Search for Ligand of N-Acetylglucosaminyl-N-Acetylmuramyl Dipeptide Using Its Peptide Mimetic
G. V. Savinov*, A. O. Shepelyakovskaya, Kh. M. Boziev, F. A. Brovko, and A. G. Laman
Pushchino Branch of Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, pr. Nauki 6, 142290 Pushchino, Moscow Region, Russia; E-mail: gsawinow@gmail.com* To whom correspondence should be addressed.
Received October 2, 2013; Revision received November 12, 2013
The method for searching for ligands exerting an adjuvant effect is described. The method involves isolation of polysomes using an immobilized peptide mimetic of N-acetylglucosaminyl-N-acetylmuramyl dipeptide (GMDP) – RN-peptide. After the affinity chromatography and washing, RN-peptide complexes with the target sequences were dissociated with guanidine hydrochloride. The obtained mRNA was used for cDNA synthesis and subsequent cloning in an expression vector. Further studies showed the effectiveness of this method. Clones interacting with the peptide were selected using biotinylated RN-peptide. It was found that all clones encode a sequence identical to the protein YB-1. Recombinant antibodies against protein YB-1 were selected from a phage display human scFv library. Using these antibodies, we determined the binding constant of RN-peptide to protein YB-1. Competitive analysis showed that RN-peptide and GMDP compete for the same portion of YB-1 at molar ratio 1 : 12.
KEY WORDS: peptide mimetic, GMDP, YB-1DOI: 10.1134/S0006297914020060
Abbreviations: GMDP, N-acetylglucosaminyl-N-acetylmuramyl dipeptide.
The search for new immune system regulators (activators), particularly
ones having adjuvant activity, is a subject for both fundamental
science, i.e. for researchers interested in the development of immune
responses, including the mechanisms of cellular differentiation of the
immune system, and for practical experts in vaccines and autoimmune
disorders. This trend has become especially relevant with reference to
the creation of a novel generation of vaccines containing not
inactivated bacterial cells or viral particles, but antigen fragments
obtained by the methods of chemical synthesis or molecular
biotechnology, which are recognized by the host immune system and
thereby protect an organism from infection with minimal side effects.
As a rule, such vaccines are weakly immunogenic and need helper
molecules to intensify and direct the immune response. This is also
true for DNA vaccines.
It should be noted that adjuvants in the modern sense of the word are compounds that not only activate the humoral/antibody immune response but also regulate cellular immunity by facilitating the creation and possible implementation of a prolonged response to the antigen. Such response is impossible without the involvement of memory T and B cells [1]. The currently available adjuvants have considerable limitations and shortcomings that necessitate the search for new drugs exerting directional effects with minimal side effect. We believe such adjuvants might be natural compounds (activators of dendritic cells) or their analogs obtained not only by sorting out the structures of known compounds that have been often and successfully used previously, but also by modern methods including the search for mimetics by means of combinatorial chemistry. Fragments of bacterial cell wall peptidoglycan and their synthetic analogs are actively studied in many laboratories worldwide as activators of nonspecific immunity, immune correctors, and adjuvant medications [2, 3].
High-specificity and high-affinity monoclonal anti-GMDP antibodies have been created in the Laboratory of Immunochemistry of the Institute of Bioorganic Chemistry, Russian Academy of Sciences. It has been shown that one type of GMDP receptor has intracellular localization [4]. Using monoclonal anti-GMDP antibodies and the peptide phage display technique, we discovered a 15-mer peptide mimetic of GMDP [5]. The pentadecapeptide RVPPRYHAKISPMVN (RN-peptide) interacts with monoclonal antibodies E6/1.2 against GMDP and has adjuvant but not pyrogenic activity, in contrast to GMDP. It also increases the expression of class II histocompatibility antigens on the surface of macrophages. Quite often, the mimetics of compounds are found to be more strictly specific and more active than the initial compounds [6]. RN-peptide is more than 10-fold (in molar ratio) active than GMDP. This work is devoted to the search for the molecular target of GMDP using its peptide mimetic RN-peptide.
MATERIALS AND METHODS
Primers, cell lines, and buffer solutions. The following primers were used: Sfi I (3211), AATTCGAGGCCCAGCCGGCCAAACACCATC; dT30NOT, CTGCGGCCGC T30.
