Arachidonic Acid Activates Release of Calcium Ions from Reticulum via Ryanodine Receptor Channels in C2C12 Skeletal Myotubes
E. R. Muslikhov, I. F. Sukhanova, and P. V. Avdonin*
Koltsov Institute of Developmental Biology, Russian Academy of Sciences, ul. Vavilova 26, 119334 Moscow, Russia; E-mail: pvavdonin@yandex.ru; eldr87@gmail.com; Irina131280@yandex.ru; idbras@bk.ru* To whom correspondence should be addressed.
Received December 4, 2013; Revision received January 15, 2014
Arachidonic acid causes an increase in free cytoplasmic calcium concentration ([Ca2+]i) in differentiated skeletal multinucleated myotubes C2C12 and does not induce calcium response in C2C12 myoblasts. The same reaction of myotubes to arachidonic acid is observed in Ca2+-free medium. This indicates that arachidonic acid induces release of calcium ions from intracellular stores. The blocker of ryanodine receptor channels of sarcoplasmic reticulum dantrolene (20 µM) inhibits this effect by 68.7 ± 6.3% (p < 0.001). The inhibitor of two-pore calcium channels of endolysosomal vesicles trans-NED19 (10 µM) decreases the response to arachidonic acid by 35.8 ± 5.4% (p < 0.05). The phospholipase C inhibitor U73122 (10 µM) has no effect. These data indicate the involvement of ryanodine receptor calcium channels of sarcoplasmic reticulum in [Ca2+]i elevation in skeletal myotubes caused by arachidonic acid and possible participation of two-pore calcium channels from endolysosomal vesicles in this process.
KEY WORDS: calcium ions, skeletal myotubes, arachidonic acid, ryanodine-sensitive channelsDOI: 10.1134/S0006297914050071
Abbreviations: AA, arachidonic acid; ARC, arachidonate-regulated calcium channels; RyR1, ryanodine receptor type I channels; TPC, two-pore channels.
Calcium ions play a key role in the regulation of a wide variety of
processes occurring in living organisms ranging from protozoa to higher
animals and plants. Since the origin of life calcium, thanks to its
chemical properties, is involved in regulation of many biochemical
reactions and physiological processes inside cells. The concentration
of calcium ions in the cytoplasm of resting cells is about 100 nM.
With its increase, energy metabolism, secretion, contraction, and many
others processes are activated. At the same time, long-term increase in
the concentration of Ca2+ in the cytoplasm leads to calcium
overload, which is toxic to cells since it can cause the aggregation of
proteins and nucleic acids, deposition of phosphate, and disruption of
cell membranes [1, 2].
Therefore, cells constantly spend energy to pump calcium to maintain
low levels of [Ca2+]i. A key role in pumping of
calcium ions from the cytoplasm is played by sarcoplasmic/endoplasmic
reticulum Ca2+-transporting ATPase (SERCA) [3]. At the same time, the reticulum serves as a depot
of calcium ions performing signaling functions. Mitochondria having
large buffer capacity for these ions are also involved in the
maintenance of calcium homeostasis. In 2002, a new intracellular source
of calcium ions, lysosomes and acid lysosome-like vesicles, was found
[4]. Release of Ca2+ from acid
endolysosomal vesicles occurs via special type of channels –
two-pore channels (TPC) activated by second messenger nicotinic acid
adenine dinucleotide phosphate (NAADP) [5]. The
physiological role of this calcium depot is not well studied. One of
the functions of TPCs in endolysosomes is to trigger calcium-induced
calcium release (CICR) from the reticulum [6].
