REVIEW: Mechanisms of Phototransformation of Protochlorophyllide into Chlorophyllide
O. B. Belyaeva* and F. F. Litvin
Lomonosov Moscow State University, Faculty of Biology, 119992 Moscow, Russia; fax: +7 (495) 939-4309; E-mail: belyaeva0104@gmail.com* To whom correspondence should be addressed.
Received January 14, 2014
The purpose of this review is to summarize and discuss data obtained in studies on the mechanisms of the primary photophysical and photochemical reactions of protochlorophyllide photoreduction in plant materials (etiolated leaves and leaf homogenates) and in model systems. Based on the results of numerous studies, it can be stated that the reduction of active forms of the chlorophyll precursor is a multistep process comprising two or three short-lived intermediates characterized by a singlet ESR signal. The first intermediate is probably a complex with charge transfer between protochlorophyllide and the hydride ion donor NADPH. The conserved tyrosine residue Tyr193 of protochlorophyllide oxidoreductase is the donor of the second proton.
KEY WORDS: chlorophyll, protochlorophyllide, photoreduction, non-fluorescent intermediates, fluorescence spectra, absorption spectraDOI: 10.1134/S0006297914040038
Abbreviations: Chl, chlorophyll; Chld, chlorophyllide; Pchl, protochlorophyll; Pchld, protochlorophyllide; POR, protochlorophyllide oxidoreductase.
The final light-dependent stage of chlorophyll biosynthesis in plant
leaves involves photoreduction of its immediate precursor
protochlorophyllide and a range of light and dark reactions leading to
the formation of the system of functional chlorophyll forms of the two
photosystems of photosynthesis. Phototransformation of
protochlorophyllide into chlorophyllide includes the addition of two
hydrogen atoms in positions C17 and C18 of the tetrapyrrole
protochlorophyllide molecule, the double bond being replaced by a
single bond (Fig. 1). The high efficiency of this
photoreaction in etiolated leaves is due to the fact that it proceeds
within the photoactive complex emerging in etiolated leaves at the
preceding stage of chlorophyll precursor synthesis in the dark. The
active complex includes protochlorophyllide, the hydrogen donor NADPH,
and the photoenzyme protochlorophyllide oxidoreductase (POR) (see
review [1]). The structure of the active ternary
complex creates spatial relations between protochlorophyllide and
hydrogen donors favorable for the photoreduction reaction. The POR
photoenzyme catalyzes the reaction. The photoenzyme as well as the
protochlorophyllide of etioplasts are in an aggregated, most likely a
dimeric state.
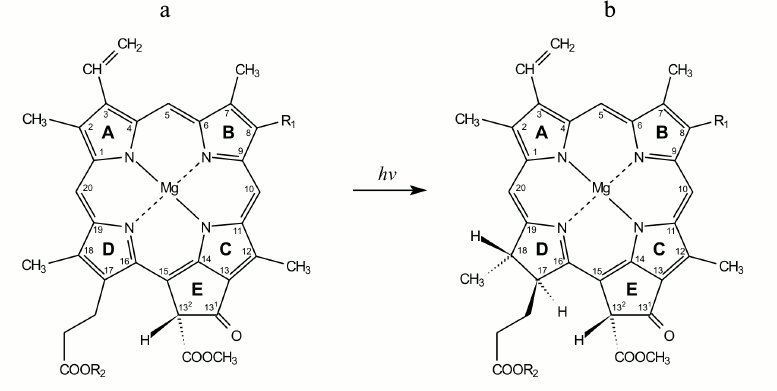
Fig. 1. Structural formulas of protochlorophyll(ide) (a) and chlorophyll(ide) (b) molecules. R1 corresponds to CH2–CH3 for the monovinyl pigment form and CH=CH2 for the divinyl form. R2 corresponds to C20H39 for protochlorophyll and chlorophyll and to hydrogen atom for protochlorophyllide and chlorophyllide.
There are several spectrally different forms of protochlorophyllide in both etiolated and green plant leaves (see review [1]). Pchld633/628, Pchld643/637, and the usually dominant Pchld655/650 are the main protochlorophyllide forms (the numbers show the position of fluorescence and absorption maxima). Several minor long-wave forms have also been found. Pchld655/650 is the main active form of protochlorophyllide; it is due to the transformation of this form that the bulk chlorophyll is synthesized.
Low temperature spectroscopy and high temporal resolution spectroscopy have shown that the photoreduction of protochlorophyllide is a rather complex process involving several fast light and dark reactions.
SHORT-LIVED INTERMEDIATES OF PROTOCHLOROPHYLLIDE
PHOTOTRANSFORMATION INTO CHLOROPHYLLIDE in vivo STABILIZED AT
LOW TEMPERATURES
Non-fluorescent intermediate X690. Studies of protochlorophyllide photoreduction in vivo (in etiolated leaves) at very low temperatures, when biochemical temperature-dependent stages are inhibited, reveal information on the mechanism of this reaction. The fluorescence quantum yield of the active protochlorophyllide form was found to decrease after leaves are illumination at low temperature (77-173 K) without the appearance of any new fluorescence bands. Subsequent increase in temperature in the dark causes the appearance of fluorescence bands characteristic for chlorophyllide. It was suggested that exciting light at low temperature causes the protochlorophyllide molecule to enter an intermediate state (non-fluorescent intermediate) that is transformed into chlorophyllide due to the next dark reaction proceeding on raising the temperature [2-14]. Photoinduced fluorescence quenching proceeds even at liquid helium temperature [12], indicating the elementary photophysical nature of the primary reaction.
The absorption band of the non-fluorescent intermediate that is stabilized at low temperatures is at about 690 nm [15]. The similar position of the maximum absorption of the primary intermediate was later registered also at room temperature [16, 17]. This intermediate is commonly called X690. In the dark increasing temperature causes X690 to turn into chlorophyllide:
![]()
Existence of several short-lived non-fluorescent intermediates. Studies of the primary stages of protochlorophyllide photoinduction at very low temperatures suggested the existence of even earlier stages of the photochemical reaction than the formation of the X690 intermediate. This is evidenced by comparison of the spectral pattern changes after illumination of etiolated leaves at different temperatures [9-11, 18-21] (Fig. 2).
Fig. 2. Fluorescence quenching as the primary act of photoreduction of the active form of protochlorophyllide. Changes in low-temperature (77 K) fluorescence spectra of etiolated bean leaves after illumination with light of 104 W/m2 at low temperatures (4.2, 77, 153 K): 1) unilluminated etiolated leaf; 2) the same leaf after illumination at low temperature (temperature indicated in the upper part of the figure); 3) the same sample after warming to 233 K (10 min) in the dark; 4) the same sample after warming to 263 K (20 min) in the dark.