Escherichia coli strain tg1 (E. coli K, Δlac-proAB, thi, r+m–, F′(lacZΔM15, proA, proB, traD36, supE44)) and two cell lines, WEHI3 (mouse monocyte cell line) and SP2/0 (mouse myeloma cell line), were used.
The 2xYT medium (5 g/liter NaCl, 16 g/liter Tryptone, 11 g/liter yeast extract, pH 7.0) and PBS (150 mM NaCl, 5.2 mM Na2HPO4, 1.7 mM KH2PO4) and PBST (1× PBS, 0.1% Tween-20) buffer solutions were used.
RNP isolation. The polysome-containing RNP fraction was isolated according to Palmiter [7]. About 109 cells were homogenized in 90 ml of homogenization buffer (25 mM Tris-HCl, pH 7.5, 25 mM NaCl, 5 mM MgCl2, 1 mg/ml heparin, and 2% Triton X-100), and the suspension was agitated for 5 min on a magnetic stirrer and centrifuged for 5 min at 27,000g in a JA20 rotor of a JA2-ME centrifuge (Beckman, USA). An equal volume of homogenization buffer containing 200 mM MgCl2 was added to the supernatant, and the sample was incubated for 1 h at 4°C. The preparation was layered on 1 M sucrose solution containing 25 mM Tris-HCl (pH 7.5), 25 mM NaCl, and 100 mM MgCl2, with a homogenate/sucrose ratio of 2 : 1, and centrifuged for 10 min in the JA20 rotor at 27,000g. The polysome precipitate was suspended in homogenization buffer without Triton X-100 (100 µl per 108 cells).
Isolation of polysomes synthesizing proteins with high affinity to RN-peptide. The RNP fraction (100 µl) in RNP isolation buffer without Triton X-100 was added to RN-peptide immobilized on TENTA-GEL (TentaGel S-NH2; Rapp Polymere GmbH). The mixture was incubated overnight at 4ºC and then washed five times with the same buffer. The RNA was eluted with 4 M guanidine thiocyanate. The RNA was precipitated by adding three volumes of C2H5OH and incubated overnight at –20ºC, harvested by centrifugation for 20 min at 10,000g, washed with 80% alcohol, and dissolved in water.
cDNA synthesis. The resulting RNA was treated with tobacco acid pyrophosphatase (Sigma-Aldrich, USA) to remove the cap structure of mRNA. Then, using T4 RNA-ligase (Fermentas, Lithuania), an “anchor” oligonucleotide Sfi I (3211) was attached using the manufacturer’s buffers and instructions.
The first cDNA strand was synthesized using the dT30NOT primer. The mRNA was dissolved in 68 µl of 1 mM solution of vanadyl ribonucleoside complexes with the addition of 5 µl of 5 mM dNTP mixture, 5 µl of 0.1 M DTT, 20 pmol of the primer, and 10 µl of 10× revertase buffer (Amersham, GB). The mixture was heated at 67°C for 5 min and then slowly cooled to room temperature with the addition of 4 µl of RNasin (Amersham) to 0.5 U/µl and 4 µl of reverse transcriptase AMV-RT (20 U/µl) (Amersham). The mixture was incubated at 42°C for 1 h.
DNA amplification. cDNA was amplified using the pair of primers dT30NOT and Sfi (3211). The first cDNA strand was precipitated with ethanol after phenol-induced deproteinization and dissolved in 30 µl of H2O. The reaction mixture for amplification contained: the first cDNA strand, primers (10 µM), 0.25 mM dNTP, 50 mM Tris-HCl, pH 9.0, 50 mM KCl, 2 mM MgCl2, 0.02% Tween-20, and 100 µl of Taq polymerase (5 U). Nonspecific amplification was reduced by five cycles of amplification in the following mode: denaturing at 94°C, 1 min; annealing, 1 min; elongation at 74°C, 2 min. The annealing stage was performed at the following temperatures: t1 = 65°C, t2 = 64°C, t3 = 63°C, t4 = 62°C, t5 = 61°C. Then 30 amplification cycles were performed at annealing temperature of 55°C and the same time parameters. The amplification products were analyzed by electrophoresis in 1% agarose gel.
cDNA restriction and ligation into a vector. DNA was restricted in buffer 4 from New England Biolabs (NEB) using 10 U of the enzyme Sfi I (NEB) per 1 µg of DNA. The total volume of the reaction mixture was 50 µl. After mixing all the components, the mixture was incubated at 50°C for 3 h. Then NaCl was added to achieve concentration 100 mM. Then restriction was performed with 10 U of enzyme Not I (NEB) at 37°C for 3 h. The vector DNA of the plasmid pHEN1 was ligated with cDNA using T4 DNA ligase (Fermentas) using the manufacturer’s buffers and instructions.