The development of cell specialization along with morphological peculiarities of functional systems regulating Ca2+ inflow into the cytoplasm specific to a particular type of cell has appeared. An example of this is skeletal myotubes – excitable cells in which release of calcium ions from the reticulum occurs through ryanodine receptor type 1 channels (RyR1) [7]. In the last few years, it has been found that in differentiated skeletal muscle cells, along with Ca2+ release from sarcoplasmic reticulum in response to depolarization of sarcolemma, receptor-dependent mechanisms of [Ca2+]i regulation are operating. C2C12 skeletal myoblasts that can under certain conditions differentiate into multinuclear myotubes is a useful and convenient object for studying calcium metabolism in striated muscle cells. As differentiation of C2C12 myoblasts into myotubes is going on, they have increased expression of all three types of inositoltrisphosphate (InsP3) receptors [8], and this is accompanied by an increase in calcium signal in response to ATP via P2Y purinergic receptors [9]. Store-operated calcium entry (SOCE) characteristic of non-excitable cells has been identified in skeletal muscle cells [10, 11], and it was demonstrated that Orai1 channels are involved in SOCE in skeletal myotubes [12, 13]. In addition, it was found that several TRP (transient receptor potential) channels are expressed in C2C12 myotubes [14]. Some of these participate in SOCE, and others participate in Ca2+ influx in response to mechanical stimuli [15, 16]. Another representative of the Orai family – Orai3 – is also expressed in skeletal muscles [17]. It is known that Orai3 is part of the ARC channels (arachidonate-regulated calcium channels) whose specific activator is arachidonic acid (AA) [18]. ARC channels are formed by three Orai1 and two Orai3 molecules as well as by protein STIM1 localized in plasma membrane. ARC channels are revealed in several types of non-excitable cells [18], but it is not clear how widespread these channels are in different types of cells.
It was established that the products of the phospholipase A2 reaction – AA and lysophosphatidylcholine – are able to regulate the activity of calcium channels in various cell types [19, 20]. It turned out that one of the isoforms of this enzyme, a calcium-independent phospholipase A2 (iPLA2), participates in the regulation of SOCE in skeletal myotubes [21]. SOCE is activated in myotubes by lysophosphatidylcholine [22]. AA is also considered to be a second messenger [20]. AA can affect neighboring cells as a paracrine regulator since it easily penetrates cell membranes and is released into the environment. There is evidence that in addition to ARC channels, AA regulates activity of several TRP channels and a number of other Ca2+-transporting channels [23]. In particular, it was shown that in some cells AA activates ryanodine receptor channels [24]. So today we have a wide list of AA targets in different types of cells, but it remains unclear whether and how it affects intracellular calcium metabolism in skeletal muscle cells.
MATERIALS AND METHODS
Experiments were performed on C2C12 myoblasts (ATCC collection). To induce differentiation of myoblasts into multinucleated myotubes culture medium DMEM with 10% fetal calf serum was replaced by DMEM with 2% horse serum. After replacement of the medium, some of the myoblasts fuse to form myotubes. When replacing the growth medium to differentiation medium, the cells were not treated to stop divisions, thus undifferentiated myoblasts continued to divide for some time and filled the free space between the myotubes.
Myotubes formed after five days growth in the differentiation medium were stained with monoclonal antibodies (Developmental Studies Hybridoma Bank, Iowa City, IA, USA) against a marker of myogenic differentiation – myosin heavy chains (MHC). Goat antibodies labeled with Alexa Fluor 594 (Molecular Probes, USA) were used as second antibodies.
Arachidonic acid (Sigma-Aldrich, USA) dissolved in 0.9% NaCl with bovine serum albumin (BSA) was used. BSA binds and stabilizes AA. The total concentration of AA in the incubation medium was 1 mM, and the concentration of free AA was less than 10 µM since more than 99% of AA binds to BSA [23]. To evaluate the effectiveness of AA, its activity was tested in the reaction of platelet aggregation. BSA solution without AA was used as a control.
Studies of the influence of AA on free cytoplasmic calcium concentration ([Ca2+]i) were performed with C2C12 cells grown in 24-well plates on the fifth day after replacement of the growth medium by differentiation medium. To load the cells with Fura-2/AM (Invitrogen, USA), they were initially washed once with physiological salt solution (PSS) containing 145 mM NaCl, 5 mM KCl, 5 mM HEPES, 1 mM MgCl2, 1 mM CaCl2, and 10 mM glucose (pH 7.4). Then the cells were incubated for 1 h in PSS with 2 µM Fura-2/AM and 0.04% Pluronic F-127 (Molecular Probes) at room temperature. Then the cells were washed free from the probe and incubated for about one hour in PSS for full hydrolysis of Fura-2/AM in the cytoplasm. Fluorescence of individual Fura-2-loaded myoblasts and myotubes was measured using a Leica DM 6000 microscope (Leica Microsystems, Germany) at 25°C. Changes in [Ca2+]i are presented as ratio of fluorescence at emission wavelength 508 nm at excitation wavelengths 340 and 387 nm. Background fluorescence was subtracted.