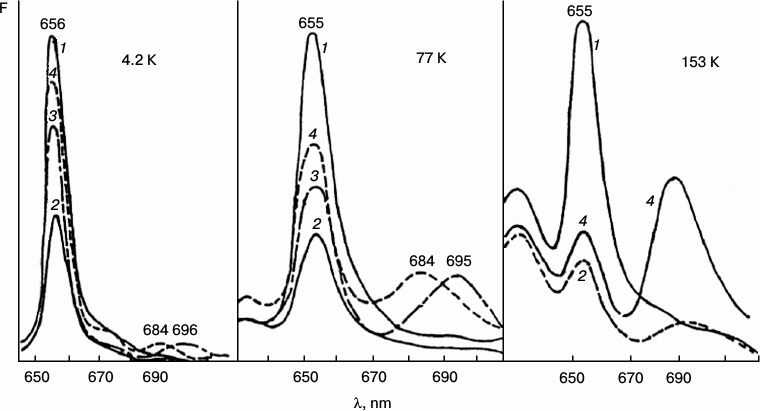
The initial reaction, photoinduced fluorescence quenching of the active form of protochlorophyllide Pchld655/650, observed at low temperatures, becomes reversible when the temperature increases. The degree of reversibility depends on the temperature conditions during the illumination (75-80% after illumination at 4.2 K; 30-50% after illumination at 77 K; 5-6% after illumination at 153 K). The quantum yield of photoinduced quenching of protochlorophyllide Pchld655/650 fluorescence on deep cooling of etiolated leaves is very low (three orders of magnitude lower than the quantum yield of its photoreduction at room temperature); its dependence on temperature is rather weak in the range from 4.2 to 180 K. It is practically unaltered by increasing the temperature of the illuminated sample from 4.2 to 77 K and is slightly increased (50%) at 153 K (Fig. 2). The quantum yield of the following reaction, formation of the primary fluorescent chlorophyllide forms after the increase in temperature of illuminated samples, is characterized by higher dependence on the temperature at which the sample was illumination. This parameter (in contrast to the quantum yield of fluorescence quenching) changes significantly with increase in temperature of the illuminated leaves from 4.2 to 153 K. The quantum yield of the overall reaction (fluorescence quenching plus subsequent chlorophyllide formation after temperature increase) in leaves illuminated at 4.2 K, is three to four times lower than in leaves illuminated at 77 K, and it is ten times lower than when illuminated at 153 K.
These facts indicate greater complexity of the primary reaction: the existence of reverse reactions in the pathway of the formation of the X690 intermediate, these reactions being less dependent on temperature than the direct process of transformation of the non-fluorescent intermediate into chlorophyllide.
Fluorescence and absorption spectroscopy revealed that the primary photoreaction of protochlorophyllide transformation into a non-fluorescent intermediate is photoreversible when affected by a powerful monochromatic laser beam with wavelength of 694 nm (corresponds to the absorption of the X690 intermediate) [22, 23]. Rate constants and quantum yields of the direct and reverse photoreactions at photostationary equilibrium were calculated. The calculations showed that the photoreversibility of the reaction of X690 formation cannot be the reason for the low quantum yield of the general process as the quantum yield of the reverse process is substantially (about 20 times) lower. Since X690 formation is not reversible in the dark, the authors suggested the existence of another intermediate preceding X690 formation, which includes a fast reverse reaction; it is this reaction that reduces the yield of the overall process. The putative intermediate was designated as R (reversible):
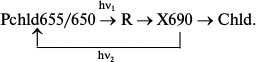
These data explain the decrease in the reaction quantum yield at low temperature compared to the quantum yield at room temperature, when the quantum yield is close to 0.5 [24, 25]. Temperature increase causes the rate constants of direct dark processes to increase more rapidly than the rate constants of the reverse reactions, which is accompanied by the increase in overall yield of the process of chlorophyllide formation.
Analysis of spectral changes (Fig. 3) suggests that the absorption spectrum of the primary intermediate R predominantly formed at 77 K is close (if not identical) to the absorption spectrum of the active protochlorophyllide form: the primary effect of light on etiolated leaves, manifested by quenching of protochlorophyllide fluorescence, is observed under the conditions when X690 is practically unnoticeable in absorption spectra (at 80% fluorescence quenching the absorption spectrum remains virtually unchanged) [18, 19]. The 10-12% decrease in the protochlorophyllide absorption band and the appearance of a long-wavelength absorption maximum at 690 nm belonging to the non-fluorescent X690 intermediate are observed in the absorption spectra of leaves illuminated at 153 K.
Fig. 3. Changes in low temperature (77 K) fluorescence (a) and absorption (b) spectra of etiolated bean leaves illuminated with intense white light (104 W/m2) at low temperatures: solid line, unilluminated etiolated leaf; dotted line, after 4 min illumination at 77 K; dashed line, after 4 min illumination at 153 K.
Data of time-resolved spectroscopy obtained at room temperature indicate the appearance of a short-lived intermediate with absorption band close to the absorption band of the active protochlorophyllide form, in the chain of protochlorophyllide transformations [26, 27].
Low-temperature (77 K) optical (absorption and fluorescence) and ESR spectra of entire etiolated and illuminated leaves were studied to elucidate the mechanism of the early stages of protochlorophyllide photoreduction [18, 19, 21]. Both short-lived intermediates, R and X690, were shown to be paramagnetic. Leaves illumination at 77 K (when non-fluorescent intermediate R is mainly formed) was found to cause the emergence of a structureless singlet ESR signal with g-factor 2.0021 characteristic of a free electron, width 1.1 mT. Increasing temperature to 200 K caused a slight increase in the amplitude of the ESR signal of etiolated leaves illuminated at 77 K (Fig. 4). This increase in the signal amplitude probably corresponds to the transformation of the primary product R into intermediate X690. Decrease in the signal width was observed along with amplitude increase. Further increase in temperature above 200 K caused a sharp drop in the ESR signal amplitude, which reached the initial dark value at the temperature of about 250 K. Absorption and fluorescence spectra showed the formation of the primary forms of chlorophyllide along with the sharp decline of the ESR signal amplitude of etiolated leaves illuminated at 77 K on increasing the temperature.
Fig. 4. ESR spectral study to clarify the nature of the non-fluorescent intermediates [18, 19, 21]. Changes in the ESR signal of an etiolated bean leaf illuminated for 4 min with white light with intensity 104 W/m2 at 77 K after gradual increase in temperature in the spectrophotometer. a) Changes in the ESR spectrum: 1) unilluminated leaf; 2) after illumination at 77 K; 3-6) after temperature increase; b) changes in the amplitude of the ESR signal of the leaf illuminated at 77 K depending on the temperature increase; c) changes in the width of the ESR signal of the leaf illuminated at 77 K depending on the temperature increase.
