Study of Wnt2 Secreted by A-549 Cells in Paracrine Activation of β-Catenin in Co-cultured Mesenchymal Stem Cells
N. S. Petrov and B. V. Popov*
Institute of Cytology, Russian Academy of Sciences, Tikhoretsky pr. 4, 194064 St. Petersburg, Russia; fax: +7 (812) 297-3541; E-mail: borisvp478@gmail.com* To whom correspondence should be addressed.
Received February 20, 2014
The canonical Wnt signal pathway is a key regulator of self-renewal and cell fate determination in various types of stem cells. The total pool of β-catenin consists of two different forms: the signaling form of the protein transmits the Wnt signals from the cell membrane to the target genes, whereas the membrane β-catenin is involved in formation of cell-to-cell contact at cadherin junctions. Earlier we developed an in vitro model of epithelial differentiation of mesenchymal stem cells (MSCs) co-cultured with epithelial A-549 cells. The purpose of the present work was to study the role of Wnt2 secreted by the A-549 cells in paracrine induction of β-catenin in co-cultured MSCs. Using the somatic gene knockdown technique, we obtained A-549 cell cultures with down-regulated WNT2. The MSCs co-cultured with the control A-549 cells displayed an increase in the levels of total cellular and signaling β-catenin and transactivation of a reporter construction containing the Lef/Tcf protein family binding sites. In contrast, β-catenin was not induced in the MSCs co-cultured with the A-549 cells with down-regulated WNT2 expression, but the total protein level was increased. We suggest that Wnt2 secreted by A-549 cells induces in co-cultured MSCs the Wnt/β-catenin signaling pathway, whereas the associated increase in total β-catenin level should be due to another mechanism.
KEY WORDS: mesenchymal stem cells, Wnt/β-catenin signaling pathwayDOI: 10.1134/S0006297914060054
Abbreviations: ABC, active (signaling) β-catenin; MSCs, mesenchymal stem cells; TBC, total β-catenin.
Wnt/β-catenin is one of few highly conserved signaling cascades
regulating the development of animals from lower multicellular ones to
mammals. The Wnt/β-catenin signaling pathway is steadily induced
during ante- and postnatal development in stem cells, and this seems to
explain the ubiquity of this protein [1]. Members
of the Wnt family form a family of extracellular regulators initiating
signal transduction through binding to Fz/Lrp receptors on the cell
surface [2]. Formation of the receptor complex
Wnt-Fz/Lrp leads to accumulation in the cytosol of the β-catenin
signaling form, which is translocated into the nucleus and transmits
signals through interactions with various transcription factors mainly
represented by proteins of the Lef/Tcf family (later designated as Tcf)
[3, 4]. Studies on the
participation of β-catenin in the regulation of development has
revealed another role: it plays a structural role during the formation
of cadherin contact junctions in the plasma membrane, which was shown
clearly in recent work by Lyashenko et al. [5]. In mice, embryonal stem cells unable to produce
β-catenin lose the ability to produce mesoderm and do not produce
neuroepithelium. The re-expression in such cells of β-catenin
deprived of the ability to transmit signals into the nucleus restores
the integrity of cadherin junctions and the ability of the cells to
produce the neuroepithelium. However, under these conditions embryonal
stem cells do not produce the mesodermal epithelial layer, which is
mediated by the β-catenin signaling form [5].
In mature organisms the β-catenin signaling form has genes
responsible for the regulation of self-renewal and differentiation of
somatic stem cells as targets [6, 7]. One such target is represented by the TCF4
gene, whose product plays a key role in proliferation of intestinal
somatic stem cells and in their differentiation into enterocytes [8]. In the epidermis the Wnt/β-catenin signals
regulate the self-renewal of bulge stem cells [9].
The role of the Wnt/β-catenin signals in the regulation of
self-renewal and differentiation of mesenchymal stem cells (MSCs)
remains unknown [10].
Populations of the signaling and membrane β-catenins are interrelated, but mechanisms regulating their levels and functions are different and uncoupled [11, 12]. The membrane β-catenin is stable, whereas the lifetime of the cytosolic form is very short [13]. A newly synthesized β-catenin interacts in the endoplasmic reticulum with cadherin and as a dimer is transported to the plasma membrane. Such interaction stabilizes β-catenin and prevents its phosphorylation by a special destructive complex in the cytosol [14]. An “excess” of the newly synthesized β-catenin is released from binding compounds, accumulated in the cytosol, and inactivated by the destructive complex. This double-pool β-catenin model is supported by existence of two different types of β-catenin homologs in C. elegans: the adhesion specific protein that binds cadherin but not Tcf, and two signaling proteins interacting with Tcf [15, 16].