Preparation of electrocompetent E. coli cells. Competent cells were prepared as follows: the cell culture of E. coli strain TG1 was grown in 2xYT medium at 37°C under intense aeration until reaching optical density A600 = 0.5. The culture was cooled on ice for 30 min, and then the cells were precipitated by centrifugation at 4°C and 3000g for 15 min. The precipitate was suspended in cooled 10% glycerol in MilliQ water at volume equal to the initial volume of the culture liquid. The procedure was repeated twice, and then the cells were suspended in 10% glycerol at volume equal to 1/1000 volume of the initial culture liquid. The cell suspension was poured into test tubes by 50-µl portions, frozen in liquid nitrogen, and stored at –70°C.
Electroporation. The cells were electroporated in 0.2-cm cuvettes with 50 µl of competent cells and 50-100 ng of DNA obtained after reprecipitation of the ligase mixture. Immediately after the electric impulse (2500 V), 1 ml of the 2xYT medium containing 1% glucose and 10 mM MgCl2 was added into the cuvette; the cells were accurately suspended, transferred into sterile tubes, and incubated at 37°C for 1 h.
Primary analysis of the library by plate screening. Escherichia coli cells transformed by the plasmid carrying a cDNA insert were inoculated on Petri dishes with agarized 2xYT medium containing 100 µg/ml ampicillin and 0.1% glucose and incubated overnight at 30°C. After counting the number of grown colonies, replicas were taken from the plates on nitrocellulose filters. The filters were shifted onto the surface of agarized 2xYT medium containing 1 mM IPTG and incubated at 30°C for 5 h. After the incubation under conditions of induction, the cells on the filter were lysed in chloroform vapor, and the plates were left overnight at 4°C. Then the filters were washed five times with TBS-T buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween-20) and placed into the solution with biotinylated RN-peptide (1 µM) in TBS-T buffer for 1 h. After fivefold washing with TBS-T buffer, the filters were incubated in TBS-T with streptavidin–horseradish peroxidase conjugate (Amersham) (dilution according to the manufacturer’s protocol) at room temperature for 30 min. The filters were developed with diaminobenzidine as a chromogenic substrate. Colonies with a marked signal were selected for further analysis.
Plasmid isolation. The cell culture (5 ml) was centrifuged at 2500 rpm for 20 min. The supernatant was decanted. PBS (100 µl) and lysozyme (up to 1 mg/ml) were added to the precipitate. The sample was incubated for 10 min. Then, 200 µl of NaOH and SDS mixture (1% SDS, 0.2 M NaOH) was added, and the sample was left on ice for 5-10 min. After the addition of 1/2 volume of 3 M potassium acetate, the mixture was centrifuged at 14,000 rpm for 10 min. After centrifugation and addition of 0.6 volume of isopropanol, the mixture was incubated for 15 min and centrifuged at room temperature at 3000 rpm for 20 min. The precipitate was dissolved in 500 µl of TE (10 mM Tris-HCl, pH 7.6, 2 mM EDTA). RNase was added into the resulting solution (10 µg/ml). The mixture was heated at 70°C for 15 min, incubated at 42°C for 1 h and, and, after the addition of an equal volume of phenol–chloroform mixture (1 : 1 v/v), centrifuged at 13,400 rpm for 5 min. The supernatant was collected without taking the interphase; 1/10 volume of 3 M sodium acetate and an equal volume of isopropanol were added to the supernatant. The precipitate formed was centrifuged at 13,400 rpm for 10 min and dissolved in 50 µl of TE.
