REVIEW: Molecular Chaperone GroEL/ES: Unfolding and Refolding Processes
N. A. Ryabova, V. V. Marchenkov, S. Yu. Marchenkova, N. V. Kotova, and G. V. Semisotnov*
Institute of Protein Research, Russian Academy of Sciences, ul. Institutskaya 4, 142290 Pushchino, Moscow Region, Russia; fax: +7 (4967) 318-435; E-mail: nina@vega.protres.ru* To whom correspondence should be addressed.
Received May 21, 2013; Revision received July 14, 2013
Molecular chaperones are a special class of heat shock proteins (Hsp) that assist the folding and formation of the quaternary structure of other proteins both in vivo and in vitro. However, some chaperones are complex oligomeric proteins, and one of the intriguing questions is how the chaperones fold. The representatives of the Escherichia coli chaperone system GroEL (Hsp60) and GroES (Hsp10) have been studied most intensively. GroEL consists of 14 identical subunits combined into two interacting ring-like structures of seven subunits each, while the co-chaperone GroES interacting with GroEL consists of seven identical subunits combined into a dome-like oligomeric structure. In spite of their complex quaternary structure, GroEL and GroES fold well both in vivo and in vitro. However, the specific oligomerization of GroEL subunits is dependent on ligands and external conditions. This review analyzes the literature and our own data on the study of unfolding (denaturation) and refolding (renaturation) processes of these molecular chaperones and the effect of ligands and solvent composition. Such analysis seems to be useful for understanding the folding mechanism not only of the GroEL/GroES complex, but also of other oligomeric protein complexes.
KEY WORDS: protein folding, molecular chaperones, GroEL, GroESDOI: 10.1134/S0006297913130038
Abbreviations: ANS, 8-anilino-1-naphthalenesulfonic acid; Hsp, heat shock protein.
Many years of research into the processes of protein unfolding and
refolding essentially confirm Anfinsen’s hypothesis suggesting
that all necessary and sufficient information about the spatial
structure of a protein is contained in its amino acid sequence [1, 2]. Nevertheless, analysis of
the processes of cell vital activity under various stress states of
cells has revealed a number of protein factors called molecular
chaperones, which are involved either in the catalysis of protein
folding or in the regulation of distribution of newly synthesized
proteins among competitive pathways of folding and aggregation [3-5]. Thus, molecular chaperones
create the optimal conditions for substrate protein folding via the
elimination of “interferences” or “undesirable”
intermolecular contacts [6, 7].
In addition, chaperones assist the assembly of oligomeric complexes,
transmembrane transport, and degradation of polypeptide chains [2, 7, 8].
Another important function of molecular chaperones is to prevent lethal
nonspecific association of proteins under stress conditions [6].
Molecular chaperones have been found in both prokaryotic and eukaryotic organisms, and many chaperones per se are complex oligomeric proteins consisting of 6-8 subunits (each with a molecular weight of 10 to 100 kDa), usually combined into one- or two-ring structures [9]. Therefore, an intriguing task is to understand the mechanisms of folding of chaperones.
The purpose of this review is to analyze the literature and our own data on denaturation (unfolding) and renaturation (refolding) of one of the most actively studied chaperones: the heat shock protein of Escherichia coli GroEL (Hsp60), as well as its partner co-chaperone GroES (Hsp10). GroEL is a complicated oligomeric protein complex consisting of 14 identical subunits (each with molecular weight of 60 kDa) combined into two ring structures interacting via their ends, each containing seven subunits [10]. This protein, when functioning, interacts with another oligomeric protein, GroES, consisting of seven identical subunits (each with molecular weight of 10 kDa) combined into a dome-like ring structure [11, 12].
Studies of denaturation and renaturation of the large oligomeric protein complex of chaperone GroEL and its co-chaperone GroES attempt to reveal two basic aspects in understanding the mechanism of protein folding. One is to answer the question “How do proteins involved in folding of other proteins fold themselves?” The other aspect is concerned with obtaining new knowledge about self-organization of oligomeric proteins, which is still poorly studied due to difficulties with selecting the conditions for their effective self-organization in vitro [2].
