.,
Preissner, K. T., and Vilcinskas, A. (2008) Host-derived extracellular
nucleic acids enhance innate immune responses, induce coagulation, and
prolong survival upon infection in insects, J. Immunol.,
181, 2705-2712.
8.Wen, F., White, G. J., VanEtten, H. D., Xiong, Z.,
and Hawes, M. C. (2009) Extracellular DNA is required for root tip
resistance to fungal infection, Plant Physiol.,
151, 820-829.
9.Hawes, M. C., Curlango-Rivera, G., Wen, F., White,
G. J., Vanetten, H. D., and Xiong, Z. (2011) Extracellular DNA: the tip
of root defenses? Plant Sci., 180, 741-745.
10.Urban, C. F., Ermert, D., Schmid, M., Abu-Abed,
U., Goosmann, C., Nacken, W., Brinkmann, V., Jungblut, P. R., and
Zychlinsky, A. (2009) Neutrophil extracellular traps contain
calprotectin, a cytosolic protein complex involved in host defense
against Candida albicans, PLoS Pathog., 5,
e1000639.
11.Yousefi, S., Mihalache, C., Kozlowski, E.,
Schmid, I., and Simon, H. U. (2009) Viable neutrophils release
mitochondrial DNA to form neutrophil extracellular traps, Cell Death
Differ., 16, 1438-1444.
12.Maianski, N. A., Geissler, J., Srinivasula, S.
M., Alnemri, E. S., Roos, D., and Kuijpers, T. W. (2004) Functional
characterization of mitochondria in neutrophils: a role restricted to
apoptosis, Cell Death Differ., 11, 143-153.
13.Pilsczek, F. H., Salina, D., Poon, K. K., Fahey,
C., Yipp, B. G., Sibley, C. D., Robbins, S. M., Green, F. H., Surette,
M. G., Sugai, M., Bowden, M. G., Hussain, M., Zhang, K., and Kubes, P.
J. (2010) A novel mechanism of rapid nuclear neutrophil extracellular
trap formation in response to Staphylococcus aureus, J.
Immunol., 185, 7413-7425.
14.Fuchs, T. A., Abed, U., Goosmann, C., Hurwitz,
R., Schulze, I., Wahn, V., Weinrauch, Y., Brinkmann, V., and
Zychlinsky, A. (2007) Novel cell death program leads to neutrophil
extracellular traps, J. Cell Biol., 176, 231-241.
15.Kessenbrock, K., Krumbholz, M., Schonermarck, U.,
Back, W., Gross, W. L., Werb, Z., Grone, H. J., Brinkmann, V., and
Jenne, D. E. (2009) Netting neutrophils in autoimmune small-vessel
vasculitis, Nature Med., 15, 623-625.
16.Garcia-Romo, G. S., Caielli, S., Vega, B.,
Connolly, J., Allantaz, F., Xu, Z., Punaro, M., Baisch, J., Guiducci,
C., Coffman, R. L., Barrat, F. J., Banchereau, J., and Pascual, V.
(2011) Netting neutrophils are major inducers of type I IFN production
in pediatric systemic lupus erythematosus, Sci. Transl. Med.,
3, 73ra20.
17.Lande, R., Ganguly, D., Facchinetti, V., Frasca,
L., Conrad, C., Gregorio, J., Meller, S., Chamilos, G., Sebasigari, R.,
Riccieri, V., Bassett, R., Amuro, H., Fukuhara, S., Ito, T., Liu, Y.
J., and Gilliet, M. (2011) Neutrophils activate plasmacytoid dendritic
cells by releasing self-DNA–peptide complexes in systemic lupus
erythematosus, Sci. Transl. Med., 3, 73ra19.
18.Neeli, I., Dwivedi, N., Khan, S., and Radic, M.
(2009) Regulation of extracellular chromatin release from neutrophils,
J. Innate Immun., 1, 194-201.
19.Lim, M. B. H., Kuiper, J. W. P., Katchky, A.,
Goldberg, H., and Glogauer, M. (2011) Rac2 is required for the
formation of neutrophil extracellular traps, J. Leukoc. Biol.,
90, 771-776.
20.Oehmcke, S., Morgelin, M., and Herwald, H. (2009)
Activation of the human contact system on neutrophil extracellular
traps, J. Innate Immun., 1, 225-230.
21.Guimaraes-Costa, A. B., Nascimento, M. T.,
Froment, G. S., Soares, R. P., Morgado, F. N., Conceicao-Silva, F., and
Saraiva, E. M. (2009) Leishmania amazonensis promastigotes
induce and are killed by neutrophil extracellular traps, Proc. Natl.
Acad. Sci. USA, 106, 6748-6753.
