Dissecting the Extended “–10” Escherichia coli rpsB Promoter Activity and Regulation in vivo
L. V. Aseev, L. S. Koledinskaya, and I. V. Boni*
Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: (495) 330-6538; E-mail: irina_boni@ibch.ru* To whom correspondence should be addressed.
Received April 21, 2014; Revision received May 15, 2014
As we have shown previously, transcription of the rpsB-tsf operon encoding essential components of the translation machinery, a ribosomal protein S2 and an elongation factor Ts, is driven by a single promoter PrpsB, which is highly conserved among γ-proteobacteria. PrpsB belongs to the extended “–10” promoter class; it comprises a TGTG-extension upstream of the “–10” hexamer TATAAA, a suboptimal “–35” region TTGGTG, and a GC-rich discriminator GCGCGC that separates the “–10” element from the transcription start site. In this work, we examined an impact of site-directed mutations in the rpsB promoter region on expression of the reporter gene PrpsB-lacZ within the E. coli chromosome as well as promoter regulation by transcription factors ppGpp and DksA upon amino acid starvation. The results show that the transcription level largely depends on both the TGTG-extension and the TTG-element in the “–35” region, as mutations in these sequences dramatically decrease the activity of the promoter. Upon induction of amino acid starvation, the rpsB promoter is negatively regulated by ppGpp due to the presence of the GC-rich discriminator, whose substitution for the AT-rich element abolished stringent control. These and other data obtained demonstrate the necessity of a natural combination of all the conserved promoter elements for efficient and regulated transcription of the essential rpsB-tsf operon.
KEY WORDS: rpsB-tsf operon, extended “–10” promoter, site-directed mutagenesis, ppGpp/DksA, GC-rich discriminator, stringent controlDOI: 10.1134/S0006297914080057
Abbreviations: bp, base pair; OD600, optical density at 600 nm; PCR, polymerase chain reaction; RBS, ribosome binding site; RT, reverse transcription; TIR, translation initiation region; 5′-UTR, 5′-untranslated region.
Transcription plays a major role in regulation of gene expression. In
bacterial cells, transcription is governed by a single DNA-dependent
RNA-polymerase that is composed of six subunits. Five subunits form a
core enzyme (α2ββ′ω) that
possesses the polymerase activity. To recognize promoter sequences and
to define the start point for RNA synthesis, the sixth subunit, a
σ-factor, joins the core RNA-polymerase to form the holoenzyme
[1-3]. Most bacteria utilize a
set of σ-factors that enable them to initiate transcription of
necessary genes in response to environmental stimuli [3]. In Escherichia coli there are seven
σ-factors. The primary factor, σ70, governs
transcription of the majority of operons during exponential growth, and
it has the highest affinity for the core enzyme; the other six
σ-factors control operons that should be activated under specific
conditions [2-6].
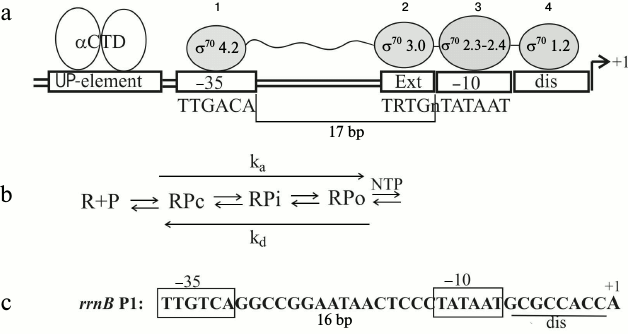
A great deal has been already learned about the modular structure of promoters and the role of each promoter element in forming specific contacts with the holoenzyme (reviewed in [5-9]). Interaction of RNA polymerase with the σ70-dependent promoters has been studied in detail and can be illustrated by a simplified layout (Fig. 1a). The process of productive transcription complex formation includes several steps (Fig. 1b): recognition and binding of the double-stranded DNA (formation of the closed complex RPc), melting of the promoter DNA and formation of the initial transcription bubble, isomerization of both the RNA polymerase and DNA via intermediates (RPi), and transition to the open complex RPo capable of NTP binding and coming into elongation [5, 7]. For productive elongation, the consecutive disruption of the contacts established between the σ-factor and promoter elements is necessary [1, 10]. Different domains of σ70 participate in recognition of the double-stranded promoter DNA: region 4.2 recognizes the –35 hexamer (–35)TTGACA(–30), and region 3 contacts the extended –10 element (–17)TRTGn(–14) (Fig. 1a). Besides the position –12, recognition of the –10 hexamer (–12)TATAAT(–7) in a duplex form is almost excluded, so that all the σ70 contacts downstream from position –12 are established with the non-template DNA strand during promoter melting. The σ70 regions 2.3 and 2.4 participate in interactions with the –10 hexamer, with base-specific interactions occurring primarily with the most conserved A(–11) and T(–7) that are buried deep in dedicated protein pockets [11]. Region σ70 1.2 contacts the discriminator that separates the –10 hexamer and the transcription start site (Fig. 1a). Besides σ70, the C-terminal domains of two α subunits (αCTD in Fig. 1a) participate in promoter DNA recognition, interacting with a so-called UP-element, an A/T-rich region located 5′ to the –35 hexamer (positions from –38 to –57); this interaction often is not essential [5].