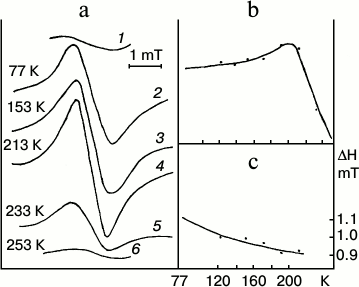
Thus, the primary intermediate R resulting from the photoreaction itself is close in its absorption spectrum to the original protochlorophyllide form, but it differs from it in having a very low fluorescence quantum yield. Intermediate R is characterized by a singlet ESR signal with the g-factor of a free electron. Increasing the temperature at which the sample is illuminated causes an increase in the rate constant of the direct reaction of the transformation of intermediate R into X690. Non-fluorescent intermediate X690 is also characterized by a singlet ESR signal with g-factor 2.002 characteristic of a free electron, but it has a narrower signal width of 0.95 mT.
Two forms of a non-fluorescent intermediate X690. It was noted in several studies that the non-fluorescent intermediate X690 is characterized by a broad absorption band at 680-710 nm [5-8]. The position of the absorption maximum was shown to depend significantly on the temperature during the illumination. Illumination at 153-158 K was accompanied by the appearance of a wide absorption band at 705 nm, while illumination at 158-163 K resulted in a band with maximum at 689 nm [8].
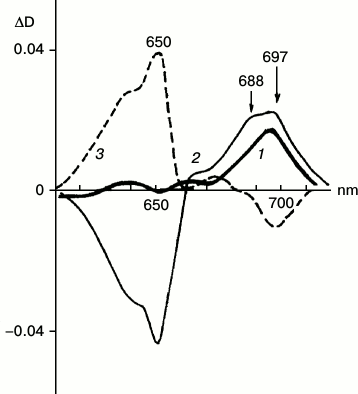
Differential and derivative spectroscopy revealed the existence of two forms of X690 characterized by absorption maxima at 697 and 688 nm and different formation rates (Fig. 5) [28]. The intermediates were named R697 and R688. The R697 intermediate could be detected after a brief illumination of etiolated leaves at 77 K. The intensity of the absorption band of the active protochlorophyllide form at 650 nm remained practically unchanged. Subsequent increase in the temperature of the sample in the dark to 233 K led to the complete reversibility of the reaction of R697 formation. The R688 intermediate was accumulated more slowly; this process was accompanied by a decrease in the absorption band of the active protochlorophyllide form at 650 nm. Reduction of the band at 650 nm indicates more profound changes in the protochlorophyllide molecule at this stage when compared to the previous stage of intermediate R697 formation, when absorption at 650 nm remains unchanged. Judging from ESR spectra, both R697 and R688 intermediates are paramagnetic. Increasing the temperature of a sample characterized by two absorption bands in the difference spectrum at 697 and 688 nm to 233 K caused both the effective reverse reaction of the intermediates into protochlorophyllide and the forward reaction of formation of the primary chlorophyllide forms with absorption maxima at 676 and 684 nm. Since the formation of the chlorophyllide primary forms (at higher temperature) required joint accumulation of two non-fluorescent intermediates R697 and R688, these intermediates were suggested to be formed in parallel photoreactions, but at different rates; increasing temperature causes interaction of their chromophores leading to the formation of a chlorophyllide molecule by a mechanism of free radical disproportionation. This assumption about the mechanism of chlorophyllide formation by free radical disproportionation was suggested earlier in [8].
Fig. 5. Two forms of non-fluorescent intermediate X690. Low-temperature differential absorption spectra of etiolated barley leaves: 1) spectrum of a sample illuminated for 5 s at 77 K minus spectrum of the same sample prior to illumination; 2) spectrum of a sample illuminated for 1 min at 77 K minus spectrum of the same sample prior to illumination; 3) spectrum of a sample after illumination for 1 min at 77 K and its subsequent incubation at 233 K in the dark for 15 min minus the spectrum of the sample illuminated for 1 min at 77 K. Light intensity 102 W/m2 [28].
At the end of this section, we conclude that protochlorophyllide photoreduction in vivo includes at least two elementary reactions that can be registered by spectroscopic methods at very low temperatures, when the products of these reactions, non-fluorescent intermediates, are stabilized:
![]()
STUDY OF FAST STAGES OF PROTOCHLOROPHYLLIDE PHOTOREDUCTION in
vivo AT PHYSIOLOGICAL TEMPERATURES
Study of chlorophyll photobiosynthesis in etiolated leaves by time-resolved spectroscopy is another way to detect labile short-lived intermediates of protochlorophyllide photoreduction. Application of this method has revealed the formation of short-lived intermediates also at physiological temperatures. Studies of the kinetics of the changes in pigment fluorescence intensity after brief illumination of etiolated leaf homogenates [29], as well as fast light-induced absorption spectral changes of leaves and isolated pigment–protein complexes [16, 17] at room temperature revealed a primary short-lived (0.2 μs) intermediate with absorption maximum at 690-695 nm.
Nanosecond and picosecond absorption spectroscopy [26] showed that the reaction of protochlorophyllide photoreduction in isolated pigment–protein complexes at room temperature involves four intermediates with time constants of formation of 5 ps, 2 ns, 35-250 ns, and 1-2 μs and the subsequent appearance of chlorophyllide at 12 μs:
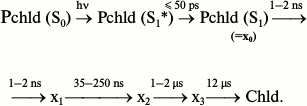
The position of the absorption maximum of the primary product X0 is virtually identical to the position of the absorption band of the initial protochlorophyllide. The time constant of X0 formation corresponds to the relaxation of the protochlorophyllide molecule from the Franck–Condon state (S1*) to the equilibrium state (S1), hence X0 cannot be an intermediate in the chemical sense. Using the flash-photolysis method, it was shown that the absorption maximum of the X1 intermediate is also close to the absorption maximum of the initial protochlorophyllide (≥640 nm). Studies of the changes in differential absorption spectra have shown that intermediates X2 and X3 are characterized by absorption maxima at 688 and 684 nm, respectively. The authors of [26] suggested that intermediate X3 corresponds to the non-fluorescent intermediate X690 discovered in etiolated leaves illuminated at low temperatures. However, according to other researchers [30, 31], intermediate X690 rather corresponds to the X2 intermediate discovered by Iwai et al., as evidenced both by the absorption spectrum of this intermediate (maximum at 688 nm) and by the time of its formation (35-250 ns), which is comparable to the formation time of the X690 intermediate recorded by other researchers [16, 32]. Intermediate X1 [26] probably corresponds to the intermediate R discovered in our work [22, 23], as evidenced by the similarity between its absorption spectrum and the absorption spectrum of the initial protochlorophyllide. Intermediate X3 can be compared to one of the primary chlorophyllide forms appearing after temperature increase of etiolated leaves that were illuminated at 77 K.
Comparison of the results of research of intermediate stages of protochlorophyllide photoreduction in vivo at physiological and low temperatures showed that this process involves several intermediate products, including two or three short-lived intermediates that, according to our studies, are characterized by significant quenching of protochlorophyllide fluorescence.