In the absence of Wnt signals the cytoplasmic β-catenin is phosphorylated with casein kinase 1α (Ck1α) on Ser45. This modification is a primary label recognized by Gsk3β kinase, which induces the subsequent phosphorylation of the protein on Ser33/37/Thr41 [17]. In addition to the above-mentioned two kinases, the destructive complex also includes the tumor suppressor Apc (adenomatous polyposis coli), axin, and phosphatase Pp2a [18, 19]. Gsk3β phosphorylates β-catenin if this protein is in the complex with Apc and axin, whose affinity for β-catenin is regulated also through their specific phosphorylation [20]. β-Catenin phosphorylated on Ser33/37 is recognized by ubiquitin ligase bTrcP, which makes the polyubiquitin chain coupled to the protein and the protein recognizable by proteasomes for degradation [19, 21, 22]. β-Catenin phosphorylated in the N-terminal region retains its binding with Apc, otherwise it is immediately dephosphorylated by phosphatase Pp2a [23]. In the case of Apc or β-catenin mutations affecting the phosphorylation sites in its N-terminal region, the lifetime of β-catenin is significantly increased, which causes activation of Tcf4 and results in the occurrence of tumors of the large intestine, lung, and other organs [24, 25].
The mechanism inducing the accumulation in the cytosol of the β-catenin signaling form by the receptor complex Wnt-Fz/Lrp is still under discussion. It seems that Wnt signals suppress the β-catenin ubiquitinylation within the destructive complex without causing its dissociation, and this leads to functional inactivation of the complex after its saturation with phospho-β-catenin [26]. On the other hand, studies on kinetics of different forms of β-catenin under conditions of Wnt/β-catenin signaling pathway induction have revealed that the formation of the protein signaling form is caused by inhibition of its phosphorylation by both Ck1α and Gsk3β [27]. Finally, the β-catenin signaling form is accumulated in the cytosol and translocated into the nucleus and forms a complex with Tcf, converting it from a suppressor of transcription to its activator due to the presence of a transactivating domain in the β-catenin structure [28, 29].
The purpose of this work was to study mechanisms of the paracrine induction of the Wnt/β-catenin signals in MSCs. Earlier we have shown that the A-549 cells of epithelial origin induce in vitro expression of epithelial differentiation markers in bone marrow MSCs co-cultured under conditions of their separation with a cell-impenetrable membrane [30]. We supposed that Wnt2 hypersecreted by the A-549 cells [31] could induce β-catenin in the co-cultured MSCs. A special purpose of this work was to detect in the MSCs different forms of β-catenin using antibodies: 1) to its C-terminal fragment recognizing the total cellular protein; 2) to β-catenin dephosphorylated on Ser37/Thr41, i.e. to its signaling form. The second purpose of this work was to realize a somatic knockdown of the WNT2 gene in the A-549 cells and to select cell cultures with inhibited expression of WNT2. The third purpose of the work included co-cultivation of the obtained A-549 line cells with MSCs and the subsequent assessment of the total cellular and signaling β-catenin levels in the latter.
MATERIALS AND METHODS
Cell lines and cell transfection. The A-549 line cells originating from human lung carcinoma were obtained from the American Type Culture Collection (ATCC). MSCs were prepared from the bone marrow of transgenic male mice C57Bl/6-Tg(ACTbEGF)1Osb/J expressing green fluorescent protein (Gfp). The mice further designated as GFP were acquired from the commercial Jackson Laboratories (USA) source. To prepare MSCs, two GFP males were killed by cervical dislocation, and the bone marrow was washed from the tibial and femoral bones with growth medium DMEM supplemented with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 µg/ml) [30]. The bone marrow cells were washed by centrifugation at 400g for 10 min, counted in a Goryaev’s chamber in the presence of Trypan Blue to assess viability, resuspended in the growth medium to cell density of 5·106/ml, and cultured at the cell concentration of 2·106/ml in a CO2-incubator under standard conditions. The cells were splitted upon reaching the 90% saturation density from one into two or more 100 mm dishes; MSCs of 12-15 passages were used during the work. For experiments with LiCl inducing the Wnt/β-catenin signaling pathway [32], the cell cultures were incubated for 4 h in growth medium containing 10 mM LiCl (Sigma, USA).