Fig. 1. Transcription initiation on σ70-dependent promoters. a) Interaction of RNA-polymerase (holoenzyme α2ββ′ωσ70) with the promoter DNA. UP-element – the A/T-rich DNA region interacting with the C-terminal domains of α-subunits (αCTD); 1, 2, 3, 4 – σ70 regions involved in recognition of the promoter elements; dis – discriminator; ext – TRTGn-extension where R = purine, n – any base. b) Scheme of transcription initiation complex formation [5]; R – RNA-polymerase; P – promoter, RPc – primary closed complex of RNA-polymerase with promoter DNA in which DNA is not yet melted; RPo – an open complex in which DNA is melted and which is capable of initiating NTP binding and coming into elongation; RPi – intermediate complexes; ka – composite association constant for open complex formation, kd – dissociation constant. c) The rrnB P1 promoter structure (the UP-element is omitted).
Despite such a clear picture of RNA polymerase interactions with the promoter region, it is not easy to predict the promoter strength only on the basis of the DNA sequence [7]. Even more difficult is to predict the regulatory mechanisms for individual promoters, since in theory they may be largely diverse. Many factors are involved in transcription regulation; they interact either with DNA as repressors or activators or directly with the RNA polymerase (e.g. the alarmone ppGpp, proteins DksA, GreA, GreB, small 6S RNA) [2, 5]. To elucidate the control mechanisms, experimental efforts are indispensable.
Several natural promoters and their regulation have been studied thoroughly [5, 7]. First, the P1 promoters of the E. coli rrn operons encoding rRNAs should be mentioned. Thus the case of rrnB P1 has been studied in detail both in vivo and in vitro [5]. This promoter is very efficient during exponential growth in rich media, but its activity quickly reacts on environmental changes. The regulation is programmed in the promoter structure (Fig. 1c). A combination of optimal and non-optimal promoter elements provides efficient formation of the closed complex RPc (Fig. 1b) and its transition to the short-lived open complex RPo. It is the short lifetime of RPo that ensures the promoter response to the environmental cues [5]. The core promoter hexamers –35 and –10 are optimal, while the non-optimal features include the distance between them as well as the distance between –10 hexamer and the transcription start (eight positions instead of six as for most other promoters), the G/C-rich discriminator and a weak interaction between the σ region 1.2, and the base two positions downstream of the –10 element (Fig. 1c). The discriminator features allow rrnB P1 to decrease its activity under nutrient starvation (so-called stringent response) [5]. This plays a major role for cell survival, providing reallocation of cellular resources under conditions when the necessity for new ribosomes decreases while requirements for amino acids and anti-stress factors emerge. Recently, it has been found that not only rRNA and tRNA operon promoters, but also a number of promoters of r-protein operons reduce their activities during starvation [12]. However, stringent control has not been studied for all r-operon promoters, and the differences in their structures do not allow the prediction of the common control mechanisms.
The main goal of this work was to study the promoter structure and transcription regulation of the rpsB-tsf operon encoding essential components of the translation machinery, r-protein S2 and elongation factor Ts. We recently localized for the first time the rpsB-promoter and suggested its structure based on comparative analysis of DNA regions preceding the rpsB genes in γ-proteobacteria [13, 14]. Phylogenetic analysis showed the conservation of the unusual promoter structure, which combines oppositely acting promoter elements (Fig. 2). Thus, according to the literature data, the TRTG-extension in front of the –10 hexamer (–12)TATAAA(–7) should stabilize the open complex [15], whereas the non-optimal positions –7 (A instead of T) and –5 (C instead of G) indicate the short-lived RPo [5, 7]. Using site-directed mutagenesis, we evaluated the contribution of different elements to the rpsB promoter efficiency and its regulation under amino acid starvation. The results revealed that the TGTG-extension provides high transcription activity in rich media. At the same time, the rpsB promoter is downregulated during stringent response due to features of the discriminator. Negative stringent control for the TRTGn-promoters is shown for the first time.
MATERIALS AND METHODS
Bacterial strains and plasmids. Escherichia coli DH5α (φ80dlacZΔM15, Δ(lacZYA-argF)U169) was used for plasmid propagation and α-complementation tests, and E. coli ENS0 (Lac–, formerly HfrG6Δ12) and a plasmid vector pEMBLΔ46 for generation of the reporter constructions with the chromosomal lacZ gene by homologous recombination [16]. The recombinant strain LABrpsB208::lacZ in which the lacZ expression is governed by the promoter and translation initiation region (TIR) of the rpsB gene (positions from –208 to +41 relative to the initiator ATG codon) was described in reference [13]. In this work, we used its mutant derivative LABrpsB208ΔGGGU::lacZ that bears a small deletion in the 5′-UTR (ΔGGGU, from –72 to –69), which completely abolished translational autogenous control [13]. The mutant alleles of relA and dksA genes were transferred by P1 transduction using donor strains RLG8124 (dksA::tet, a gift of R. L. Gourse) and CF1693 (MG1655 relA251::kan, spoT207::Cm; from collection of E. Hajnsdorf; described in reference [17]). Plasmid pES2TIR208ΔGGGU is a derivative of pEMBLΔ46 [13] where the lacZ expression is governed by the rpsB promoter and the rpsB TIR bears the deletion ΔGGGU described above. This plasmid was used to obtain the mutant variants of the rpsB promoter.