Preparation of single-stranded Fv fragments of antibodies (scFv) against YB-1. The recombinant YB-1 kindly provided by S. G. Guryanov (Institute of Protein Research, Russian Academy of Sciences) at concentration of 10 µg/ml was adsorbed to the surface of the immunoplate and incubated with 1012 phage particles in 100-µl volume of the human scFv library overnight at 4°C. Nonspecifically adsorbed phage particles were washed off 15 times with PBST, and the remaining phages were used to infect the cell culture of E. coli strain TG-1. For the second round of selection, the infected cells were grown to OD600 = 0.6, contaminated with the helper phage M13-K07, and incubated under intense aeration overnight. The cellular supernatant containing 1012 phage particles per milliliter was used for the second round. In this case, YB-1 was adsorbed to the plate surface at concentration 1 µg/ml and incubated with phage particles at room temperature for 1 h. Then, the E. coli culture was infected with specific phages, and the cells were inoculated to the density of 300 colonies per 85-mm diameter Petri dish. Individual colonies were used for further analysis.
Determination of Kd. The Kd value of the protein and RN-peptide complex was determined by indirect ELISA. Streptavidin (100 µl) at concentration 2 µg/ml was applied to a 96-well immunoplate. The plate was incubated overnight at 4°C. Then the plate was washed five times with PBS and five times with PBS + 0.1% Tween-20 (PBST). The same washing was performed after each stage of incubation. Biotinylated RN-peptide in binary dilutions was incubated with YB-1 in solution at room temperature for 1 h. The YB-1 concentration was constant (150 ng/ml). The initial RN-peptide concentration was 50 ng/ml. The RN-peptide/YB-1 complex in PBST was applied to the plate in a 100-µl aliquot. The plate was incubated for 1 h, then the plate wells were washed, and the anti-YB-1 phages were applied at concentration of 1012 phage particles/ml. The plate was developed with anti-M13–peroxidase conjugate and o-phenylenediamine (OPD) as a chromogenic substrate. The staining intensity of the resulting RN-peptide and YB-1 complex was measured at wavelength λ = 492 nm.
Competitive analysis of RN-peptide and GMDP binding to YB-1. YB-1 (1 µg/ml) was incubated with biotinylated RN-peptide (3 ng/ml) and decreasing concentrations of GMDP in PBST at room temperature for 1 h. Then the reaction mixture was adsorbed on immobilized streptavidin, and the formed biotinylated RN-peptide/YB-1 complex was detected using monoclonal phage scFv and anti-M13–peroxidase conjugate with OPD as a chromogenic substrate.
RESULTS AND DISCUSSION
RN-peptide is a pentadecapeptide: RVPPRYHAKISPMVN. It does not contain acidic (Asp, Glu) amino acid residues, but is rich in basic (Arg, Lys) and proline residues. The N-terminal part of the peptide is particularly rich in basic amino acid residues. The C-terminal part contains hydrophobic and neutral amino acids. We studied the functional role of individual amino acids using Ala-scan, the substitution of alanine for the amino acid at each position. Previously, it was shown that the necessary components for manifestation of the adjuvant activity of RN-peptide are the RVPPRYH region, K9, I10, and M13. The PRYH sequence is necessary for the binding with the anti-GMDP antibodies (E6/1.2), and arginine and tyrosine residues make the maximum contribution to the interaction with the antibodies. Accordingly, C-terminal modification was shown to have no substantial effect on manifestation of biological activity, while N-terminal modification reduced biological activity several-fold [8].
RN-peptide interacts with monoclonal antibodies E6/1.2 against GMDP and has adjuvant but not pyrogenic activity, in contrast to GMDP. It also enhances the expression of class II histocompatibility antigens on the surface of macrophages [5].
Since GMDP and RN-peptide have similar biological properties, we supposed that they interact with the same receptor. Hence, RN-peptide can be used as a probe in the search for the GMDP receptor.
Search for a protein interacting with GMDP. The analysis of expression libraries requires more recombinant clones than are necessary in the case of DNA and oligonucleotide probes.
Therefore, we decided first to enrich the RNA preparation with the target mRNA sequences. For this purpose, RNA was isolated from the RNP fraction enriched in polysomes that synthesize the hypothetical receptor protein.
The sample was enriched by affinity chromatography directly on the peptide synthesized on TENTA-GEL. The schematic diagram of the experiment is shown in Fig. 1.
Fig. 1. Scheme of polysome isolation by affinity chromatography.