MSCs were transfected with a 293Fectin™ reagent (Invitrogen, GB) in a well of a 24-well plate according the producer’s protocol. On the next day the MSCs were trypsinized, transferred into a well of a 12-well plate, and co-cultured with the A-549 cells during 72 h; then luciferase and β-galactosidase activities were determined in the MSCs. The A-549 cells were transfected with a pSuper vector (OligoEngine, USA) expressing the specific or the control small interfering RNA (siRNA) to WNT2 by electroporation on an X-cell Pulser device (BioRad, USA) with the following parameters: 360 V, 950 µF, 200 Ω. The buffer used for the electroporation contained 30.8 mM NaCl, 120.7 mM KCl, 1.46 mM KH2PO4, 8.1 mM Na2HPO4, and 10 mM MgCl2 [33].
Protein electrophoresis and immunoblotting. Proteins were subjected to electrophoresis in 8% polyacrylamide gel with SDS. After the electrophoresis, the proteins were transferred from the gel onto a PVDF membrane (Millipore, USA) and visualized using specific antibodies and a reagent enhancing the chemiluminescence (ECL) (Sigma) as described earlier [30].
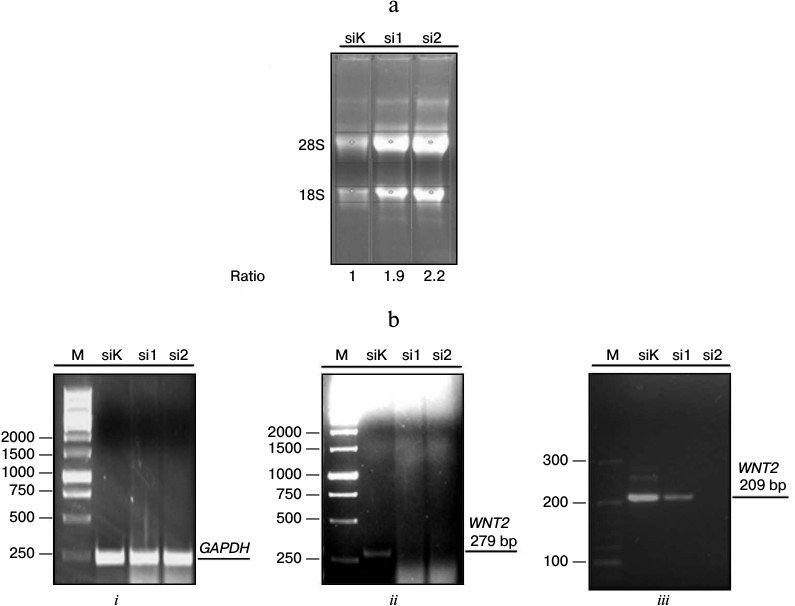
Preparation and electrophoresis of RNA in denaturing gel and its quantitative evaluation. RNA was extracted from the cultured cells with acidic water-saturated phenol. The RNA integrity was monitored and RNA amount was normalized by electrophoresis in a denaturing agarose gel supplemented with formaldehyde. Upon the electrophoresis, RNA was visualized in the gel by luminescence of the 18S and 28S bands stained with ethidium bromide. The RNA amounts in different specimens were normalized upon the determination of the 28S band intensity on the corresponding lanes using the TotalLab Quant program product.
Synthesis of cDNA on the RNA template and amplification of gene fragments using RT-PCR. To synthesize cDNA, 5 µg total RNA dissolved in 3-5 µl of deionized water were mixed with 0.5 µg oligo-dT18 and 2 µl of 10 mM dNTP, and the mixture volume was adjusted to 12 µl, incubated for 5 min at 65°C, cooled on ice, supplemented with 2 µl of 0.1 M dithiothreitol, 2 µl of 10-fold buffer for reverse transcriptase, 20 U of ribonuclease inhibitor, incubated for 2 min at 37°C, supplemented with 200 U of reverse transcriptase M-Mul V (Fermentas, Lithuania), incubated for 1 h, and the reaction was stopped by heating at 75°C for 15 min. The gene fragments were amplified by PCR. The mixture for PCR was prepared on ice and included 2.5 µl of 10-fold buffer for Taq polymerase, 1 µl of 5 mM mixture of dNTPs, 0.5 µl of 100 µM forward and reverse primers, 0.5 µl of cDNA or 0.5 µl of the PCR product, 0.125 µl of Taq polymerase with the activity of 5 U/µl (Beagle, Russia), and 20 µl of water to the reaction mixture total volume of 25 µl. Various gene fragments were amplified on a PCR Script cycler (Hybade, GB); the WNT2 was amplified using the same temperature regimen for both pairs of primers to WNT2: the initial denaturation – 94°C, 1 min (1 cycle); denaturation – 94°C, 15 s; annealing – 58°C (GAPDH) or 64°C (WNT2), 30 s; elongation – 72°C, 30 s (35 cycles for the amplification with cDNA, 20 cycles for the repeated amplification); elongation – 72°C, 10 min (1 cycle). For the amplification, primers were used that were synthesized by Beagle (Russia) (table).