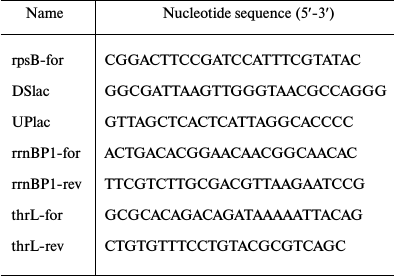
Site-directed mutagenesis. To introduce mutations in the rpsB promoter region within pES2TIR208ΔGGGU, a two-step PCR technique was exploited. In the first step, two overlapping PCR fragments were generated using pES2TIR208ΔGGGU as a template and two couples of primers. Overlapping “internal” primers comprised a desirable mutation, and the “external” primers corresponded to the invariant plasmid regions flanking the rpsB insert. Oligonucleotides UPlac and DSlac (table) described in reference [18] were used as the external primers. In the second step, the two PCR fragments were mixed and amplified in the presence of UPlac and DSlac; the resulting product was treated by BamHI and HindIII and cloned in pEMBLΔ46 in frame with the lacZ gene. Using this approach, we constructed variants of pES2TIR208ΔGGGU with a mutated rpsB-promoter. The presence of the desired mutations was checked by sequencing; then the plasmids were used for transformation of ENS0 to generate the strains LABPrpsBmut::lacZ by homologous recombination as described earlier [13, 14]. The chromosome of ENS0 (phenotype Lac–) lacks the lac-promoter and the lacZ RBS [16]. As a result of homologous recombination between the lac-regions of the plasmid and the chromosome, the cloned fragment bearing the mutant rpsB promoter and the rpsB TIR become embedded into the chromosome to drive the lacZ expression, thus allowing the selection of the recombinant clones on McConkey agar.
Oligonucleotides
Measurements of β-galactosidase activities in resulting strains. Bacterial cultures were grown with shaking in LB medium at 37ºC to the mid-log phase (OD600 = 0.4-0.5) and then chilled on ice. Cells (3 ml) were centrifuged in an Eppendorf MiniSpin, and the pellets were kept at –20ºC before use. For each strain at least three independent samples were prepared. Preparation of protein extracts and analysis of β-galactosidase activities were described earlier [18]. Specific β-galactosidase activities calculated according to Miller’s formula [19] are expressed in nmol of ο-nitrophenyl-β-D-galactopyranoside (ONPG) hydrolyzed per minute per mg of total soluble cell protein. Protein concentrations in extracts were determined using Protein Assay reagent (Bio-Rad, USA).
Induction of stringent response and isolation of total cellular RNA. Cells were grown in LB medium supplemented with antibiotics if necessary: kanamycin (50 µg/ml final concentration) in the case of relA mutants (relA::kan) and tetracycline (10 µg/ml final concentration) for dksA mutants (dksA::tet). At OD600 ~ 0.4-0.5, a 2-ml aliquot was taken and mixed with 4 ml of RNAprotect bacterial reagent (Qiagen, USA). To induce amino acid starvation, freshly prepared solution of L-serine hydroxamate (SHX; Sigma, USA) was added to the remaining culture (0.5 mg/ml final concentration). SHX causes growth arrest. After 30 min, a 2-ml aliquot was taken and mixed with 4 ml RNAprotect bacterial reagent. Total RNA was isolated from non-treated and SHX-treated samples using the RNeasy Mini Kit (Qiagen) according to recommendations of the manufacturer. RNA concentrations were measured by spectrophotometric analysis, assuming that 1 optical unit at 260 nm corresponds to 40 µg RNA.
RT-PCR analysis of transcripts. To investigate the changes in the rpsB promoter activity upon induction of ppGpp synthesis by adding SHX, we evaluated relative amounts of the rpsB-lacZ transcripts in samples isolated before and 30 min after SHX treatment. RT-PCR analysis was used for this purpose. To distinguish the rpsB-lacZ from the rpsB-tsf transcripts, we used DSlac as a reverse primer corresponding to the lac region of the fusion. In parallel, changes in transcription from rrnB P1 and thrL control promoters were determined, since their response to amino acid starvation has been studied in detail [5, 20, 21]. Primers used for RT-PCR are listed in the table. In each case, the forward primer corresponded to the beginning of the transcript, and the reverse primer was designed taking into account the possibility of separating different amplification products in agarose gel. AMV reverse transcriptase (Promega, USA) and 1 µg total RNA was taken for the RT reaction (20 µl volume, 42ºC, 1 h) in the presence of the two reverse primers specific for the rpsB-lacZ and thrL or rpsB-lacZ and rrnB P1 transcripts. The resulting cDNA (4 µl RT mix) was amplified by PCR (25 µl volume) in the presence of two couples of promoter-specific primers (table) using ready-for-use ScreenMix (Evrogen). In the PCR programs, 20 cycles were used for analysis of rpsB-lacZ and thrL transcripts in one reaction and 16 cycles for the combination of rpsB-lacZ and rrnB P1 transcripts. For the control samples (“–RT”), initial total RNA (0.2 µg) was taken. This control is indispensable to ensure the absence of DNA contaminations in total RNA preparations. DNA products were separated in 2% agarose gel (5 µl PCR mix per well).