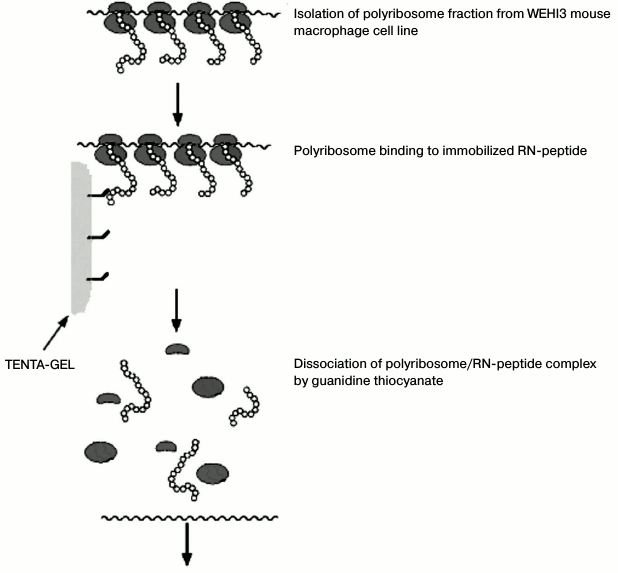
After affinity chromatography, we used the strategy of cDNA synthesis, which is schematically presented in Fig. 2.
Fig. 2. cDNA isolation and amplification based on the full-sized mRNA: 1) full-sized matrix; 2) matrix lacking polyadenylated region; 3) matrix lacking cap structure; 4) matrix lacking both polyadenylated region and cap structure.
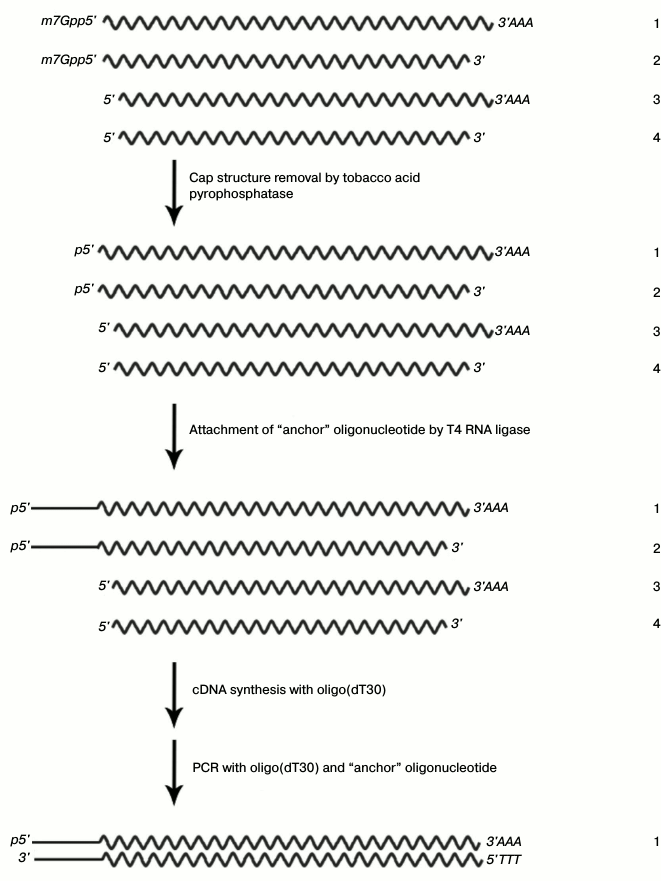
This approach is based on the removal of the cap structure of mRNA by pyrophosphatase, the attachment of “anchor oligonucleotide” instead of the removed cap structure, and cDNA synthesis followed by PCR amplification. DNA fragments encoding the protein interacting with RN-peptide were ligated with the pHEN-1 vector (Pharmacia Biotech) at the SfiI and NotI restriction sites. The conditions of ligation were selected so that its efficiency would be no less than 90% according to the data of electrophoretic analysis of the products. The strain E. coli TG1 with transformation efficiency no less than 109 colony-forming units (CFU) per 1 µg pHEN-1 was used for cloning.
A library of 105 clones was obtained. Its primary analysis was performed by screening with the biotinylated peptide.
The library was inoculated on Petri dishes (200 colonies per 9-cm diameter dish). Five clones with the most marked signal were selected for further analysis (Fig. 3).
Fig. 3. Nitrocellulose filter replica developed using biotinylated RN-peptide.

Primary structure analysis data. According to DNA sequencing data, the protein interacting with RN-peptide is identical to the Y-box-binding protein (YB-1) of vertebrates (Fig. 4). One out of five selected clones contained a full-sized YB-1 protein. The other four clones apparently contain different shortened variants of YB-1.