Nucleotide sequences used for amplification of fragments of the
WNT2 gene and its somatic inactivation in A-549 cells

Note: bp, base pairs; nt, nucleotides.
Somatic knockdown of WNT2 using siRNA. The expression of the WNT2 gene in the A-549 cells was inhibited using RNA interference with the pSuper vector (OligoEngine, USA) as described in [34]. Sixty-meric oligonucleotides that contained sequences of 19 bases complementary to mRNA of the WNT2 gene inserted twice in inverted orientation, hairpin sequence in the oligonucleotide middle, and sticky ends for ligation into the BglII site on the 5′-end and XhoI site on the 3′-end were synthesized by Sigma (Germany) and inserted into the pSuper vector by the sites indicated above. The vectors carrying oligonucleotides for synthesis of siRNA against WNT2 mRNA or the control insertion were used for transfecting A-549 line cells by electroporation. After the transfection, the cells were selected for 3 days in the growth medium supplemented with puromycin (1.5 µg/ml). To analyze the efficiency of inhibition of the WNT2 gene expression, total RNA and cDNA were obtained from the cells and the gene WNT2 regions were amplified using two pairs of primers (table). The second pair of primers recognizing the DNA sequence within the PCR product 279 bp in size amplified with the first pair of primers was used for synthesis of a WNT2 fragment of 209 bp size.
Luciferase reporter assay. Reporter constructions containing seven Lef/Tcf binding sites in the regulatory region (CCTTTGATC) and the encoding sequence of the luciferase gene (TopFlash or Top), the corresponding control construction with mutation of the Lef/Tcf binding sites (CCTTTGGCC) (FopFlash or Fop) [24], and the expression vector containing β-catenin with the S33A mutation [35] were kindly presented by E. N. Tolkunova (Institute of Cytology, Russian Academy of Sciences). The plasmid containing the β-galactosidase gene was kindly presented by Dr. L-S. Chang (Ohio State University, USA) and was described earlier [36]. The transfection efficiency was normalized by the level of β-galactosidase expression, and the ratio of light activity in the cells transfected with Topflash and Fopflash was used as the initial activity of the reporter construction.
To analyze the luciferase and β-galactosidase activities, the transfected cells grown on a 24-well culture plate were washed twice with PBS and lysed by addition into every well of 150 µl of Glo-lysis buffer (Promega, USA). To determine the luciferase activity, to 50 µl of the lysate 50 µl of a BrightGlo reagent (Promega) was added, and the luminescence intensity was measured with a luminometer. To determine the β-galactosidase activity, 20 µl of the lysate was supplemented with 60 µl of Z-buffer containing 40 mM Na2HPO4, 60 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, and 20 µl of ONPG solution (4 mg/ml). The mixture was incubated for 1 h at room temperature, the reaction was stopped by addition of 50 µl of 1 M Na2CO3, and the optical density was measured at 420 nm. The activation of the Tcf-dependent expression was determined by the change in the ratio of the luciferase activity normalized by the β-galactosidase activity in the cells transfected with the Topflash construction to the normalized luciferase activity in the cells transfected with Fopflash.
RESULTS
Preparation of A-549 line cell cultures with inhibited expression of WNT2. Published data have indicated that the malignant transformation of A-549 cells is associated with hyperproduction of Wnt2 [31]. Because Wnt proteins are released into the extracellular space, we supposed that under conditions of co-cultivation this protein could induce in MSCs activation of the Wnt/β-catenin signaling pathway. Therefore, the inhibition of its production in A-549 cells could lead to a decrease in the activation of β-catenin in MSCs co-cultured under conditions of separation with a cell-impenetrable membrane. To test this hypothesis, we obtained stable cultures of A-549 cells with decreased expression of WNT2 caused with a pSuper system (OligoEngine). The stable expression in the A-549 cells of vectors producing siRNA to WNT2 (si1 and si2) induced, respectively, a decreased or completely abolished expression of this gene (Fig. 1).