Phylogenetic analysis of the rpsB promoter. Sequences of the rpsB genes and their preceding regions in bacterial genomes are available from databases NCBI Entrez Gene (www.ncbi.nlm.nih.gov/). To illustrate conservation of the rpsB promoter regions, the WebLogo program was used [22].
RESULTS
Dissecting the promoter of the rpsB-tsf operon by site-directed mutagenesis. Previously, we localized the promoter of the E. coli rpsB-tsf operon (PrpsB) by mapping the 5′-ends of the in vivo rpsB transcripts [13]. According to rigorous criteria, mapping the transcript 5′-termini is not sufficient for irreproachable promoter localization because they can originate from processing of primary transcripts, hence direct evidence based on promoter mutagenesis should be provided. Analysis of the DNA sequence preceding the mapped 5′-end of the rpsB transcript allowed us to propose a presumed structure of PrpsB that has a distinctive pattern (Fig. 2): the extended –10 element TGTGGTATAAA, a GC-rich discriminator GCGCGC, a suboptimal –35 region TTGGTG (matches with consensus –10 and –35 elements are in bold). Phylogenetic analysis revealed high conservation of these features in γ-proteobacteria (Fig. 2), thus arguing in favor of the proposed structure.
Fig. 2. Structure of the rpsB-tsf operon promoter and its conservation in γ-proteobacteria. For conservation analysis, the WebLogo program [22] and rpsB promoter sequences from E. coli, Salmonella enterica, Yersinia pestis, Erwinia carotovora, Pseudomonas aeruginosa, Haemophilus influenzae, Vibrio cholerae, Shewanella oneidensis, and Saccharophagus degradans were used.
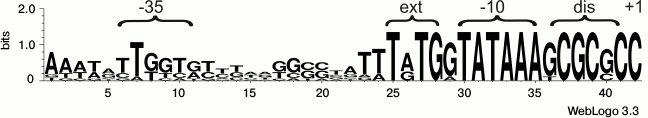
To evaluate the contribution of the conserved elements of PrpsB to its activity, we exploited site-directed mutagenesis. The impact of mutations was determined by changes in β-galactosidase activities by comparing expression of the chromosomal reporter rpsB-lacZ under the control of the mutant promoter with the expression of the reporter bearing the wild-type PrpsB whose activity was taken as 100%. To exclude the influence on post-transcriptional regulation, the experiments were done with strain LABrpsB208ΔGGGU::lacZ that bears a small deletion in the rpsB 5′-UTR (ΔGGGU, from –72 to –69), abolishing autogenous control [13]. The rpsB-lacZ activity in this strain (100% activity of the wild-type PrpsB) was estimated as 11,500 ± 750 nmol ONPG hydrolyzed per minute per mg of total soluble proteins (see “Materials and Methods”).
Using mutagenesis, we obtained strong evidence that the rpsB-promoter does belong to the extended –10 promoter class, where activities are largely determined by the TRTG-extension (R = purine) [15, 23-25]. Mutations in the TGTG-motif of PrpsB dramatically reduced the reporter expression – by 50-fold in the case of TCTC and by more than two orders of magnitude for the TCAC variant (Fig. 3). Such reduction was not compensated by changing the suboptimal –35 region (TTGgtg) for the consensus TTGACA (Fig. 3), thus indicating a key role of the TGTG-element in the PrpsB transcription activity.
Fig. 3. Mutations introduced in the rpsB promoter sequence within the reporter PrpsB-lacZ and corresponding activities of the mutant promoters as measured by β-galactosidase level (% relative to wild-type promoter activity).

As shown earlier, the presence of the –35 element could be nonessential for the TRTGn-promoters, in contrast to the classic –10/–35 promoters [26]. To elucidate whether this rule holds true in the case of PrpsB, we changed the suboptimal –35 element TTGgtg for the sequence aacgtg that bears no matches with the consensus. As a result of this change, the PrpsB activity dropped by about one order of magnitude; however, this decrease was much smaller than upon mutating the TGTG-extension (Fig. 3). Thus, though a high transcription level of PrpsB primarily depends on the extended –10 element, the TTG sequence in the –35 region also contributes to the promoter activity. It is worth mentioning that the TTG triplet in the –35 hexamer is highly conserved in E. coli σ70 promoters [8], which may reflect the importance of its contacts with σ70 region 4.2 for stabilization of σ70 interactions with double-stranded promoter DNA (Fig. 1a).
A unique feature of the rpsB promoters in γ-proteobacteria is the universal presence of A in the –7 position (Fig. 2), while T(–7) together with A(–11) are the most conserved bases in E. coli σ70-dependent promoters [8, 11]. To elucidate the role of such a unique abnormality, we changed A(–7) for T, thus generating the “ideal” extended –10 element TGTGgTATAAT (Fig. 3). It turned out that this mutation did not strengthen the promoter; on the contrary, it caused a decrease in its efficiency by about one order of magnitude (Fig. 3). This prima facie unexpected result has a rational explanation. Transcription is a dynamic process, and too tight interactions of the promoter with RNA-polymerase stalls the holoenzyme on the promoter, slows promoter clearance, and increases the abortive transcription yield, thereby leading to decreased promoter efficacy [27-30]. It was shown that even single deviations in consensus hexamers were able to augment transcription yield by decreasing the abortive rate at early elongation events [29]. Moreover, too tight binding of RNA-polymerase with the promoter reduces the capacity for regulating transcription under conditions when the cell needs to change the transcription profile in response to stress by switching from expression of genes involved in growth in favor of genes responsible for survival. We supposed that a combination of the TGTG-extension that strengthens σ70 binding to double-stranded promoter region [15, 23-25] with the non-optimal A(–7) [8, 11] and a GC-rich discriminator [31, 32], which impede the formation of the long-lived open complex RPo, allows the rpsB-promoter to be simultaneously active and regulable.