GroEL/ES AS A MOLECULAR CHAPERONE
The first large oligomeric chaperone (GroEL) was found in E. coli cells in the early 1970s as a protein necessary for the assembly of phage λ [13, 14]. It was shown that this protein could be noncovalently bound to the monomeric form of protein B of phage λ, providing the assembly of the phage head. GroEL is one of the dominant proteins of E. coli cells. However, the concentration of this protein in cells drastically increases under heat shock conditions [15]. In cells, GroEL can interact both with newly synthesized proteins, which have had no time to acquire a rigid tertiary structure during their biosynthesis, and with proteins that have lost their rigid structure for some reason [16-20]. Disturbances in the GroEL gene expression result in cell death [21]. GroEL has no marked specificity for protein targets. In vitro studies have shown that about 50% of proteins from the cell extract of E. coli, when denatured, interact with GroEL [22]. It has also been shown that more than 30% of various proteins are unable to fold into their native state in the absence of GroEL in the cell [18]. Experiments on protein renaturation in vitro show that GroEL enhances the yield of native protein by inhibiting the aggregation of unfolded protein molecules [23]. In some cases, GroEL must interact with another oligomeric heat shock protein, GroES (Hsp10), to perform its function [12, 24]. Data of electron microscopy [9] and X-ray structure analysis [10, 25] show that GroEL consists of 14 identical subunits (57 kDa each). The subunits form two heptameric rings overlying each other. The crystal structure of GroEL is a cylinder 145 Å in height and 135 Å in diameter, with a central channel of about 45 Å in diameter [10, 25] (Fig. 1). Each subunit of GroEL (547 amino acid residues) has three distinct domains: apical, middle (intermediate), and equatorial (Fig. 1). The apical domains (residues 191-376) form the ends of the GroEL cylinder and are involved in binding protein substrates and co-chaperone GroES [26-30]. According to the data of electron microscopy, the non-native substrate protein is bound just at the ends of the central channel [27, 28].
Fig. 1. Spatial structures of components of the GroEL/ES chaperone system of E. coli cells. A) 14-subunit two-ring chaperone GroEL (side view (top) and end view (bottom)). Three subunits of each ring were eliminated on the side view to reveal the inner cavity. B) GroEL subunit structure (a, apical; i, intermediate; e, equatorial domains). C) 7-subunit co-chaperone GroES (side view (top) and top view (bottom)). D) Asymmetrical complex of chaperone GroEL and co-chaperone GroES. Three subunits were eliminated from each ring of GroEL and GroES to reveal the inner cavity of the complex. Different shades of gray show the domains of GroEL subunits and GroES subunit. The figure was prepared using Ras Win software (based on RasMol 2.6 by Roger Sayle, Biomolecular Structures Group, Glaxo Wellcome Research & Development, Stevenage, Hertfordshire, UK) from the files IDER [86] and IAON [12] available in the Protein Data Bank (PDB).
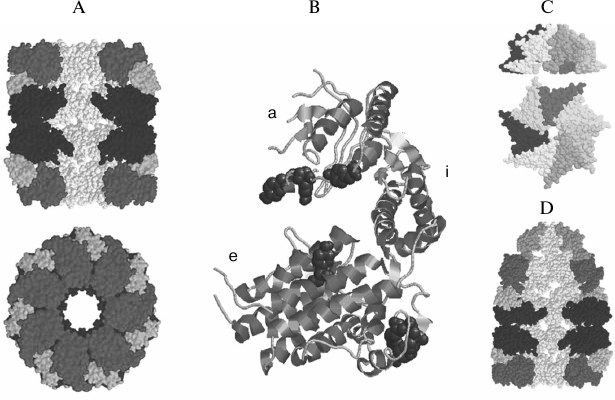
The equatorial domain (residues 6-133 and 409-523, 243 residues in all) is the largest domain of the GroEL subunit. This domain is formed mainly by α-helical regions, and it provides the contacts between subunits in the heptameric ring and the contacts between heptameric rings of the GroEL-particle [10, 25].