22.Clark, S. R., Ma, A. C., Tavener, S. A.,
McDonald, B., Goodarzi, Z., Kelly, M. M., Patel, K. D., Chakrabarti,
S., McAvoy, E., Sinclair, G. D., Keys, E. M., Allen-Vercoe, E.,
Devinney, R., Doig, C. J., Green, F. H., and Kubes, P. (2007) Platelet
TLR4 activates neutrophil extracellular traps to ensnare bacteria in
septic blood, Nature Med., 13, 463-469.
23.Neeli, I., Khan, S. N., and Radic, M. (2008)
Histone deimination as a response to inflammatory stimuli in
neutrophils, J. Immunol., 180, 1895-1902.
24.Vorobjeva, N. V. (2013) NADPH oxidase of
neutrophils and diseases associated with its dysfunction,
Immunologiya, 34, 232-238.
25.Pinegin, B. V., and Mayansky, A. N. (2007)
Neutrophils: structure and function, Immunologiya, 6,
374-382.
26.Bianchi, M., Hakkim, A., Brinkmann, V., Siler,
U., Seger, R. A., Zychlinsky, A., and Reichenbach, J. (2009)
Restoration of NET formation by gene therapy in CGD controls
aspergillosis, Blood, 114, 2619-2622.
27.Hakkim, A., Fuchs, T. A., Martinez, N. E., Hess,
S., Prinz, H., Zychlinsky, A., and Waldmann, H. (2011) Activation of
the Raf-MEK-ERK pathway is required for neutrophil extracellular trap
formation, Nature Chem. Biol., 7, 75-77.
28.Papayannopoulos, V., Metzler, K. D., Hakkim, A.,
and Zychlinsky, A. (2010) Neutrophil elastase and myeloperoxidase
regulate the formation of neutrophil extracellular traps, J. Cell
Biol., 191, 677-691.
29.Metzler, K. D., Fuchs, T. A., Nauseef, W. M.,
Reumaux, D., Roesler, J., Schulze, I., Wahn, V., Papayannopoulos, V.,
and Zychlinsky, A. (2011) Myeloperoxidase is required for neutrophil
extracellular trap formation: implications for innate immunity,
Blood, 117, 953-959.
30.Palmer, L. J., Cooper, P. R., Ling, M. R.,
Wright, H. J., Huissoon, A., and Chapple, I. L. C. (2012) Hypochlorous
acid regulates neutrophil extracellular trap release in humans,
Clin. Exp. Immunol., 167, 261-268.
31.Wang, Y., Li, M., Stadler, S., Correll, S., Li,
P., Wang, D., Hayama, R., Leonelli, L., Han, H., Grigoryev, S. A.,
Allis, C. D., and Coonrod, S. A. (2009) Histone hypercitrullination
mediates chromatin decondensation and neutrophil extracellular trap
formation, J. Cell Biol., 184, 205-213.
32.Li, P., Li, M., Lindberg, M. R., Kennett, M. J.,
Xiong, N., and Wang, Y. (2010) PAD4 is essential for antibacterial
innate immunity mediated by neutrophil extracellular traps, J. Exp.
Med., 207, 1853-1862.
33.Hemmers, S., Teijaro, J. R., Arandjelovic, S.,
and Mowen, K. A. (2011) PAD4-mediated neutrophil extracellular trap
formation is not required for immunity against influenza infection,
PLoS ONE, 6, e22043.
34.Remijsen, Q., Vanden Berghe, T., Wirawan, E.,
Asselbergh, B., Parthoens, E., De Rycke, R., Noppen, S., Delforge, M.,
Willems, J., and Vandenabeele, P. (2011) Neutrophil extracellular trap
cell death requires both autophagy and superoxide generation, Cell
Res., 21, 290-304.
35.Farley, K., Stolley, J. M., Zhao, P., Cooley, J.,
and Remold-O’Donnell, E. (2012) A serpin B1 regulatory mechanism
is essential for restricting neutrophil extracellular trap generation,
J. Immunol., 189, 4574-4581.
36.Monfregola, J., Johnson, J. L., Meijler, M. M.,
Napolitano, G., and Catz, S. D. (2012) MUNC13-4 protein regulates the
oxidative response and is essential for phagosomal maturation and
bacterial killing in neutrophils, J. Biol. Chem.,
287, 44603-44618.
37.Hakkim, A., Furnrohr, B. G., Amann, K., Laube,
B., Abed, U. A., Brinkmann, V., Herrmann, M., Voll, R. E., and
Zychlinsky, A. (2010) Impairment of neutrophil extracellular trap
degradation is associated with lupus nephritis, Proc. Natl. Acad.
Sci. USA, 107, 9813-9818.
38.Brinkmann, V., and Zychlinsky, A. (2007)
Beneficial suicide: why neutrophils die to make NETs, Nat. Rev.
Microbiol., 5, 577-582.