The promoter of the rpsB-tsf operon is negatively regulated by ppGpp due to the presence of the GC-rich discriminator. Our assumption about the rpsB promoter regulation in response to stress was experimentally validated. Stringent control is a global regulatory mechanism of cell adaptation to stressful conditions, in particular to changes in nutrient availability [33]. In response to starvation the cell synthesizes the low molecular weight regulator (p)ppGpp (a mix of guanosine tetra- and pentaphosphates, further referred to as ppGpp), which is a major transcription “switch” in stringent control and which acts in concert with transcriptional factor DksA [5, 20, 21, 34]. Promoters negatively regulated by ppGpp under starvation usually bear a GC-rich discriminator [5, 12, 30, 31]. It is just the case of the studied rpsB promoter. However, this feature alone is not sufficient for judging a priori to what extent the promoter will be sensitive to increased ppGpp concentration in a cell.
Serine starvation was induced by a standard treatment of exponential cultures with serine hydroxamate (SHX) [12, 35]. SHX inhibits seryl-tRNA-synthetase [36], resulting in cessation of protein synthesis, growth arrest, and induction of ppGpp synthesis by the ribosome-associated RelA protein. The occurrence of uncharged tRNA (in this case tRNASer) in the ribosomal A-site triggers the activation of relA-dependent ppGpp synthesis [33]. The impact of the induced serine starvation on the rpsB promoter activity was studied by RT-PCR. Two RNA samples were obtained from one cellular culture – one sample was isolated before SHX addition, and another one 30 min after treatment (“Materials and Methods”). To make sure that the RT-PCR technique adequately reflects the changes in transcription profile during stringent response, we evaluated alterations in the transcript levels for two reference promoters – rrnB P1 and thrL (Fig. 4, a and b). It is well known that upon starvation the activity of rrnB P1 declines, while that of the thrL promoter, which governs threonine biosynthesis, increases [20, 21]. The data obtained by RT-PCR are fully consistent with this (Fig. 4, a and b).
Fig. 4. Changes in the transcription level upon induction of serine starvation by treating exponential E. coli cultures by serine-hydroxamate (SHX). The transcript level was estimated by RT-PCR using promoter-specific couples of primers. a) Changes in transcription from rpsB (within the PrpsB-lacZ reporter) and thrL promoters. b) Changes in transcription from PrpsB and rrnB P1 after SHX treatment of wild-type, RelA-deficient (relA::kan), and DksA-deficient (dksA::tet) strains. c, d) Loss of stringent response of PrpsB after changing the GC-rich discriminator for the AT-rich sequence (PrpsB-ATdis) or as a result of a single mutation C(–5)-G. M, marker of DNA fragment lengths (bp).
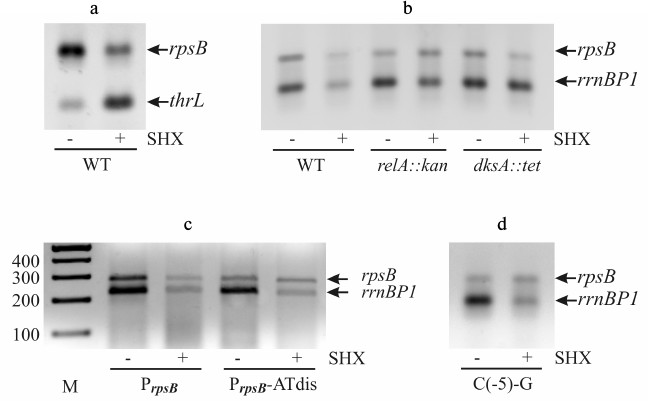
We evaluated changes in rpsB-lacZ and rrnB P1 transcript yields after SHX treatment of the wild-type strain as well as of the strains deficient in RelA (relA::kan) or transcription factor DksA (dksA::tet). The crucial role of DksA in ppGpp-dependent repression of rrnB P1 was established both in vivo and in vitro [5, 20]. Like rrnB P1, the rpsB promoter showed decrease in its activity after addition of SHX to the wild-type cells, but not to the relA mutant where the transcript levels did not change visibly (Fig. 4b). However, in the DksA-deficient strain the PrpsB activity was weakened, suggesting that DksA-dependence of PrpsB during stringent response is less apparent than in the case of rrnB P1.
To determine how the negative stringent response of PrpsB depends on the GC-rich discriminator, we changed it for the AT-rich sequence (Fig. 3). As shown in Fig. 4c, this mutation abolished stringent control. Thus, the ppGpp-dependent regulation of PrpsB upon starvation requires the presence of the GC-rich discriminator.