Fig. 1. Inhibition of WNT2 gene expression in A-549 line cells with pSuper system (OligoEngine, USA). a) Electrophoresis of RNA in denaturing gel. Total RNA from A-549 cells transfected with plasmids containing pSuper vector with control oligonucleotide (siK) or with oligonucleotides recognizing specific sequences in the human WNT2 gene mRNA (si1, si2). The transfected cells were selected for 3 days using puromycin followed by extraction of total RNA, its denaturing gel electrophoresis, and determining the relative amount of RNA in the different samples to normalize them in the subsequent preparation of cDNA. b) Assessment of the efficiency of the inhibition of WNT2 gene expression using RT-PCR. i, ii, iii) electrophoresis of DNA in 1% (i, ii) and 3% (iii) agarose to characterize the PCR-amplification products, respectively, of the control gene GAPDH; fragments of the WNT2 gene with 279 and 209 bp size. M, markers of molecular mass; bp, base pairs.
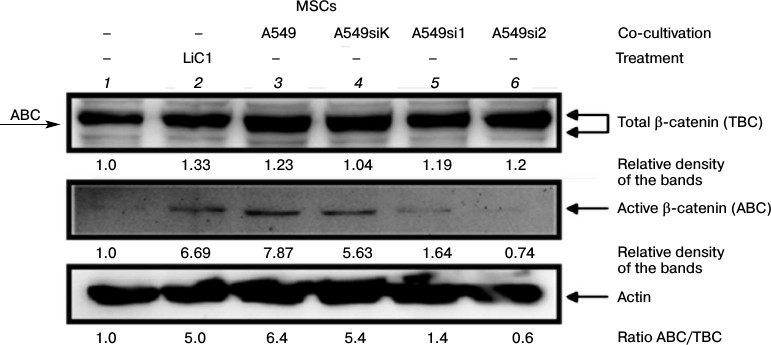
Characteristics of signaling and membrane β-catenin in MSCs co-cultured with A-549 cells with different levels of WNT2 expression. To elucidate the mechanism of β-catenin activation in MSCs co-cultured with A-549 cells, we co-cultured MSCs with A-549 line cells that had normal or decreased expression of the WNT2 gene and assessed by immunoblotting the β-catenin state in the MSCs. The control untreated MSCs produced β-catenin recognizable by immunoblotting as the major and minor bands with lower and higher electrophoretic mobility, respectively (Fig. 2, lane 1). The treatment of MSCs with lithium ions increased in these cells the amount of both total β-catenin (TBC) and its active (signaling) form (ABC) (Fig. 2, lane 2). On co-cultivation of MSCs with the normal cells or with those transfected with the pSuper vector with non-complementary oligonucleotide to the WNT2 gene mRNA, the total β-catenin level increased insignificantly, but the level of its signaling form (ABC) increased noticeably (Fig. 2, lanes 3 and 4, respectively). Determination of the ratio of the signaling β-catenin to the total β-catenin (ABC/TBC) in different groups showed that this value increases in the MSCs on their treatment with lithium ions or on co-cultivation with A-549 cells, either normal or transfected with the control vector (siK). We think that the ABC/TBC ratio more adequately characterizes the β-catenin activation than the TBC level. Moreover, this ratio also indicates that inhibition of the WNT2 gene expression in A-549 cells abolishes the β-catenin activation in the co-cultured MSCs (Fig. 2).
Fig. 2. Assessment by immunoblotting of active and total β-catenin expression in MSCs co-cultured with A-549 line cells with different levels of WNT2 expression. The A-549 line cells with stable expression of the pSuper vector containing the complementary (si1, si2) or non-complementary (siK) siRNA against the WNT2 gene mRNA and control untreated A-549 line cells were co-cultured for 72 h with MSCs of the 10th passage. The MSCs treated with LiCl were used as a positive control. After the co-cultivation, extracts from the MSCs were subjected to electrophoresis and immunoblotting, and electrophoretic replicas were stained with antibodies to active or total fraction of β-catenin. ABC, active β-catenin; TBC, total β-catenin; arrow “ABC” indicates the active β-catenin position relatively to that of the total protein on electrophoresis.