39.Wartha, F., Beiter, K., Albiger, B., Fernebro,
J., Zychlinsky, A., Normark, S., and Henriques-Normark, B. (2007)
Capsule and D-alanylated lipoteichoic acids protect Streptococcus
pneumoniae against neutrophil extracellular traps, Cell.
Microbiol., 9, 1162-1171.
40.Sumby, P., Barbian, K. D., Gardner, D. J.,
Whitney, A. R., Welty, D. M., Long, R. D., Bailey, J. R., Parnell, M.
J., Hoe, N. P., Adams, G. G., Deleo, F. R., and Musser, J. M. (2005)
Extracellular deoxyribonuclease made by group A Streptococcus
assists pathogenesis by enhancing evasion of the innate immune
response, Proc. Natl. Acad. Sci. USA, 102, 1679-1684.
41.Buchanan, J. T., Simpson, A. J., Aziz, R. K.,
Liu, G. Y., Kristian, S. A., Kotb, M., Feramisco, J., and Nizet, V.
(2006) DNase expression allows the pathogen group A Streptococcus
to escape killing in neutrophil extracellular traps, Curr.
Biol., 16, 396-400.
42.Berends, E. T. M., Horswill, A. R., Haste, N. M.,
Monestier, M., Nizet, V., and von Kockritz-Blickwede, M. (2010)
Nuclease expression by Staphylococcus aureus facilitates escape
from neutrophil extracellular traps, J. Innate Immun.,
2, 576-586.
43.Beiter, K., Wartha, F., Albiger, B., Normark, S.,
Zychlinsky, A., and Henriques-Normark, B. (2006) An endonuclease allows
Streptococcus pneumoniae to escape from neutrophil extracellular
traps, Curr. Biol., 16, 401-407.
44.Weinrauch, Y., Drujan, D., Shapiro, S. D., Weiss,
J., and Zychlinsky, A. (2002) Neutrophil elastase targets virulence
factors of enterobacteria, Nature, 417, 91-94.
45.Averhoff, P., Kolbe, M., Zychlinsky, A., and
Weinrauch, Y. (2008) Single residue determines the specificity of
neutrophil elastase for Shigella virulence factors, J. Mol.
Biol., 377, 1053-1066.
46.Parker, H., Albrett, A. M., Kettle, A. J., and
Winterbourn, C. C. (2012) Myeloperoxidase associated with neutrophil
extracellular traps is active and mediates bacterial killing in the
presence of hydrogen peroxide, J. Leukoc. Biol.,
91, 369-376.
47.Bianchi, M., Niemiec, M. J., Siler, U., Urban, C.
F., and Reichenbach, J. (2011) Restoration of anti-Aspergillus
defense by neutrophil extracellular traps in human chronic
granulomatous disease after gene therapy is calprotectin dependent,
J. Allergy Clin. Immunol., 127, 1243-1252.
48.Saitoh, T., Komano, J., Saitoh, Y., Misawa, T.,
Takahama, M., Kozaki, T., Uehata, T., Iwasaki, H., Omori, H., Yamaoka,
S., Yamamoto, N., and Akira, S. (2012) Neutrophil extracellular traps
mediate a host defense response to human immunodeficiency virus-1,
Cell Host Microbe, 12, 109-116.
49.Fadeel, B. (2009) Babies born without safety NET,
Blood, 113, 6270-6271.
50.Yost, C. C., Cody, M. J., Harris, E. S.,
Thornton, N. L., McInturff, A. M., Martinez, M. L., Chandler, N. B.,
Rodesch, C. K., Albertine, K. H., Petti, C. A., Weyrich, A. S., and
Zimmerman, G. A. (2009) Impaired neutrophil extracellular trap (NET)
formation: a novel innate immune deficiency of human neonates, Blood,
113, 6419-6427. 51.Yost, C. C., and Zimmerman, G. A. (2009) Response:
gestational age as a factor in neutrophil extracellular trap formation,
Blood, 114, 4911-4912.
52.Yoo, D. G., Winn, M., Pang, L., Moskowitz, S. M.,
Malech, H. L., Leto, T. L., and Rada, B. (2014) Release of cystic
fibrosis airway inflammatory markers from Pseudomonas
aeruginosa-stimulated human neutrophils involves NADPH
oxidase-dependent extracellular DNA trap formation, J. Immunol.,
192, 4728-4738.
53.Marcos, V., Zhou, Z., Yildirim, A. O., Bohla, A.,
Hector, A., Vitkov, L., Wiedenbauer, E. M., Krautgartner, W. D.,
Stoiber, W., Belohradsky, B. H., Rieber, N., Kormann, M., Koller, B.,
Roscher, A., Roos, D., Griese, M., Eickelberg, O., Doring, G., Mall, M.
A., and Hartl, D. (2010) CXCR2 mediates NADPH oxidase-independent
neutrophil extracellular trap formation in cystic fibrosis airway
inflammation, Nat. Med., 16, 1018-1023.