Previous studies of interactions between σ70 and promoter DNA have shown that region σ70 1.2 participates in recognition of the discriminator by forming a specific contact with the base in the non-template DNA strand two positions downstream from the –10 hexamer, which is –7 for rrn P1 and –5 for most other promoters [37, 38]. In rrnB P1, as well as in other stable RNA promoters, there is C in this position, which is the least favorable for interaction with σ70 1.2 [37, 38]. The change of C(–7) for G in the rrnB P1 discriminator region made the promoter insensitive to negative control by ppGpp/DksA [37]. This finding prompted us to study the role of C(–5) in regulation of PrpsB during stringent response. Using site-directed mutagenesis, we changed C(–5) for G, leaving the remaining sequence intact (Fig. 3). The resulting promoter variant became insensitive to increased ppGpp concentration upon starvation induced by SHX (Fig. 4d). Thus, despite the differences in overall structures, both rrnB P1 and PrpsB are negatively regulated during stringent response due to specific features of their discriminator regions.
DISCUSSION
Specific features of the rpsB promoter. In this work, using site-directed mutagenesis we confirmed the modular structure of the rpsB promoter, which was earlier proposed on the basis of mapping of the 5′-ends of the rpsB in vivo transcripts and by phylogenetic analysis [13, 14]. PrpsB does belong to the rare class of extended –10 TRTGn-promoters whose activity depends on the (–17)TRTG(–14) extension in front of the –10 hexamer, which is recognized by the σ70 region 3 [23-25]. It is worth mentioning that in E. coli the TRTGn-promoters are much less abundant than TGn-promoters: about 20% of promoters bear TGn, but only 18% among them have an additional 5′-TR-extension [24, 25]. As to the r-protein operon promoters, PrpsB represents a unique case. Mutational analysis revealed that high activity of PrpsB depends not only on the extended –10 element, but it also requires the (–35)TTG(–33) subsequence. According to the literature, the TRTGn-promoters bear significant deviations in the –35 consensus hexamer (3/6 and fewer matches) more often than classical –10/–35 promoters [25]. Moreover, it was argued that the –35 region might be nonessential for the extended –10 promoters [26]. However, this is not a general rule, e.g. in the –35 hexamer of the aroF promoter only one position is different from the consensus (TTGAaA), and this promoter was the strongest in a subset of 11 natural extended –10 E. coli promoters [25]. Thus, combinations of promoter modules may be largely diverse, which reinforces interest in studying individual natural promoters and relationships between their structure, transcriptional activity, and its regulation.
Regulation of r-protein operons upon stringent response. For a long time, studies of ribosome biogenesis regulation were focused on transcriptional control of rRNA operons and translational autogenous control of r-protein synthesis. Serious interest in promoters of r-protein operons emerged only recently [12]. It has been shown that not only rrn-promoters but also a number of r-protein operon promoters reduce their activities during stringent response in vivo as well as upon increasing ppGpp concentration in vitro, and that in both cases the effects require transcription factor DksA [12]. These data lead to the very important conclusion that transcription regulation contributes much to coordination of synthesis of all ribosomal components, both rRNA and r-proteins. Interestingly, in a subset of the studied r-protein promoters there were TGn-extended promoters, viz rplN, rpsA P1, rpsT P1, which were also able to decrease their activities in ppGpp/DksA-dependent manner despite the stabilization of the RNA polymerase–promoter complex by additional interactions with σ70 region 3 [12]. In these promoters, the TGn-extension is combined with the GC-rich discriminator, which, according to authors, could ensure negative stringent control; however, direct evidence was not provided. In our work, we directly showed that even the TRTGn-extended promoter (where the extension should stabilize the open complex to a higher extent [15, 24, 25]) was negatively regulated upon stringent response. Moreover, we provided evidence that the discriminator context is responsible for ppGpp-dependent negative regulation. Indeed, replacement of the rpsB GC-rich discriminator for an AT-rich sequence abolished the negative stringent control of PrpsB upon induction of starvation (Fig. 4c). It is of interest that a similar effect was achieved by a single change of C(–5) for G (Fig. 4d), which favors the optimal contact with σ70 region 1.2 [37, 38]. The overall results allowed us to conclude that optimal interactions of σ70 with the double-stranded promoter DNA (σ70 4.2 with TTG in the –35 element and σ70 3 with the TGTG-extension) are responsible for PrpsB efficiency, while the weakness of σ70 interactions with the non-template strand downstream from position –11 (due to the absence of contacts between A(–7) with σ70 2.3 and C(–5) with σ70 1.2) underlies the negative regulation of PrpsB during stringent response.
The sequence between the –10 hexamer and the transcription start is important not only for negative control by ppGpp/DksA, but also for positive regulation of some stress promoters, e.g. of the promoter of the uspA gene encoding universal stress protein A [39]. Activation of the uspA promoter was shown to require the AT-rich discriminator, and its alteration for the GC-rich sequence of rrnB P1 resulted in decreased promoter activity under stringent response [39]. The authors interpreted these data in terms of influences of ppGpp/DksA on kinetic parameters of promoters. If the lifetime of RPo (Fig. 1b) is short (the case of promoters with a GC-rich discriminator), its further destabilization by ppGpp/DksA negatively impacts the promoter activity. However, if RPo is stable (the case of the uspA promoter), the shortening of its lifetime has a positive effect by increasing the rate of promoter clearance and hence the promoter efficacy.