Overall, our data have confirmed the literature data indicating that A-549 line cells secret the Wnt2 protein [31]. Our data also indicated that the mechanism of activation of the Wnt/β-catenin signaling pathway in MSCs co-cultured with A-549 cells is underlain by secretion by the A-549 cells of Wnt2, which is responsible for paracrine activation of β-catenin in the MSCs. Results of this and the preceding experiments have shown that the activation of the Wnt/β-catenin signaling pathway is manifested not only by an increase in the level of total protein, but mainly in the increase in production of the active signaling β-catenin. It seems that the level of signaling β-catenin detected with specific antibodies to the protein dephosphorylated on Ser37/Thr41 most convincingly indicates the activation of the Wnt/β-catenin signaling pathway [37].
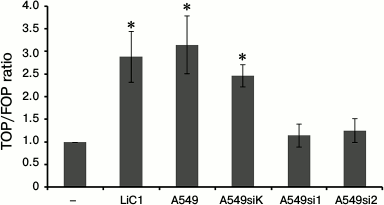
Functional assessment of the state of the Wnt/β-catenin signaling pathway in co-cultured MSCs using luciferase reporter assay. The transfection of MSCs with the reporter constructions TOP or FOP containing Tcf binding sites induces a 2.5-3.0-fold increase in reporter activation in MSCs co-cultured with A-459 cells – intact, treated with LiCl, or transfected with siRNA with the control insertion (Fig. 3). On the contrary, the A-549 cells with inhibited WNT2 do not transactivate in the MSCs the TOP reporter that contains binding sites of the Tcf family transcriptional factors (Fig. 3). These results correspond to data of immunoblotting presented in Fig. 2 and show the loss of the Wnt/β-catenin signaling pathway induction in the MSCs co-cultured with the A-549 cells deprived of WNT2 expression.
Fig. 3. Somatic knockdown of WNT2 in A-549 cells prevents transactivation of the reporter TOPflash in co-cultured MSCs. The MSCs were transfected with the reporters TOPflash/FOPflash and co-cultured with A-549 cells expressing or not expressing WNT2; MSCs treated for 4 h with 10 mM LiCl were used as a positive control. The luciferase activity was determined 96 h after the transfection and normalized by its efficiency using β-galactosidase as a control. The reporter vector activity was assessed as the ratio of conventional light units in specimens containing TOPflash to such in the control specimens containing FOPflash. The data are presented as the mean arithmetic value and standard deviation. * P < 0.05 compared to results in groups designated as A549si1 and A549si2.
DISCUSSION
In the present work, we studied the paracrine induction mechanism of the Wnt/β-catenin signaling pathway in MSCs co-cultured with A-549 line cells under conditions of cell separation with a cell-impenetrable membrane. The stable expression of the pSuper vector containing siRNA against WNT2 allowed us to obtain stable cultures of A-549 cells with decreased (si1) or completely inhibited (si2) expression of this gene (Fig. 2). The co-cultivation of the A-549 cells expressing the normal level of WNT2 with the MSCs was accompanied by a 6-8-fold increase in the level of the signaling β-catenin form (ABC), which was similar to the increase observed on the cells treated with LiCl (Fig. 2, lanes 2-4). Under these conditions, the level of total β-catenin (TBC) was increased only slightly, similar to the increase in MSCs co-cultured with A-549 cells with inhibited expression of WNT2 (Fig. 2, lanes 5 and 6). On the contrary, the level of ABC and the ABC/TBC ratio increased only in the MSCs co-cultured with A-549 producing the normal level of WNT2. Therefore, we suppose that an increase in the total β-catenin (TBC) level is not obligatory for activation of the canonical Wnt signaling pathway. Current publications have shown that the TBC level can increase not only as a result of induction of the canonical Wnt signaling pathway but can be a consequence of the interaction between β-catenin and signaling cascades of insulin, TGFβ, MAPK, etc. [38-40].