PRIMARY FLUORESCENT CHLOROPHYLLIDE FORMS in vivo
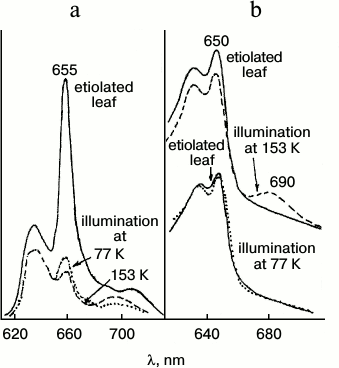
The non-fluorescent intermediate X690 formed by light at low temperatures transforms into chlorophyllide following a dark path after increasing the temperature of the illuminated sample. When using white light for illumination of etiolated leaves at 77 K, subsequent increase in temperature caused almost simultaneous formation of two primary chlorophyllide forms with fluorescence maxima at 684 and 695 nm and corresponding absorption bands at 676 and 684 nm [9, 10, 33]. Then the Chld685/684 form turns into a shorter-wavelength form Chld684/676 during a dark reaction. The ratio of the bands at 695 and 684 nm appearing after illumination at low temperature depends on the spectral composition of the light. Blue light (with maximum at 470 nm) used for illumination resulted (after temperature increase) in the formation of only one band at 695 nm, which gradually shifts to 684 nm. Red light (>600 nm) used for illumination of leaves resulted (after temperature increase) in the appearance of only one shorter-wavelength maximum at 684 nm. Formation of only one, shorter-wavelength primary chlorophyllide form was observed in etioplast preparations. These data suggest the existence of two types of active protochlorophyllide–protein complex with nearly identical absorption and fluorescence bands in the red spectral region in etiolated leaves. Both forms are converted to chlorophyllide through the formation of a non-fluorescent intermediate. One of the precursors can be transformed directly into the shorter-wavelength chlorophyllide form Chld684/676. The hypothetical reaction scheme is as follows:
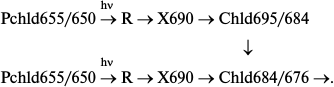
An additional active protochlorophyllide form Pchld653/648, close in its spectral characteristics to the main active form, was later discovered in vivo [34, 35]. Moreover, it is possible that the second pathway is based on the transformation of a shorter-wavelength form of the precursor Pchld643/637, whose fluorescence is quenched as a result of highly effective energy migration to the main active form.
The primary reactions of protochlorophyllide photoreduction in plant leaves were studied by means of time-resolved fluorescence spectroscopy [32]. The results of these studies have shown that dark transformation of non-fluorescent intermediates leads to the formation of four primary chlorophyllide forms characterized by fluorescence maxima at 684, 690, 695-697, and 706 nm. Similar results were obtained when studying primary fluorescent chlorophyllide forms by differential spectroscopy and dividing difference fluorescence spectra into Gaussian components [33]. It was found that the long-wavelength primary chlorophyllide forms with fluorescence maxima at 696 and 706 nm are later transformed at room temperature into shorter-wavelength forms with fluorescence maxima, respectively, at 675 and 684 nm. The form with fluorescence maximum at 684 nm is probably the chlorophyllide (Chld684/676) form, which serves as the initial form in the biosynthetic pathway of the pigments of the reaction center of photosystem II and the light-harvesting complex (see review [36]). Apparently, the formation of several primary labile chlorophyllide forms indicates the early differentiation of the pathways of the formation of functionally different native pigment forms.
PROTOCHLOROPHYLL(IDE) PHOTOREDUCTION IN MODEL SYSTEMS
Photoreactions in protochlorophyll solutions. Studying the reaction of protochlorophyll photoreduction in simple systems has become another way to elucidate the mechanism of the primary stages of chlorophyll biosynthesis from the precursor and the role of biological structures in this process. Studying protochlorophyll(ide) phototransformation in vitro reveals the spectral changes associated with the transformation of the actual chromophore molecule.
Many researchers have tried to photoreduce protochlorophyll(ide) to chlorophyll(ide) in vitro. The earliest systematic studies in this direction were carried out in the laboratory of Academician A. A. Krasnovsky using different model systems (solutions, solid films, pigment micellar solutions). As a result, rather high photoreaction product yield could be achieved (up to 20%), the product being identified as chlorophyll [37-39]. However, in these experiments the reaction was complicated by side processes, in particular, by photoreduction in the system of conjugated bonds of the chromophore. The primary reaction stages and its mechanism remained unclear. The singlet ESR signal, which was thought to belong to the pigment radical-anion, was discovered when studying protochlorophyll photoreduction by strong reducing agents in solutions at low temperatures (243-183 K) [40]. However, the character of the absorption spectrum changes indicated that in this case the reduction occurred in the system of conjugated bonds (with the formation of a “red” intermediate product).
To obtain information about the mechanism of the earliest stages of protochlorophyll(ide) photoreduction reactions, previously studied in whole leaves, we together with K. N. Timofeev and M. I. Bystrova studied this process in model systems [18, 19, 41]. Experimental conditions were as close as possible to those in the studies on plant leaves, when primary stages of photoreduction of the protochlorophyllide molecule could be observed (high intensity short illumination at 77 K). It was shown that primary photophysical stages of protochlorophyll reduction in solutions and films could proceed also without the use of reducing agents. Apparently, it is the solvent (ethanol, ethyl ether, pyridine) that serves as the hydrogen donor in this case.
Photoreduction of monomeric protochlorophyll(ide). The effect of quenching of fluorescence at 626 nm belonging to the monomeric pigment form was discovered when studying dilute solutions of protochlorophyll and protochlorophyllide in ethyl ether (10–6 M) after illumination of the samples at 77 K. This effect could be practically completely reversed by subsequent temperature increase to 273 K. The absorption spectrum remained unchanged [41]. A singlet signal with g-factor of 2.0013 and singlet band-width of 1.2 mT could be registered in the ESR spectrum simultaneously with protochlorophyllide fluorescence quenching; increasing temperature caused rapid disappearance of this signal and reappearance of fluorescence [18, 19]. Addition of reducing agents accelerated the fluorescence quenching several-fold.
Thus, illumination at 77 K caused the formation of a product close in the position of its red absorption band to the initial pigment, but different from it in low fluorescence yield and the appearance of a singlet ESR signal in low-concentration solutions of monomeric protochlorophyll. Increasing temperature caused disappearance of the product, which turned back into the initial compound. Comparison with the precursor photoreduction in whole etiolated leaves indicates that apparently the very first stage of chlorophyll precursor photoreduction is reproduced in the model systems with monomeric protochlorophyll; this stage is manifested in vivo in the quenching of fluorescence of the active protochlorophyllide form and the formation of the paramagnetic intermediate R.