The mechanism for amplification of ppGpp action by DksA is still debated, and many things remain unclear and require further investigations. These two factors interact with different sites on the RNA-polymerase: DksA binds in the secondary channel [20] that is more than 30 Å away from the site of the ppGpp binding on the interface between ω and β′ subunits [40, 41]. While our data directly confirm the negative ppGpp-mediated regulation of the rpsB promoter under stringent response in vivo, the role of DksA as a cofactor of ppGpp in this case still remains unclear, as the activity of PrpsB decreased even in the DksA-deficient cells (Fig. 4b). Although the possibility of DksA-independent impact of ppGpp on the kinetic features of individual promoters is not theoretically excluded [42], quantitative data obtained in a purified in vitro transcription system are necessary to validate this assumption, which is an interesting task for future studies.
The authors are grateful to R. L. Gourse and E. Hajnsdorf for sharing strains from their collections.
This work was supported by the Russian Foundation for Basic Research grants 09-04-01014 and 12-04-01138 and by the Program of the Presidium of RAS “Molecular and Cell Biology”.
REFERENCES
1.Young, B. A., Gruber, T. M., and Gross, C. A.
(2002) Views of transcription initiation, Cell, 109,
417-420.
2.Browning, D. F., and Busby, S. J. (2004) The
regulation of bacterial transcription initiation, Nat. Rev.
Microbiol., 2, 57-65.
3.Gruber, T. M., and Gross, C. A. (2003) Multiple
sigma subunits and the partitioning of bacterial transcription space,
Ann. Rev. Microbiol., 57, 441-466.
4.Grigorova, I. L., Phleger, N. J., Mutalik, V. K.,
and Gross, C. A. (2006) Insights into transcriptional regulation and
sigma competition from an equilibrium model of RNA polymerase binding
to DNA, Proc. Natl. Acad. Sci. USA, 103, 5332-5337.
5.Haugen, S. P., Ross, W., and Gourse, R. L. (2008)
Advances in bacterial promoter recognition and its control by factors
that do not bind DNA, Nat. Rev. Microbiol., 6,
507-519.
6.Busby, S. (2009) More pieces in the promoter
jigsaw: recognition of –10 regions by alternative sigma factors,
Mol. Microbiol., 72, 809-811.
7.Hook-Barnard, I. G., and Hinton, D. M. (2007)
Transcription initiation by mix and match elements: flexibility for
polymerase binding to bacterial promoters, Gene Regul. Syst.
Biol., 1, 275-293.
8.Schultzaberger, R. K., Chen, Z., Lewis, K. A., and
Schneider, T. D. (2007) Anatomy of Escherichia coli
σ70 promoters, Nucleic Acids Res., 35,
771-788.
9.Feklistov, A. (2013) RNA polymerase: in search of
promoters, Ann. N. Y. Acad. Sci., 1293, 25-32.
10.Revyakin, A., Liu, C., Ebright, R. H., and
Strick, T. R. (2006) Abortive initiation and productive initiation by
RNA polymerase involve DNA scrunching, Science, 314,
1139-1143.
11.Feklistov, A., and Darst, S. A. (2011) Structural
basis for promoter –10 element recognition by the bacterial RNA
polymerase σ subunit, Cell, 147, 1257-1269.
12.Lemke, J. J., Sanchez-Vazquez, P., Burgos, H. L.,
Hedberg, G., Ross, W., and Gourse, R. L. (2011). Direct regulation of
Escherichia coli ribosomal protein promoters by the
transcription factors ppGpp and DksA, Proc. Natl. Acad. Sci.
USA, 108, 5712-5717.
13.Aseev, L. V., Levandovskaya, A. A., Tchufistova,
L. S., Skaptsova, N. V., and Boni, I. V. (2008) A new regulatory
circuit in ribosomal protein operons: S2-mediated control of the
rpsB-tsf expression in vivo, RNA,
14, 1882-1894.
14.Aseev, L. V., Levandovskaya, A. A., Skaptsova, N.
V., and Boni, I. V. (2009) Conservation of regulatory elements
controlling the expression of the rpsB-tsf operon in
γ-proteobacteria, Mol. Biol., 43, 101-107.
15.Voskuil, M. I., and Chamblis, G. H. (2002) The
TRTGn motif stabilizes the transcription initiation open complex, J.
Mol. Biol., 322, 521-532.
16.Dreyfus, M. (1988) What constitutes the signal
for the initiation of protein synthesis on Escherichia coli
mRNAs? J. Mol. Biol., 204, 79-94.
17.Xiao, H., Kalman, M., Ikehara, K., Zemel, S.,
Glaser, G., and Cashel, M. (1991) Residual guanosine
3′,5′-bispyrophosphate synthetic activity of relA
null mutants can be eliminated by spoT null mutations, J.
Biol. Chem., 266, 5980-5990.
18.Komarova, A. V., Tchufistova, L. S., Supina, E.
V., and Boni, I. V. (2002) Protein S1 counteracts the inhibitory effect
of the extended Shine–Dalgarno sequence on translation,
RNA, 8, 1137-1147.
19.Miller, J. H. (1972) Experiments in Molecular
Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
N.Y.
20.Paul, B. J., Barker, M. M., Ross, W., Schneider,
D. A., Webb C., Foster, J. W., and Gourse, R. L. (2004) DksA: a
critical component of the transcription initiation machinery that
potentiates the regulation of rRNA promoters by ppGpp and the
initiating NTP, Cell, 118, 311-322.