Fig. 4. Structure of YB-1 and cDNA clones demonstrating the degree of homology of nucleotide (nt) and amino acid (aa) sequences to YB-1 protein.
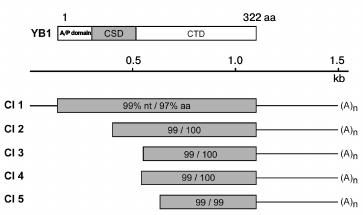
YB-1 is a member of the domain family of cold shock proteins, and it participates in transcription and translation control [9-14].
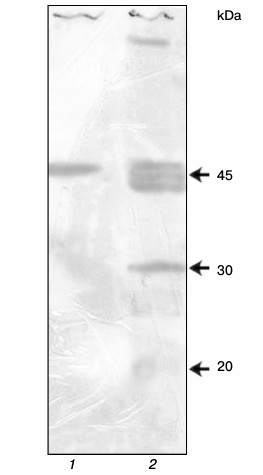
Preparation of antibodies against YB-1. In Kd determination, it is desirable that the protein–ligand interaction occurs in solution. Therefore, the anti-YB-1 antibodies are necessary for detection of the formed complex. For this purpose, recombinant scFv were derived using the scFv human nonimmune library with a repertoire of 6·109 obtained previously in our laboratory [15]. Positive clones were selected after two rounds of affinity selection and characterized by enzyme immunoassay and immunoblotting. The sensitivity of the resulting scFv in the phage display format was 5 ng YB-1 per milliliter. The 5 ng/ml sensitivity in an indirect enzyme immunoassay is very high. These antibodies show several bands on immunoblotting (Fig. 5) but, nevertheless, they can be used for characterization of the purified protein.
Fig. 5. YB-1 immunoblotting: 1) recombinant YB-1; 2) SP2/0 mouse myeloma cell lysate.
Determination of dissociation constant and competitive analysis. The dissociation constant of the YB-1/RN-peptide complex was determined by the method of indirect enzyme-linked immunosorbent assay (ELISA).
We have shown that modification of the C-terminal amino acid of RN-peptide has no effect on its biological activity. On the contrary, modification of the N-terminal part of the peptide or lysine-9 results in the loss of biological activity [8]. Therefore, we determined the dissociation constant using the RN-peptide with C-terminal biotinylation.
The interaction between the peptide and YB-1 occurred in solution, ensuring the correctness of Kd determination. Then the formed complex was adsorbed to the immunoplate with streptavidin. The YB-1/RN-peptide complex was detected using monoclonal phage scFv and horseradish peroxidase conjugate with anti-M13 bacteriophage antibodies (Fig. 6).
Fig. 6. Scheme of enzyme immunoassay for Kd determination. The interaction between biotinylated RN-peptide and YB-1 occurred in solution, ensuring correct Kd determination. The formed complex interacted with streptavidin adsorbed on the immunoplate. The YB-1/RN-peptide complex was detected using monoclonal phage scFv and horseradish peroxidase conjugate with anti-M13 bacteriophage antibodies.
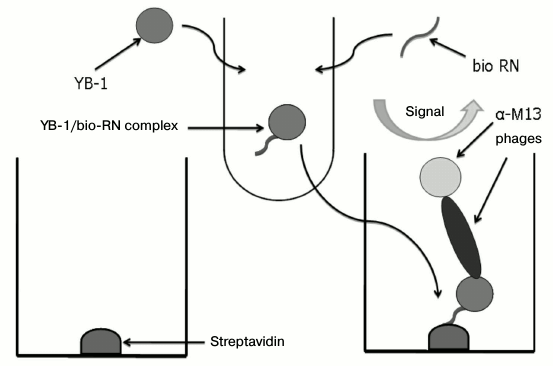
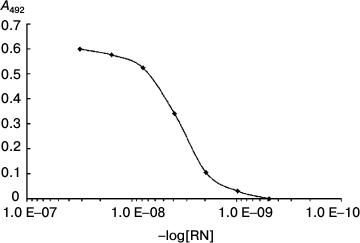
The dissociation constant of the complex was 4·10–9 (Fig. 7).
Fig. 7. Determination of the dissociation constant of RN-peptide relative to the YB-1 protein. YB-1 concentration was 150 ng/ml. RN-peptide titration by binary dilutions from 50 ng/ml. Kd = 4·10–9.