Photoreduction of aggregated protochlorophyll. Absorption and fluorescence spectra of the three main native forms of chlorophyll precursor are rather well simulated in concentrated (10–4 M) ethanolic solutions of protochlorophyll: absorption bands at 625, 635, and 645-647 nm and fluorescence bands at 627, 637, and 651-652 nm are observed. According to [42, 43], the short-wavelength maxima at 627 and 636 nm belong to singly and doubly solvated pigment, respectively. Measurements of the circular dichroism spectra of protochlorophyll in model systems [44, 45] indicate that the longest-wavelength spectral band at 651-652 nm results from the formation of pigment dimer.
Illumination of the concentrated ethanolic solution at 77 K leads to the quenching of all the fluorescence bands [18, 19, 41]. This quenching proved to be reversible on increasing the temperature for two shorter-wavelength bands (similar to the experiments with the pigment monomer), whereas fluorescence intensity for the band at 651 nm could not be fully recovered. Light caused the appearance of a singlet signal with g-factor of 2.0015 and width of 1.1 mT in the ESR spectrum (these parameters match those of the signal caused by illumination of etiolated leaves). Along with fluorescence quenching, a small maximum at about 685-690 nm is observed in the low-temperature absorption spectrum; this maximum is close in its position to the absorption maximum of the non-fluorescent intermediate X690 discovered in plant leaves. Hence, deeper protochlorophyll transformation is apparently taking place in the systems with aggregated protochlorophyll (when compared to the monomeric pigment), comprising the formation of a chlorophyll-like product, which may be identical to the X690 intermediate. The aggregated long-wavelength form of the pigment is obviously involved in this stage of the process.
Thus, the similarities between the primary stages of protochlorophyll(ide) photoreduction in vivo and its most simple photoreactions in model systems suggest that primary photoreactions observed in whole leaves are really connected to the changes in the actual pigment molecule.
PROTOCHLOROPHYLLIDE PHOTOREDUCTION IN RESTORED TERNARY
COMPLEXES
In recent years, a growing number of researchers have been using artificial ternary complexes to study the mechanism of the protochlorophyllide photoreduction reaction; these complexes include protochlorophyllide, NADPH, and the photoenzyme protochlorophyllide oxidoreductase. Pchld644/642 [14, 27, 46, 47] is the main active protochlorophyllide form in such systems; its spectral properties are close to those of the active protochlorophyllide form Pchld643/639 from whole etiolated leaves and to the main form that can be found in isolated pigment–protein complexes.
Similarly to a whole etiolated leave, the formation of an unstable non-fluorescent intermediate was registered on protochlorophyllide photoreduction in artificial ternary complexes at low temperatures; this intermediate proved to be the primary product that appears after illumination of samples at low temperatures [14, 47, 48], with the absorption band at about 696 nm [14]. Fluorescence quenching was accompanied by the appearance of a singlet signal with g-factor of a free electron in the ESR spectrum [46, 48]. Increasing temperature caused the non-fluorescent intermediate to change spontaneously into chlorophyllide, which was identified both by spectral characteristics [48] and by liquid chromatography [46]. The primary fluorescent intermediate was transformed into a shorter-wavelength chlorophyllide form in a dark reaction:
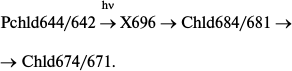
Formation of a non-fluorescent intermediate X696 can proceed at temperatures below 200 K, a critical temperature below which there are no dynamic enzyme changes. Hence, enzyme movement or reorganization is not involved in this stage of the catalytic mechanism. The temperature limit of X696 formation is 120 K. These data correlate with the results of studies on the temperature dependence of X690 formation in whole etiolated leaves; it was already mentioned that in these leaves frozen to 77 K there could be observed only protochlorophyllide fluorescence quenching accompanied by the formation of the first intermediate R with spectral properties virtually identical to those of protochlorophyllide.
Impulse techniques revealed the primary processes in reconstructed active complexes and at room temperature [27]. A slight increase in the initial protochlorophyllide absorption (maximum at 642 nm) with its shift to 636 nm could be observed in the time interval between 3 and 400 ps after illumination of the sample with a 50 fs laser flash (475 nm). At the same time, there also appeared a weak maximum at about 677 nm. A short-lived intermediate with absorption bands at 636 and 677 nm was transformed into chlorophyllide:
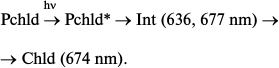
As mentioned above, the primary non-fluorescent intermediate formed by the action of light in living systems is similar to the initial protochlorophyllide in terms of the location of the main absorption maximum and its intensity. This could be observed both at low temperature – intermediate R formed at 77 K in whole leaves [19, 23] – and at room temperature in isolated pigment–protein complexes – formation of intermediate X1 [26]. It should be noted that a very slight short-wavelength shift of absorption maximum of the active protochlorophyllide form (650 nm) can also be found in absorption spectra of whole etiolated leaves briefly illuminated at 77 K [19] (this shift at first remained unnoticed). Probably, the Int (636, 677 nm) intermediate found by Heyes et al. [27] at room temperature in the picosecond time range corresponds to the simultaneous observation of two primary intermediates found in vivo: intermediate R (X1) and the next intermediate X690 (X2); the formation time of the latter at room temperature (35-250 ns) corresponds to the rate of primary intermediate formation in reconstructed complexes.
Spectral differences between the short-lived intermediate with absorption maximum at 675-677 nm discovered at room temperature and the X696 intermediate observed at low temperatures can be probably explained by different states of the protein part of the complex at low and high temperatures. The non-fluorescent intermediate X696 is formed on the deep freezing of the complex to a temperature below the critical value of 200 K, when dynamic changes in the enzyme start. At room temperature the pigment photoreduction proceeds along with protein conformational changes.
Thus, protochlorophyllide photoreduction in reconstructed ternary complexes proceeds in at least in two stages: light-induced formation of the short-lived non-fluorescent intermediate characterized by a weak ESR signal with g-factor of a free electron, and its subsequent dark transformation into chlorophyll. The observed processes are similar to those in whole etiolated leaves.