The curves presented in the figures were obtained by averaging the data of five independent experiments in each of which the fluorescence of 5-10 individual myoblasts or myotubes were registered. The means ± SEM are presented. For calculations, the LAS AF Lite and Microsoft Excel programs were used. Statistical significance was calculated according to Student’s t-test.
RESULTS
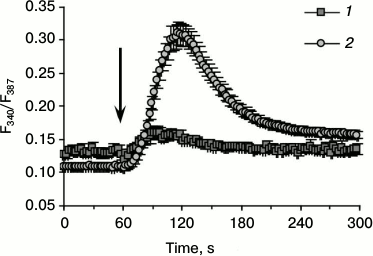
Figure 1 shows C2C12 cells after five days incubation in differentiation medium stained with antibodies to myosin heavy chains (MHC). Besides stained multinucleated cells, undifferentiated mononuclear cells containing no MHC are observed in the field of view. The fluorescence of Fura-2-loaded multinucleated myotubes and undifferentiated myoblasts was registered. The averaged curves depicting kinetics of [Ca2+]i changes in response to AA in individual myotubes and myoblasts are presented in Fig. 2.
Administration of AA caused a rapid and abundant inflow of Ca2+ into the cytoplasm of myotubes. This effect was not observed when control solution of BSA without AA was added. In undifferentiated C2C12 myoblasts, the increase in [Ca2+]i under the action of AA was much less pronounced (Fig. 2).
Fig. 1. Multinucleated myotubes and undifferentiated myoblasts on the fifth day of incubation in differentiation medium. The cells were stained with antibodies against heavy myosin chains and nuclear specific stain Hoechst.

Fig. 2. Changes in the ratio of fluorescence of Fura-2-loaded myoblasts (1) and myotubes (2) in response to AA at 340 and 387 nm excitation wavelengths. The arrow indicates administration of AA.
It was demonstrated that elevation of [Ca2+]i in skeletal myotubes might be due to its entry via voltage-gated [25, 26] and store-operated Orai1 channels [12, 13]. Furthermore, as noted, the presence of ARC channels in the myotubes might be expected, since its component Orai3 is expressed in skeletal muscle [17]. To determine whether calcium ions enter skeletal myotubes under the action of AA from the extracellular space, we studied the effect of AA on cells placed in medium without Ca2+. Comparison of the responses of myotubes to AA in medium with or without calcium shows lack of significant differences in kinetics and in the amplitude of [Ca2+]i elevations (Figs. 3 and 4). This indicates that AA induces [Ca2+]i increase in myotubes due to release of calcium ions from intracellular stores.
Fig. 3. Kinetics of [Ca2+]i elevation in C2C12 myotubes in response to AA in standard PSS with Ca2+ (1), in PSS without Ca2+ (2), and in PSS with Ca2+ after 10 min preincubation with 20 µM dantrolene (3). The arrow indicates administration of AA.
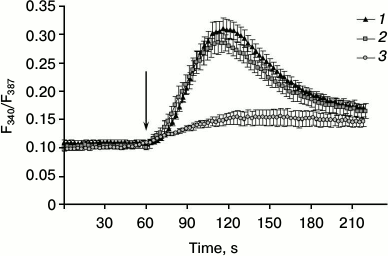
Fig. 4. Amplitudes of [Ca2+]i elevation in C2C12 myotubes in response to AA under control conditions in PSS with Ca2+ (1), in PSS without Ca2+ (2), in PSS with Ca2+ after preincubation for 10 min with 20 µM trans-NED19 (3), 10 µM U73122 (4), or 20 µM dantrolene (5). Significance of the difference from control: * p < 0.05; ** p < 0.001. The data from 3-5 independent experiments are presented.