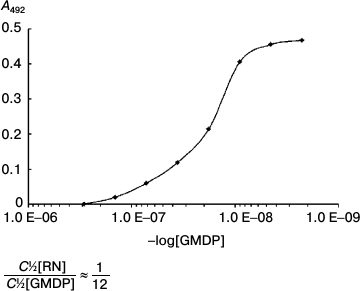
Competitive analysis showed that GMDP (2·10–8 M) displaces 50% of the RN-peptide (1.5·10–9 M) from the complex with the antibodies (Fig. 8). This implies than RN-peptide and GMDP compete for the same protein region. This type of competition suggests that the YB-1 protein is involved in the implementation of biological activity of both GMDP and RN-peptide.
Fig. 8. Competitive analysis of RN-peptide and GMDP. The RN-peptide concentration was 3 ng/ml. GMDP titration by tertiary dilutions from 150 ng/ml. YB-1 concentration was 1 µg/ml.
In the present work, the protein interacting with RN-peptide has been cloned. The primary structure analysis of the cloned protein showed its identity to Y-box-binding protein 1. The Kd value of the peptide relative to this protein was 4·10–9. In addition, we have shown that GMDP and RN-peptide compete for binding to the same region of YB-1. The method used has demonstrated its efficiency in the search for ligands of biologically active compounds using their peptide mimetics.
The authors are grateful to researcher of the Institute of Protein Research (Russian Academy of Sciences) S. G. Guryanov for his assistance and consultations during the work.
This work was supported by the Federal Targeted Program (grant No. 14.132.21.1314).
REFERENCES
1.Audibert, F. (2003) Int. Immunopharmacol.,
3, 1187-1193.
2.Cipriano, C., Giacconi, R., Muzzioli, M.,
Gasparini, N., Orlando, F., Corradi, A., Cabassi, E., and Mocchegiani,
E. (2003) Mech. Ageing Dev., 124, 877-886.
3.Steiner, H. (2004) Immunol. Rev.,
198, 83-96.
4.Golovina, T., Fattakhova, G., Swiderek, K.,
Makarov, E., Bovin, N., Shively, J., and Nesmeyanov, V. (1999) FEBS
Lett., 454, 152-156.
5.Laman, A. G., Shepelyakovskaya, A. O., Boziyev, Kh.
M., Savinov, G. V., and Brovko, F. A. (2010) Bioorg. Khim.,
36, 141-148.
6.Monzavi-Karbassi, B., Cunto-Amesty, G., Luo, P.,
and Kieber-Emmons, T. (2002) Trends Biotechnol., 20,
207-214.
7.Palmiter, R. D. (1974) Biochemistry (Wash.),
13, 3606.
8.Laman, A. G., Shepelyakovskaya, A. O., Boziev, Kh.
M., Savinov, G. V., Baidakova, L. K., Chulin, A. N., Chulina, I. A.,
Korpela, T., Nesmeyanov, V. A., and Brovko, F. A. (2011)
Vaccine, 29, 7779-7784.
9.Sullivan, K. E., and Peterlin, B. M. (1987) Mol.
Cell Biol., 7, 3315-3319.
10.Dorn, A., Durand, B., Marfing, C., Lemeur, M.,
Benoist, C., and Mathis, D. (1987) Proc. Natl. Acad. Sci. USA,
84, 6249-6253.
11.Boss, J. M., and Strominger, J. L. (1986)
Proc. Natl. Acad. Sci. USA, 83, 9139-9143.
12.Sherman, P. A., Basta, P. V., and Ting, J. P.-Y.
(1987) Proc. Natl. Acad. Sci. USA, 84, 4254-4258.
13.Miwa, K., Doyle, C., and Strominger, J. L. (1987)
Proc. Natl. Acad. Sci. USA, 84, 4939-4943.
14.Eliseeva, I. A., Kim, E. R., Guryanov, S. G.,
Ovchinnikov, L. P., and Lyabin, D. N. (2011) Biochemistry
(Moscow), 76, 1402-1433.
15.Batanova, T. A., Ulitin, A. B., Morozova, V. V.,
Laman, A. G., Zhirakovskaia, E. V., Dubrovskaia, V. V., Brovko, F. A.,
and Tikunova, N. V. (2006) Mol. Gen. Mikrobiol. Virusol.,
3, 35-41.