POSSIBLE MECHANISMS OF PHOTOREDUCTION OF THE PROTOCHLOROPHYLLIDE
MOLECULE
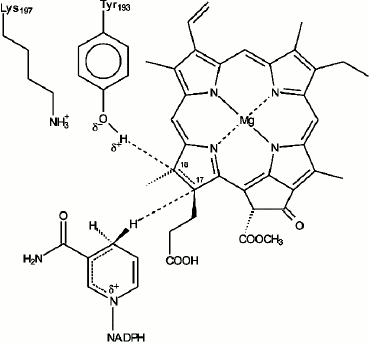
In our early studies [9, 10] we suggested that the primary reactions of protochlorophyllide photoreduction in vivo characterized by the formation of paramagnetic intermediates correspond to the sequential transfer of two electrons and two protons to the semi-isolated double bond C17=C18 of the protochlorophyllide molecule, the process leading to the formation of an intermediate semi-reduced form similar to the mechanism of the stepwise reduction of the double bond described for the photoreaction of porphyrin reduction in solutions [49]. However, after it became clear that it was NADPH that served as an electron donor [50, 51], it was suggested that the sequential transfer of two electrons was rather unlikely due to the high energy of nicotinamide radicals [52]. Since the structure of the photoenzyme POR is similar to that of short-chain alcohol dehydrogenases, transfer of the hydride-ion from NADPH by a mechanism analogous to the mechanism of substrate reduction by alcohol dehydrogenases seemed to be a more likely option for the reaction of protochlorophyllide photoreduction. The reaction of protochlorophyllide photoreduction in the reaction mixture including protochlorophyllide, NADPH, and etioplast membranes was studied by NMR spectroscopy using NADPH isomers with radiolabeled hydrogen (3H). These studies showed that the hydride-ion from the 4S position in the nicotinamide nucleus of the NADPH molecule is transferred to position C17 of the protochlorophyllide molecule; the pigment molecule undergoes trans-reduction [53, 54]. The conservative amino acid residue Tyr193 of POR located near the bond C17=C18 is probably the source of the second proton [55-57]. According to the structural model of the POR enzyme [58], the D ring of the protochlorophyllide molecule is fixed against NADPH and the tyrosine residue to ensure the hydride-ion and proton transfer. POR was shown to lose its activity when the conservative amino acid residues Tyr193 and Lys197 were changed to Cys and Arg, respectively [55]. This constitutes a difference between POR and dehydrogenases, as such replacement is possible in the latter case. It was suggested that Tyr193 could be the donor of the second protein (Fig. 6). The proximity of Lys197 is necessary to reduce the pKa of Tyr193, which facilitates deprotonation of its phenol group [55]. Conservative residues Lys197 and Cys226 play an important role in the formation of the optimal structure of the ternary complex, providing the necessary steric conditions for the reaction of protochlorophyllide photoreduction [55-57, 59]. The proton from the tyrosine phenol group is transferred to the C18 atom of the protochlorophyllide molecule. In the case of the reconstructed ternary complexes with mutant POR where Tyr193 was replaced by Phe, the protochlorophyllide reduction stopped at the stage of the intermediate photoproduct and the second (light-independent) reaction stage was inactivated [56]. These data indicate that the hydride-ion is transferred first, before the proton transfer. The existence of several short-lived intermediates also supports the suggestion of non-simultaneous transfer of hydride-ion and proton during the photoreduction of the protochlorophyllide molecule.
Fig. 6. Hypothetical model of the mechanism of protochlorophyllide photoreduction by protochlorophyllide oxidoreductase in the active center of the ternary complex Pchld–POR–NADPH [55].
Paramagnetism of the molecules of electron donor or acceptor, which is manifested in the appearance of the light-induced ESR signal, is one of the most convincing evidences of the formation of charge transfer complexes (CTC) in a donor–acceptor system [60]. The formation of the charge transfer complex is accompanied by the appearance of a new absorption band when the redox potentials of the reaction partners are sufficient for the complete electron transfer from one molecule of the complex to the other [60]. However, if the difference in redox potentials of the molecule partners in the main state is rather small and insufficient for the complete electron transfer from the donor molecule to the acceptor molecule, a complex with partial charge transfer is formed. This is accompanied by fluorescence quenching, but the absorption spectrum remains unchanged without the appearance of new bands [60]. The assumption that protochlorophyllide photoreduction in vivo involves the stage of formation of a charge transfer complex has been expressed in a number of studies [8, 15, 20, 21, 30, 31, 61]. Based on the spectral characteristics of two non-fluorescent intermediates in vivo (R and X690 according to [11, 18, 19, 21]; X1 and X2 according to [26]), we can assume that their photoinduced formation corresponds to the formation of the complexes with partial [Dδ+ Aδ–] and complete [D+ A–] charge transfer [20, 21, 31]:
![]()
Raskin and Schwartz analyzing work [26] suggested that the rather long lifetime of the intermediate X1 (35-250 ns) indicates the existence of two intermediates at this stage: singlet exciplex and triplet exciplex of protochlorophyllide and hydrogen donor. The results of the studies of low-temperature phosphorescence spectra of whole etiolated leaves illuminated at 77 K also favor the formation of not only singlet, but also triplet exciplexes of protochlorophyllide and hydrogen donor, since under these conditions phosphorescence quenching of the active protochlorophyllide form was observed simultaneously with its fluorescence quenching [62]; the quantum yield of phosphorescence prior to illumination was very high (about 50%) [43, 62]. However, we think that additional experiments are required to answer the question of the involvement of the protochlorophyllide molecule triplet state in its photoreduction.
Heyes et al. conducted a comparative study of the reaction of protochlorophyllide molecule reduction in ternary complexes at 180 K using absorption, ESR, ENDOR, and Stark spectroscopies to elucidate the mechanism of this reaction [47]. ESR spectra indicating the existence of two types of paramagnetic products were registered for illuminated samples. However, quantitative evaluations of the ESR spectra indicated that only about 5% of the pigment was transformed into free radicals during almost complete transformation of protochlorophyllide active form (estimated by the changes in absorption spectra). A wide Stark effect characteristic of the charge transfer corresponded to the formation of a non-fluorescent intermediate with an absorption band at 696 nm. Stark spectroscopy data indicated the existence of two components in the non-fluorescent intermediate. So it was suggested that a charge transfer complex is formed at the initial stage of the protochlorophyllide photoreduction reaction. Temperature dependences of intermediate formation and NADPH oxidation proved to be identical. Therefore, it was suggested that the formation of the non-fluorescent intermediate involves the hydride-ion transfer to create a charge transfer complex. It was suggested that photon absorption by a protochlorophyllide molecule leads to a temporary separation of charges in the double bond C17=C18, promoting ultra-rapid transfer of the hydride-ion from NADPH to the C17 atom of protochlorophyllide [47, 63]. The resulting charge transfer complex facilitates proton transfer to the C18 atom in the subsequent dark reaction.
The importance of flexibility and dynamics of the POR enzyme for its catalytic function was illustrated in the studies on protochlorophyllide photoreduction in reconstructed ternary complexes using femtosecond absorption spectroscopy in the visible and infrared regions [64]. The results of the measurements showed that the first photon absorption activates the enzyme; as a result a high quantum yield of the Int 675 intermediate is observed after the second photon absorption in the picosecond time range. The authors suggested that minor structural changes may be involved in the primary light-dependent stage. These changes are supposed to optimize the alignment of the position of the NADPH nicotinamide ring and the tyrosine residue relative to the protochlorophyllide D ring.