In our experiments the accumulation of the β-catenin signaling form in MSCs treated with lithium ions or co-cultured with A-549 cells corresponded to the functional activation of β-catenin targets. We found that the reporter containing binding sites of the Tcf family transcriptional factors is transactivated in MSCs treated with LiCl or co-cultured with A-549 cells expressing the normal but not decreased level of WNT2 (Fig. 3). Note that the Li+-caused induction of the total and signaling β-catenin was similar to the induction under conditions of cell co-cultivation. From our standpoint, the only important difference between β-catenin detected by immunoblotting on treatment of MSCs with lithium ions and β-catenin under conditions of cell co-cultivation is the increased electrophoretic mobility of the latter (Fig. 2, lanes 3-4 and 2, respectively). The electrophoretic mobility of a protein is known to depend on the number of phosphorylated groups in its structure, and an increase in their number even by one residue decreases the mobility [41]. We suppose that the signaling form of β-catenin produced in the MSCs under co-culture conditions has greater electrophoretic mobility than the form produced on treatment of MSCs with LiCl because of an additional dephosphorylation of the β-catenin molecule. The literature data show differences in mechanisms of β-catenin activation when the cells are treated with LiCl or Wnt. Thus, LiCl transactivates the Tcf reporter much more strongly than Wnt-1, whereas the two factors equally increase levels of total and active β-catenin in 293T cells [37]. It seems that the increased electrophoretic mobilities of ABC and TBC in MSCs co-culture detected by us compared to the increase caused by lithium ions can be a result of a modification giving β-catenin additional abilities in the regulation of cell-to-cell contacts and cell differentiation, but such studies were beyond the purpose of the present work. In the future, studies on functional features of differently dephosphorylated forms of signaling β-catenin may be promising for understanding the pathogenesis of diseases in which a constitutive induction of Wnt/β-catenin signals has been revealed, e.g. lung and intestine carcinomas, pulmonary fibrosis, acute lymphoid leukemia, etc. [24, 42, 43].
The authors are deeply grateful to E. N. Tolkunova (Institute of Cytology, Russian Academy of Sciences) and to Dr. L-S. Chang (Ohio State University, USA) for the presented constructions.
This work was supported by the Russian Foundation for Basic Research (projects Nos. 12-04-00252 and 14-04-31115).
REFERENCES
1.Schuijers, J., and Clevers, H. (2012) EMBO
J., 31, 2685-2696.
2.Nusse, R., and Varmus, H. (2012) EMBO J.,
31, 2670-2684.
3.Behrens, J., von Kries, J. P., Kuhl, M., Bruhn, L.,
Wedlich, D., Grosschedl, R., and Birchmeier, W. (1996)
Nature, 382, 638-642.
4.Huber, O., Korn, R., McLaughlin, J., Ohsugi, M.,
Herrmann, B. G., and Kemler, R. (1996) Mech. Dev., 59,
3-10.
5.Lyashenko, N., Winter, M., Migliorini, D.,
Biechele, T., Moon, R. T., and Hartmann, C. (2011) Nat. Cell
Biol., 13, 753-761.
6.Reya, T., and Clevers, H. (2005) Nature,
434, 843-850.
7.Ying, Q. L., Wray, J., Nichols, J., Batlle-Morera,
L., Doble, B., Woodgett, J., Cohen, P., and Smith, A. (2008)
Nature, 453, 519-523.
8.Van Es, J. H., van Gijn, M. E., Riccio, O., van den
Born, M., Vooijs, M., Begthel, H., Cozijnsen, M., Robine, S., Winton,
D. J., Radtke, F., and Clevers, H. (2005) Nature, 435,
959-963.
9.Alonso, L., and Fuchs, E. (2003) Proc. Natl.
Acad. Sci. USA, 100, Suppl. 1, 11830-11835.
10.Ling, L., Nurcombe, V., and Cool, S. M. (2009)
Gene, 433, 1-7.
11.Gottardi, C. J., and Gumbiner, B. M. (2004) J.
Cell Biol., 167, 339-349.
12.Heuberger, J., and Birchmeier, W. (2010) Cold
Spring Harb. Perspect. Biol., 2, a002915.
13.Kimelman, D., and Xu, W. (2006) Oncogene,
25, 7482-7491.
14.Huber, A. H., and Weis, W. I. (2001) Cell,
105, 391-402.
15.Korswagen, H. C., Herman, M. A., and Clevers, H.
C. (2000) Nature, 406, 527-532.
16.Liu, J., Phillips, B. T., Amaya, M. F., Kimble,
J., and Xu, W. (2008) Dev. Cell, 14, 751-761.
17.Liu, C., Li, Y., Semenov, M., Han, C., Baeg, G.
H., Tan, Y., Zhang, Z., Lin, X., and He, X. (2002) Cell,
108, 837-847.
18.Papkoff, J., Rubinfeld, B., Schryver, B., and
Polakis, P. (1996) Mol. Cell Biol., 16, 2128-21234.
19.Hart, M. J., de los Santos, R., Albert, I. N.,
Rubinfeld, B., and Polakis, P. (1998) Curr. Biol., 8,
573-581.
20.Wu, D., and Pan, W. (2010) Trends Biochem.
Sci., 35, 161-168.
21.Kishida, S., Yamamoto, H., Ikeda, S., Kishida,
M., Sakamoto, I., Koyama, S., and Kikuchi, A. (1998) J. Biol.