54.Papayannopoulos, V., Staab, D., and
Zychlinsky, A. (2011) Neutrophil elastase enhances sputum
solubilization in cystic fibrosis patients receiving DNase therapy,
PLoS One, 6, e28526.
55.Fuchs, T. A., Brill, A., Duerschmied, D.,
Schatzberg, D., Monestier, M., Myers, D. D., Jr., Wrobleski, S. K.,
Wakefield, T. W., Hartwig, J. H., and Wagner, D. D. (2010)
Extracellular DNA traps promote thrombosis, Proc. Natl. Acad. Sci. USA,
107, 15880-15885. 56.Geddings, J. E., and Mackman, N. (2014) New
players in hemostasis and thrombosis, Thromb. Haemost.,
111, 570-574.
57.Farquharson, D., Butcher, J. P., and Culshaw, S.
(2012) Periodontitis, porphyromonas, and the pathogenesis of rheumatoid
arthritis, Mucosal Immunol., 5, 112-120.
58.Vitkov, L., Klappacher, M., Hannig, M., and
Krautgartner, W. D. (2009) Extracellular neutrophil traps in
periodontitis, J. Periodontal Res., 44, 664-672.
59.Yarilin, A. A. (2010) Immunology [in
Russian], GEOTAR-Media, Moscow.
60.Lande, R., Gregorio, J., Facchinetti, V.,
Chatterjee, B., Wang, Y. H., Homey, B., Cao, W., Wang, Y. H., Su, B.,
Nestle, F. O., Zal, T., Mellman, I., Schroder, J. M., Liu, Y. J., and
Gilliet, M. (2007) Plasmacytoid dendritic cells sense self-DNA coupled
with antimicrobial peptide, Nature, 449, 564-569.
61.Ganguly, D., Chamilos, G., Lande, R., Gregorio,
J., Meller, S., Facchinetti, V., Homey, B., Barrat, F. J., Zal, T., and
Gilliet, M. (2009) Self-RNA-antimicrobial peptide complexes activate
human dendritic cells through TLR7 and TLR8, J. Exp. Med.,
206, 1983-1994.
62.Chamilos, G., Gregorio, J., Meller, S., Lande,
R., Kontoyiannis, D. P., Modlin, R. L., and Gilliet, M. (2012)
Cytosolic sensing of extracellular self-DNA transported into monocytes
by the antimicrobial peptide LL37, Blood, 120,
3699-3707.
63.Singh, D., Qi, R., Jordan, J. L., San Mateo, L.,
and Kao, C. C. (2013) The human antimicrobial peptide LL-37, but not
the mouse ortholog, mCRAMP, can stimulate signaling by poly(I:C)
through a FPRL1-dependent pathway, J. Biol. Chem., 288,
8258-8268.
64.Lai, Y., Adhikarakunnathu, S., Bhardwaj, K.,
Ranjith-Kumar, C. T., Wen, Y., Jordan, J. L., Wu, L. H., Dragnea, B.,
San Mateo, L., and Kao, C. C. (2011) LL37 and cationic peptides enhance
TLR3 signaling by viral double-stranded RNAs, PLoS One,
6, e26632.
65.Chuang, C. M., Monie, A., Wu, A., Mao, C. P., and
Hung, C. F. (2009) Treatment with LL-37 peptide enhances antitumor
effects induced by CpG oligodeoxynucleotides against ovarian cancer,
Hum. Gene Ther., 20, 303-313.
66.Kahlenberg, J. M., and Kaplan, M. J. (2013)
Little peptide, big effects: the role of LL-37 in inflammation and
autoimmune disease, J. Immunol., 191, 4895-4901.
67.Kaplan, M. J. (2011) Neutrophils in the
pathogenesis and manifestations of SLE, Nature Rev. Rheumatol.,
27, 691-699.
68.Denny, M. F., Yalavarthi, S., Zhao, W., Thacker,
S. G., Anderson, M., Sandy, A. R., McCune, W. J., and Kaplan, M. J.
(2010) A distinct subset of proinflammatory neutrophils isolated from
patients with systemic lupus erythematosus induces vascular damage and
synthesizes type I IFNs, J. Immunol., 184, 3284-3297.
69.Budikhina, A. S., and Pinegin, B. V. (2008)
Defensins – multifunctional human cationic peptides,
Immunopatol. Allergol. Infektol., 2, 31-40.
70.Leffler, J., Martin, M., Gullstrand, B.,
Tyden, H., Lood, C., Truedsson, L., Bengtsson, A. A., and Blom, A. M.
(2012) Neutrophil extracellular traps that are not degraded in systemic
lupus erythematosus activate complement exacerbating the disease, J.
Immunol., 188, 3522-3531.