Comparison of the results of studies of the molecular mechanism of protochlorophyllide photoreduction in vivo and in various model systems suggests that the primary stages characterized by fluorescence quenching and paramagnetic product formation are identical for the whole etiolated leaves and for all the model systems, including the simplest ones – the low concentration solutions. However, a number of facts suggest that the mechanism of protochlorophyllide photoreduction in vivo is somewhat different from the reaction mechanism in the model systems. First, the active complex in leaves probably has a more complex structure than the ternary complex reconstructed in vitro out of the three main components: Pchld, NADPH, and POR. This is evidenced, in particular, by differences in spectral characteristics between the artificial and natural complexes. There are several spectrally different protochlorophyllide active forms in etiolated leaves; the main forms are Pchld643/639, Pchld655/650, and Pchld653/648, which are accumulated and transformed in very young leaves; there are also several minor long-wave forms participating in the processes of protochlorophyllide photoreduction (see review [1]). One or two protochlorophyllide forms are usually found in reconstructed active ternary complexes: Pchld633/630 (active only at room temperature) and Pchld644/641 (photoactive also at low temperatures). The mechanism of protochlorophyllide photoreduction in vivo is apparently more complex, as evidenced by the results of research on this process in whole etiolated leaves at low temperature [19, 23], as well as the results obtained by high time resolution spectroscopy at physiological temperatures [26], which showed the presence of multiple stages in the reaction of protochlorophyllide photoreduction, including two or three short-lived intermediates. Only one short-lived intermediate is found when studying the reaction of protochlorophyllide phototransformation in artificial ternary complexes.
There is evidence of the presence of flavins in the active pigment–protein complex of etiolated leaves and their possible involvement in the primary reactions of protochlorophyllide photoreduction [65-67]. The results of our research support this possibility: fluorescence at 520 nm characteristic of flavins decreases after the leaves are illuminated with white or red (>600 nm) light at 77 K, the process being reversible after the temperature is increased [67].
Redox reactions between nicotinamides and flavins proceed via hydride-ion transfer [52]; the single-electron mechanism of redox reactions is characteristic of many flavoenzymes.
In plant leaves flavins can serve as an intermediate in the reaction, receiving the hydride-ion from NADPH and giving the electron to the pigment molecule. In this case, the reaction can follow the mechanism of multi-stage transfer of two electrons and two protons to form a semi-reduced pigment molecule with one attached electron (intermediate semi-reduced form) at the first stage, similar to the reactions of porphyrin photoreduction in solution [49]. It is possible that two variants of the mechanism of hydrogen transfer from NADPH to protochlorophyllide can exist in whole etiolated leaves: hydride-ion transfer directly from NADPH to the C17 position in the protochlorophyllide molecule followed by proton connection and/or flavin-mediated electron transfer from NADPH to the pigment. It is also possible that the mechanisms of the primary reactions of spectrally different active protochlorophyllide forms vary.
The results of studies of protochlorophyllide photoreduction in whole living cells and in the closest model systems – artificial ternary complexes comprising protochlorophyllide, NADPH, and POR, provided deeper understanding of the mechanism of this reaction. Both in the whole etiolated leaves and in reconstructed complexes, photoreduction of the chlorophyll precursor proceeds via the formation of short-lived intermediates of free-radical nature; NADPH is the hydride-ion donor and a tyrosine residue of the POR enzyme is the donor of the second proton. The charge transfer complex is the primary product of the reaction of protochlorophyllide photoreduction. The stage of primary intermediate formation could be modeled also in simple systems, e.g. protochlorophyll(ide) solutions. However, as mentioned above, the photoprocess is more complex in living systems than in artificially created ternary active complexes. Furthermore, in leaves there are several parallel reactions of spectrally different forms of chlorophyll precursor that play an important role in the creation of the system of the pigment forms of the photosynthetic apparatus. We believe that further studies using whole functional cells and tissues are essential. This approach is particularly important for further understanding of the still understudied regulation of the final stage of chlorophyll biosynthesis.
REFERENCES
1.Belyaeva, O. B., and Litvin, F. (2007)
Biochemistry (Moscow), 72, 1458-1477.
2.Rubin, A. B., Minchenkova, L. E., Krasnovsky, A.
A., and Tumerman, L. A. (1962) Biofizika, 7, 571-577.
3.Goedheer, J., and Verhulsdonk, C. (1970)
Biochem. Biophys. Res. Commun., 39, 260-266.
4.Sironval, C., and Kuyper, P. (1972)
Photosynthetica, 6, 254-275.
5.Dujardin, E., and Sironva, C. (1977) Plant Sci.
Lett., 10, 347-353.
6.Dujardin, E., and Correia, M. (1979)
Photobiochem. Photobiophys., 1, 25-32.
7.Dujardin, E. (1984) in Protochlorophyllide
Reduction and Greening (Sironval, C., and Brouers, M., eds.)
Martinus Nijhoff/Dr. W. Junk Publisher, The Hague, pp. 87-98.
8.Losev, A. P., and Lyalkova, N. D. (1979) Mol.
Biol. (Moscow), 13, 837-844.
9.Belyaeva, O. B., and Litvin, F. F. (1980)
Biofizika (Moscow), 25, 617-623.
10.Belyaeva, O. B., and Litvin, F. F. (1981)
Photosynthetica, 15, 210-215.
11.Litvin, F. F., Ignatov, N. V., and Belyaeva, O.
B. (1981) Photobiochem. Photobiophys., 2, 233-237.
12.Belyaeva, O. B., Personova, E. R., and Litvin, F.
F. (1983) Photosynth. Res., 4, 81-85.
13.Heyes, D. J., Ruban, A. V., Wilks, H. M., and
Hunter, C. N. (2002) PNAS, 99, 11145-11150.
14.Heyes, D. J., Ruban, A. V., and Hunter, C. N.
(2003) Biochemistry, 42, 523-528.
15.Raskin, V. I. (1976) Vestnik Akad. Nauk
BSSR, 5, 43-46.
16.Franck, F., and Mathis, P. (1980) Photochem.
Photobiol., 32, 799-803.
17.Inoue, Y., Kobayashi, T., Ogawa, T., and Shibata,
K. (1981) Plant Cell Physiol., 22, 197-204.
18.Belyaeva, O. B., Timofeev, K. N., and Livin, F.
F. (1987) Biofizika (Moscow), 32, 104-109.
19.Belyaeva, O. B., Timofeev, K. N., and Litvin, F.
F. (1988) Photosynth. Res., 15, 247-256.
20.Belyaeva, O. B. (1994) Final Stages of
Chlorophyll Biosynthesis in Plant Leaves: Abstract of the doctoral
dissertation [in Russian], Institute of Soil Science and
Photosynthesis, Pushchino.
21.Belyaeva, O. B. (2009) Light-Dependent
Chlorophyll Biosynthesis [in Russian], BINOM, Moscow.
22.Litvin, F. F., and Ignatov, N. V. (1980)
Doklady AN SSSR, 250, 1463-1465.