21.Paul, B. J., Berkmen, M. B., and Gourse, R. L.
(2005) DksA potentiates direct activation of amino acid promoters by
ppGpp, Proc. Natl. Acad. Sci. USA, 102, 7823-7828.
22.Crooks, G. E., Hon, G., Chandonia, J.-M., and
Brenner, S. E. (2004) WebLogo: a sequence logo generator, Genome
Res., 14, 1188-1190.
23.Barne, K. A., Bown, J. A., Busby, S. J., and
Minchin, S. D. (1997) Region 2.5 of the Escherichia coli RNA
polymerase σ70 subunit is responsible for the
recognition of the “extended −10” motif at promoters,
EMBO J., 16, 4034-4040.
24.Burr, T., Mitchell, J., Kolb, A., Minchin, S.,
and Busby, S. (2000) DNA sequence elements located immediately upstream
of the −10 hexamer in Escherichia coli promoters: a
systematic study, Nucleic Acids Res., 28, 1864-1870.
25.Mitchell, J. E., Zheng, D., Busby, S. J., and
Munchin, S. D. (2003) Identification and analysis of “extended
–10” promoters in Escherichia coli, Nucleic Acids
Res., 31, 4680-4695.
26.Kumar, A., Malloch, R. A., Fujita, N., Smillie,
D. A., Ishihama, A., and Hayward, R. S. (1993) The –35
recognition region of Escherichia coli σ70 is
inessential for initiation of transcription at an “extended
–10” promoter, J. Mol. Biol., 232,
406-418.
27.Ellinger, T., Behnke, D., Bujard, H., and Gralla,
J. D. (1994) Stalling of Escherichia coli RNA polymerase in
the +6 to +12 region in vivo is associated with tight
binding to consensus promoter elements, J. Mol. Biol.,
239, 455-465.
28.Hsu, L. M. (2002) Promoter clearance and escape
in prokaryotes, Biochim. Biophys. Acta, 1577,
191-207.
29.Vo, N. V., Hsu, L. M., Kane, C. M., and
Chamberlin, M. J. (2003) In vitro studies of transcript
initiation by Escherichia coli RNA polymerase. 3. Influences of
individual DNA elements within the promoter recognition region on
abortive initiation and promoter escape, Biochemistry,
42, 3798-3811.
30.Goldman, S. R., Ebricht, R. H., and Nickels, B.
E. (2009) Direct detection of abortive RNA transcripts in vivo,
Science, 324, 927-928.
31.Travers, A. A. (1984) Conserved features of
coordinately regulated E. coli promoters, Nucleic Acids
Res., 12, 2605-2618.
32.Pemberton, I. K., Muskhelishvili, G., Travers, A.
A., and Buckle, M. (2000) The G+C-rich discriminator region of the
tyrT promoter antagonizes the formation of stable preinitiation
complex, J. Mol. Biol., 16, 859-864.
33.Potrykus, K., and Cashel, M. (2008) (p)ppGpp:
still magical? Annu. Rev. Microbiol., 62, 35-51.
34.Aberg, A., Fernandez-Vazquez, J., Cabrer-Panes,
J. D., Sanchez, A., and Balsalobre, C. (2007) Similar and divergent
effects of ppGpp and DksA deficiencies on transcription in
Escherichia coli, J. Bacteriol., 191,
3226-3236.
35.Durfee, T., Hansen, A.-M., Zhi, H., Blattner, F.
R., and Jin, D. J. (2008) Transcription profiling of the stringent
response in Escherichia coli, J. Bacteriol.,
190, 1084-1096.
36.Toza, T., and Pizer, L. I. (1971) Biochemical
bases for the antimetabolite action of L-serine hydroxamate, J.
Bacteriol., 106, 972-982.
37.Haugen, S. P., Berkmen, M. B., Ross, W., Gaal,
T., Ward, C., and Gourse, R. L. (2006) rRNA promoter regulation by
non-optimal binding of sigma region 1.2: an additional recognition
element for RNA polymerase, Cell, 125, 1069-1082.
38.Haugen, S. P., Ross, W., Manrique, M., and
Gourse, R. L. (2008) Fine structure of the promoter–sigma region
1.2 interaction, Proc. Natl. Acad. Sci. USA, 105,
3292-3297.
39.Gummesson, B., Lovmar, M., and Nystrom, T. (2013)
A proximal promoter element required for positive transcriptional
control by ppGpp and DksA during the stringent response, J. Biol.
Chem., 288, 21055-21064.
40.Ross, W., Vrentas, C. E., Sanchez-Vazquez, P.,
Gaal, T., and Gourse, R. L. (2013) The magic spot: a ppGpp binding site
on E. coli RNA polymerase responsible for regulation of
transcription initiation, Mol. Cell, 50, 420-429.
41.Zuo, Y., Wang, Y., and Steitz, T. A. (2013) The
mechanism of E. coli RNA polymerase regulation by ppGpp is
suggested by the structure of their complex, Mol. Cell,
50, 430-436.
42.Magnusson, L. U., Gummesson, B., Joksmovic, P.,
Farewell, A., and Nystrom, T. (2007) Identical, independent, and
opposing roles of ppGpp and DksA in Escherichia coli, J.
Bacteriol., 189, 5193-5202.