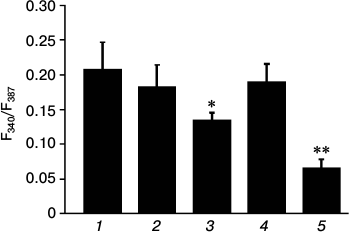
The major intracellular depot of Ca2+ in skeletal myotubes is sarcoplasmic reticulum. Ryanodine receptor channels type 1 (RyR1) and, as previously shown [8], three types of InsP3-sensitive channels are located in its membrane. We checked the possible involvement of phospholipase C, which catalyzes synthesis of InsP3, in the effect of AA on [Ca2+]i in myotubes. For this purpose, the inhibitor of phospholipase C U73122 [27, 28] was used. Phospholipase C inhibitor and its inactive analog U73343 at 10 µM concentration do not affect [Ca2+]i increase in myotubes in response to AA (Fig. 4).
Contraction of skeletal muscle occurs after depolarization of the plasma membrane, the activation of dihydropyridine receptors, and transfer of excitation on RyR1 channels of sarcoplasmic reticulum. Dantrolene is a blocker of RyR1 channels [29]. Incubation of C2C12 myotubes for 10 min with 25 µM dantrolene resulted in a decrease in [Ca2+]i elevation in response to AA by 68.7 ± 6.3% (Figs. 3 and 4). These data indicate that RyR1 calcium channels are involved in the increase in [Ca2+]i induced by AA. ATP is a powerful stimulator of calcium metabolism in skeletal myotubes [9]. In our experiments, dantrolene had no effect on ATP-induced increase in [Ca2+]i in myotubes (data not shown). This suggests that Ca2+-signaling mechanisms of AA and ATP are different.
Endolysosomes were recently found to be an intracellular source of calcium ions [4]. The pool of calcium ions in endolysosomes is relatively small but sufficient for the realization of their physiological function: calcium signal propagation via Ca2+-induced Ca2+ release (CICR). The exit of calcium ions from endolysosomal vesicles occurs through a special type of channels – two-pore calcium channels (TPC), which are activated by the second messenger nicotinic acid adenine dinucleotide phosphate (NAADP) [5]. To determine the role of TPC channels in AA-induced increase in [Ca2+]i, we used their specific inhibitor trans-NED19 [30]. Preincubation of myotubes with 10 µM trans-NED19 decreased the amplitude of the increase in [Ca2+]i in response to AA by 35.8 ± 5.4% (Fig. 4).
DISCUSSION
In the present work we have shown that AA activates release of calcium ions from the intracellular stores in skeletal myotubes. Dantrolene considerably suppresses the effect of AA. This indicates that ryanodine receptor calcium channels in sarcoplasmic reticulum are involved in AA-induced increase in [Ca2+]i in C2C12 myotubes. The effect of AA on calcium metabolism in myoblasts is much less pronounced. The reason for this might be insufficient development of sarcoplasmic reticulum and RyR1 channels in undifferentiated progenitor muscle cells.
The increase in [Ca2+]i in response to AA was shown in HEK293 cells, where AA stimulates both Ca2+ mobilization from internal stores and Ca2+ influx [31]. The mechanism of AA-induced [Ca2+]i elevation in HEK293 cells is different from the mechanisms Ca2+-signaling in response to carbachol, caffeine, or thapsigargin. In several types of cells, AA activates calcium ions entry via ARC channels, whose components are Orai1, Orai3, and STIM1 [18]. We did not observe such a mechanism of calcium ion entry into skeletal myotubes. However, it might be that Ca2+ influx via ARC channels if they are present in myotubes causes increase in Ca2+ concentration locally near the plasma membrane ([Ca2+]pm), and the Fura-2 method is not sensitive enough to detect changes in [Ca2+]pm as shown in [32].
Activation of Ca2+ release in response to 150 µM AA was demonstrated in pancreatic β-cells, which also possess ryanodine receptor channels [24]. Increase in [Ca2+]i was due to release of calcium ions from intracellular depots and influx from the extracellular environment. Elevation of [Ca2+]i induced by AA was attenuated by the SERCA inhibitor thapsigargin [24]. Application of the blockers of InsP3 receptors and RyR channels demonstrated that AA selectively affects the latter [24]. 5,8,11,14-Eicosatetraenoic acid, a non-metabolizable analog of AA, mimicked the effect of AA, indicating that AA itself stimulates ryanodine receptors in β-cells. AA-induced Ca2+ influx in β-cells is apparently due to subsequent activation of store-operated channels after emptying of the reticulum.