23.Litvin, F. F., Ignatov, N. V., and Belyaeva, O.
B. (1981) Photobiochem. Photobiophys., 2, 233-237.
24.Smith, J. H. C. (1960) in Comparative
Biochemistry of Photoreactive Systems (Allen, M. B., ed.) Academic
Press, New-York-London, pp. 257-277.
25.Nielsen, O. F. (1975) Biochem. Physiol.
Pflansen, 167, 195-206.
26.Iwai, J., Ikeuchi, M., Inoue, Y., and Kobayashi,
T. (1984) in Protochlorophyllide Reduction and Greening
(Sironval, C., and Brouers, M., eds.) Martinus Nijhoff/Dr. W. Junk
Publisher, The Hague, pp. 99-112.
27.Heyes, D. J., Hunter, C. N., van Stokkum, I. H.
M., Grondelle, R., and Groot, M. L. (2003) Nat. Struct. Biol.,
10, 491-492.
28.Ignatov, N., Belyaeva, O., and Litvin, F. (1993)
Photosynth. Res., 38, 117-124.
29.Frank, F., Dujardin, E., and Sironval, C. (1980)
Plant. Sci. Lett., 18, 375-380.
30.Belyaeva, O. B., and Litvin, F. F. (1989)
Chlorophyll Photobiosynthesis [in Russian], MGU, Moscow.
31.Raskin, V. I., and Schwartz, A. (2002)
Photosynth. Res., 74, 181-186.
32.Dobek, A., Dujardin, E., Franck, F., Sironval,
C., Breton, J., and Roux, E. (1981) Photobiochem. Photobiophys.,
2, 35-44.
33.Belyaeva, O. B., and Sundqvist, C. (1998)
Photosynth. Res., 55, 41-48.
34.Schoefs, B., Bertrand, M., and Franck, F. (2000)
Photochem. Photobiol., 72, 85-93.
35.Ignatov, N. V., and Litvin, F. F. (2002)
Photosynth. Res., 71, 195-207.
36.Belyaeva, O. B., and Litvin, F. F. (2009)
Biochemistry (Moscow), 74, 1535-1544.
37.Krasnovsky, A. A., Bystrova, M. I., and Lang, F.
(1970) Doklady AN SSSR, 194, 1441-1444.
38.Krasnovsky, A. A., and Bystrova, M. I. (1974)
Chlorophyll (Shlyk, A. A., ed.) [in Russian], Nauka i Tekhnika,
Minsk, pp. 139-153.
39.Bystrova, M. I., Safronova, I. A., and
Krasnovsky, A. A. (1983) Doklady AN SSSR, 270,
1227-11231.
40.Bublichenko, N. V., Umrikhina, A. V., and
Krasnovsky, A. A. (1979) Biofizika (Moscow), 24,
588-593.
41.Belyaeva, O. B., Bystrova, M. I., Safronova, I.
A., Litvin, F. F., and Krasnovsky, A. A. (1985) Mol. Biofiz.
(Moscow), 30, 933-938.
42.Krasnovsky, A. A., Jr., Shuvalov, V. A., Litvin,
F. F., and Krasnovsky, A. A. (1971) Doklady AN SSSR, 199,
1181-1184.
43.Krasnovsky, A. A., Jr. (1982) Photochem.
Photobiol., 36, 733-741.
44.Brouers, M. (1975) Photosynthetica,
9, 304-310.
45.Boddi, B., Soos, J., and Lang, F. (1980) Biochim. Biophys.
Acta, 593, 158-165.
46.Lebedev, N., and Timko, M. (1999) Proc. Natl.
Acad. Sci. USA, 96, 17954-17959.
47.Heyes, D. J., Heathcote, P., Rigby, S. E. J.,
Palacios, M. A., Grondelle, R., and Hunter, C. N. (2006) J. Biol.
Chem., 281, 26847-26853.
48.Belyaeva, O. B., Griffits, V. T., Kovalev, J. V.,
Timofeev, K. N., and Litvin, F. F. (2001) Biochemistry (Moscow),
66, 173-177.
49.Terenin, A. N. (1967) Photonics of Dye
Molecules and Related Compounds [in Russian], Nauka, Leningrad, pp.
406-432.
50.Griffiths, W. T. (1974) FEBS Lett.,
46, 301-304.
51.Griffiths, W. T. (1975) Biochem. J.,
152, 623-635.
52.Blenkenhorn, G. (1976) Eur. J. Biochem.,
67, 67-80.
53.Valera, V., Fung, M., Wessler, A. N., and
Richards, W. R. (1987) Biochem. Biophys. Res. Commun.,
148, 515-520.
54.Begley, J. R., and Young, M. (1989) J. Am.
Chem. Soc., 111, 3095-3096.
55.Wilks, H. M., and Timko, M. P. (1995) Proc.
Natl. Acad. Sci. USA, 92, 724-728.
56.Lebedev, N., Karginova, O., McIvor, W., and
Timko, M. (2001) Biochemistry, 40, 12562-12574.
57.Menon, B. R. K., Waltho, J. P., Scrutton, N. S., and Heyes, D.
J. (2009) J. Biol. Chem., 284, 18160-18166.
58.Townley, H. E., Sessions, R. B., Clarke, A. R.,
Dafforn, T. R., and Griffiths, W. T. (2001) Proteins:
Structure, Function, and Genetics, 44,
329-335.
59.Menon, B. R. K., Davison, P. A., Hunter, C. N.,
Scrutton, N. S., and Heyes, D. J. (2010) J. Biol. Chem.,
285, 2113-2119.
60.Gurinovich, G. P., Sevchenko, A. N., and
Solovyov, K. N. (1968) Spectroscopy of Chlorophyll and Related
Compounds [in Russian], Nauka i Tekhnika, Minsk, pp. 468-475.
61.Litvin, F. F., Belyaeva, O. B., and Ignatov, N.
V. (2000) Uspekhi Biol. Khim., 40, 3-42.
62.Krasnovsky, A. A., Jr., Belyaeva, O. B., Kovalev,
Yu. V., Ignatov, N. V., and Litvin, F. F. (1999) Biochemistry
(Moscow), 64, 587-591.
63.Griffiths, W. T., McHugh, T., and Blankenship, R.
E. (1996) FEBS Lett., 398, 235-238.
64.Sytina, O. A., Heyes, D. J., Hunter, C. N.,
Alexandre, M. T., van Stokkum, I. H. M., van Grondelle, R., and Groot,
M. L. (2008) Nature, 456, 1001-1005.
65.Walke, C. J., and Griffiths, W. T. (1988) FEBS
Lett., 239, 259-262.
66.Nayar, P., Brun, A., Harriman, A., and Begley, T.
P. (1992) J. Chem. Soc. Chem. Commun., 395-397.
67.Ignatov, N. V., Belyaeva, O. B., and Litvin, F.
F. (1993) Photosynthetica, 29, 235-241.