Chem., 27, 10823-10826.
22.Sakanaka, C., Weiss, J. B., and Williams, L. T.
(1998) Proc. Natl. Acad. Sci. USA, 95, 3020-3023.
23.Su, Y., Fu, C., Ishikawa, S., Stella, A., Kojima,
M., Shitoh, K., Schreiber, E. M., Day, B. W., and Liu, B. (2008)
Mol. Cell, 32, 652-661.
24.Korinek, V., Barker, N., Morin, P. J., van
Wichen, D., de Weger, R., Kinzler, K. W., Vogelstein, B., and Clevers,
H. (1997) Science, 275, 1784-1787.
25.Morin, P. J., Sparks, A. B., Korinek, V., Barker,
N., Clevers, H., Vogelstein, B., and Kinzler, K. W. (1997)
Science, 275, 1787-1790.
26.Li, V. S., Ng, S. S., Boersema, P. J., Low, T.
Y., Karthaus, W. R., Gerlach, J. P., Mohammed, S., Heck, A. J.,
Maurice, M. M., Mahmoudi, T., and Clevers, H. (2012) Cell,
149, 1245-1256.
27.Hernandez, A. R., Klein, A. M., and Kirschner, M.
W. (2012) Science, 338, 1337-1340.
28.Nguyen, H., Rendl, M., and Fuchs, E. (2006)
Cell, 127, 171-183.
29.Archbold, H. C., Yang, Y. X., Chen, L., and
Cadigan, K. M. (2012) Acta Physiol. (Oxford), 204,
74-109.
30.Popov, B. V., Serikov, V. B., Petrov, N. S.,
Izusova, T. V., Gupta, N., and Matthay, M. A. (2007) Tissue
Eng., 13, 2441-2450.
31.You, L., He, B., Xu, Z., Uematsu, K., Mazieres,
J., Fujii, N., Mikami, I., Reguart, N., McIntosh, J. K., Kashani-Sabet,
M., McCormick, F., and Jablons, D. M. (2004) Cancer Res.,
64, 5385-5389.
32.Stambolic, V., Ruel, L., and Woodgett, J. R.
(1996) Curr. Biol., 6, 1664-1668.
33.Ray, K. P., Farrow, S., Daly, M., Talabot, F.,
and Searle, N. (1997) Biochem. J., 328, 707-715.
34.Brummelkamp, T. R., Bernards, R., and Agami, R.
(2002) Science, 296, 550-553.
35.Aberle, H., Bauer, A., Stappert, J., Kispert, A.,
and Kemler, R. (1997) EMBO J., 16, 3797-3804.
36.Popov, B. V., Watt, S. W., Rozanov, Iu. M., and
Chang, L. S. (2010) Mol. Biol., 44, 323-334.
37.Staal, F. J., Noort, M. M., Strous, G. J., and
Clevers, H. C. (2002) EMBO Rep., 3, 63-68.
38.Fuxe, J., Vincent, T., and Garcia de Herreros, A.
(2010) Cell Cycle, 9, 2363-2374.
39.Krejci, P., Aklian, A., Kaucka, M., Sevcikova,
E., Prochazkova, J., Masek, J. K., Mikolka, P., Pospisilova, T.,
Spoustova, T., Weis, M., Paznekas, W. A., Wolf, J. H., Gutkind, J. S.,
Wilcox, W. R., Kozubik, A., Jabs, E. W., Bryja, V., Salazar, L.,
Vesela, I., and Balek, L. (2012) PLoS One, 7, e35826.
40.Palsgaard, J., Emanuelli, B., Winnay, J. N.,
Sumara, G., Karsenty, G., and Kahn, C. R. (2012) J. Biol. Chem.,
287, 12016-12026.
41.Knudsen, E. S., and Wang, J. Y. J. (1996) J.
Biol. Chem., 271, 8313-8320.
42.Roman-Gomez, J., Cordeu, L., Agirre, X.,
Jimenez-Velasco, A., San Jose-Eneriz, E., Garate, L., Calasanz, M. J.,
Heiniger, A., Torres, A., and Prosper, F. (2007) Blood,
109, 3462-3469.
43.Lam, A. P., Flozak, A. S., Russell, S., Wei, J.,
Jain, M., Mutlu, G. M., Budinger, G. R., Feghali-Bostwick, C. A.,
Varga, J., and Gottardi, C. J. (2011) Am. J. Respir. Cell Mol.
Biol., 45, 915-922.