The ability of AA to stimulate the release of calcium ions through the ryanodine receptor channels was demonstrated in cardiomyocytes [33]. This effect was reproduced by 5,8,11,14-eicosatetraenoic acid and was not blocked by inhibitors of cyclooxygenase, epoxygenase, and lipoxygenase. The latter exclude involvement of AA metabolites in its action on calcium metabolism in cardiac muscle cells. According to this work [33], increase in Ca2+ release via ryanodine receptor channels induced by AA occurs due to enhancement of Ca2+ content in sarcoplasmic reticulum and potentiation of caffeine-induced Ca2+ mobilization. It was demonstrated that in cardiomyocytes AA strengthens calcium signals and contractions in response to glucagon [33], catecholamines [34], and TNF-α [35].
We have shown that the NAADP receptor antagonist trans-NED19 causes slight inhibition of the effect of AA on [Ca2+]i in myotubes. NAADP receptors are coupled with two-pore calcium channels located in endolysosomal vesicles, and our data suggest that AA also activates this mechanism of intracellular calcium metabolism. It is interesting that in pancreatic β-cells acidic lysosome-like vesicles are also somehow involved in the effect of AA. This is evidenced by the fact that disruption of acidic stores with the inhibitor of vacuolar H+-ATPase bafilomycin A1 or glycyl-phenylalanyl-β-naphthylamide (GPN) decreases calcium signals in response to AA [36].
These data support the idea that AA functions as a second messenger regulating calcium metabolism in cells expressing ryanodine receptor calcium channels. To evaluate the physiological role of AA in regulation of skeletal muscle cell contractions and their other functions, further investigations are necessary.
The study was financially supported by the Russian Foundation for Basic Research (grant Nos. 11-04-01520 and 14-04-00951) and by the Swiss National Science Foundation (SCOPES No. IB74AO-110940).
REFERENCES
1.Case, R. M., Eisner, D., Gurney, A., Jones, O.,
Muallem, S., and Verkhratsky, A. (2007) Cell Calcium, 42,
345-350.
2.Clapham, D. E. (2007) Cell, 131,
1047-1058.
3.Rubtsov, A. M. (2005) Ros. Fiziol. Zh. im. I. M.
Sechenova, 91, 141-151.
4.Churchill, G. C., Okada, Y., Thomas, J. M.,
Genazzani, A. A., Patel, S., and Galione, A. (2002) Cell,
111, 703-708.
5.Brailoiu, E., Churamani, D., Cai, X., Schrlau, M.
G., Brailoiu, G. C., Gao, X., Hooper, R., Boulware, M. J., Dun, N. J.,
Marchant, J. S., and Patel, S. (2009) J. Cell Biol., 186,
201-209.
6.Calcraft, P. J., Ruas, M., Pan, Z., Cheng, X.,
Arredouani, A., Hao, X., Tang, J., Rietdorf, K., Teboul, L., Chuang, K.
T., Lin, P., Xiao, R., Wang, C., Zhu, Y., Lin, Y., Wyatt, C. N.,
Parrington, J., Ma, J., Evans, A. M., Galione, A., and Zhu, M. X.
(2009) Nature, 459, 596-600.
7.Rubtsov, A. M., and Batrukova, M. A. (1997)
Biochemistry (Moscow), 62, 933-945.
8.Muslikhov, E. R., Surkov, K. V., Sukhanova, I. F.,
and Avdonin, P. V. (2010) Biol. Membr. (Moscow), 27,
505-511.
9.Henning, R. H., Duin, M., van Popta, J. P.,
Nelemans, A., and den Hertog, A. (1996) Br. J. Pharmacol.,
117, 1785-1791.
10.Gutierrez-Martin, Y., Martin-Romero, F. J., and
Henao, F. (2005) Biochim. Biophys. Acta, 1711, 33-40.
11.Kurebayashi, N., and Ogawa, Y. (2001) J.
Physiol., 533, 185-199.
12.Avdonin, P. V., Surkov, K. V., Sukhanova, I. F.,
and Ruegg, U. T. (2008) Biol. Membr. (Moscow), 25,
348-354.
13.Lyfenko, A. D., and Dirksen, R. T. (2008) J.
Physiol., 586, 4815-4824.
14.Gailly, P. (2012) Curr. Opin. Pharmacol.,
12, 326-334.
15.Clapham, D. E. (2003) Nature, 426,
517-524.
16.Pedersen, S. F., Owsianik, G., and Nilius, B.
(2005) Cell Calcium, 38, 233-252.
17.Kiviluoto, S., Decuypere, J. P., De Smedt, H.,
Missiaen, L., Parys, J. B., and Bultynck, G. (2011) Skeletal
Muscle, 1, 16.
18.Shuttleworth, T. J. (2009) Cell Calcium,
45, 602-610.
19.Singaravelu, K., Lohr, C., and Deitmer, J. W.
(2008) Cerebellum, 7, 467-481.
20.Van der Zee, L., Nelemans, A., and den Hertog, A.
(1995) Biochem. J., 305 (Pt. 3), 859-864.
21.Boittin, F. X., Petermann, O., Hirn, C., Mittaud,
P., Dorchies, O. M., Roulet, E., and Ruegg, U. T. (2006) J. Cell
Sci., 119, 3733-3742.
22.Boittin, F. X., Shapovalov, G., Hirn, C., and
Ruegg, U. T. (2010) Biochem. Biophys. Res. Commun., 391,
401-406.
23.Meves, H. (2008) Br. J. Pharmacol.,
155, 4-16.
24.Woolcott, O. O., Gustafsson, A. J., Dzabic, M.,
Pierro, C., Tedeschi, P., Sandgren, J., Bari, M. R., Nguyen, K. H.,
Bianchi, M., Rakonjac, M., Radmark, O., Ostenson, C. G., and Islam, M.
S. (2006) Cell Calcium, 39, 529-537.
25.Fleischer, S., and Inui, M. (1989) Annu. Rev.
Biophys. Biophys. Chem., 18, 333-364.
26.Rios, E., and Pizarro, G. (1991) Physiol.
Rev., 71, 849-908.
27.Bleasdale, J. E., Thakur, N. R., Gremban, R. S.,
Bundy, G. L., Fitzpatrick, F. A., Smith, R. J., and Bunting, S. (1990)
J. Pharmacol. Exp. Ther., 255, 756-768.
28.Muto, Y., Nagao, T., and Urushidani, T. (1997)
J. Pharmacol. Exp. Ther., 282, 1379-1388.
29.Krause, T., Gerbershagen, M. U., Fiege, M.,
Weisshorn, R., and Wappler, F. (2004) Anaesthesia, 59,
364-373.
30.Naylor, E., Arredouani, A., Vasudevan, S. R.,
Lewis, A. M., Parkesh, R., Mizote, A., Rosen, D., Thomas, J. M., Izumi,
M., Ganesan, A., Galione, A., and Churchill, G. C. (2009) Nat. Chem.
Biol., 5, 220-226.
31.Luo, D., Sun, H., Lan, X., Xiao, R., and Han, Q.
(2005) Prostaglandins Other Lipid Mediat., 75,
141-151.
32.Basset, O., Boittin, F. X., Dorchies, O. M.,
Chatton, J. Y., van Breemen, C., and Ruegg, U. T. (2004) J. Biol.
Chem., 279, 47092-47100.
33.Sauvadet, A., Rohn, T., Pecker, F., and Pavoine,
C. (1997) J. Biol. Chem., 272, 12437-12445.
34.Magne, S., Couchie, D., Pecker, F., and Pavoine,
C. (2001) J. Biol. Chem., 276, 39539-39548.
35.Amadou, A., Nawrocki, A., Best-Belpomme, M.,
Pavoine, C., and Pecker, F. (2002) Am. J. Physiol. Cell
Physiol., 282, 1339-1347.
36.Yen-Yam-Wah, V., Lee, A. K., and Tse, A. (2012)
Cell Calcium, 51, 140-148.