The middle (intermediate) domain of the GroEL subunit is the smallest one (residues 134-190 and 377-408, 89 residues in all). It forms a kind of articulated joint between the apical and equatorial domains [10]. Analysis of ligand-induced conformational changes in GroEL by electron microscopy [9, 28, 31] and comparison of crystal structures of GroEL and the GroEL–GroES complex [10-12] (Fig. 1) confirm that the main ligand-induced changes in the GroEL structure occur due to mobility of the middle domain. Mutations in the middle domain can suppress GroEL functions partially or completely [30].
The co-chaperone GroES consists of seven identical subunits (each with molecular weight of 10 kDa) and is a dome about 30 Å in height and 70-80 Å in diameter, with a hole of about 10 Å. The internal cavity is 20 Å in height and 30 Å in diameter. The structure of GroES has the same symmetry as that of GroEL [11] (Fig. 1).
Systems similar to the GroEL/ES chaperone system of E. coli cells consisting of two components (heat shock proteins hsp60 and hsp10) have been found in the chloroplasts and mitochondria of eukaryotic cells [16, 32]. These proteins have a very high homology to the analogous proteins of E. coli cells (GroEL and GroES) [16], which suggests similarity between their spatial structures.
The protein substrate of molecular chaperone GroEL is a more or less unfolded, non-native polypeptide chain. Chaperones are able to bind both small (~2 kDa) [33, 34] and large (up to 100 kDa) unstructured polypeptides in their denatured states [35-38]. Proteins may be in non-native state both immediately after completion of their synthesis on the ribosome, and under the influence of enhanced temperature or other denaturing factors on mature proteins. GroEL interacts well with different polypeptides of both prokaryotic and eukaryotic organisms [17, 22, 24, 32, 35]. The complex with substrate proteins is stabilized mostly by hydrophobic interactions [30, 38-40]. Electrostatic interactions also make an additional contribution (the affinity of GroEL to its protein substrates varies depending on ionic conditions of the medium) [41-44].
The functioning of GroEL as a chaperone is provided by its interaction with a number of ligands: K+ and Mg2+ ions, adenine nucleotides (ADP and ATP), and the smaller (compared to GroEL) co-chaperone GroES [18, 20, 24, 45, 46]. The free GroEL has a weak ATPase activity (the rate of hydrolysis is 0.1 s–1 per subunit) in the presence of K+ in the solution [45]. Tight cooperative binding of Mg-ATP (Kdiss = 10 µM) leads to conformational changes in the GroEL particle [31, 47].
The low molecular weight ligands (adenine nucleotides) interacting with GroEL in the presence of Mg2+ can reduce the binding constant for protein targets. In the presence of Mg-ATP, GroEL and GroES rapidly form a GroEL14/ATP7/GroES7 complex. The subsequent ATP hydrolysis leads to formation of a highly stable GroEL14/ADP7/GroES7 complex with a dissociation constant of ~0.3 nM [48, 49]. The GroEL particle has a high affinity to one heptameric GroES oligomer and a low affinity to the other GroES oligomer, so that originally the presence of an asymmetric complex of one GroEL particle with one GroES oligomer was shown [9, 12, 49, 50] (see Fig. 1). However, the GroEL particle can also form a complex with two GroES oligomers, and the portion of symmetric complex directly depends on the ATP/ADP ratio and K+ concentration in the solution [51].
DENATURATION AND STABILITY OF GroEL AND GroES
Information about stability of the structure of oligomeric proteins gives a key to estimation of their ability for effective self-organization in vitro. Oligomeric components of the chaperone system of E. coli cells (tetradecameric GroEL and heptameric GroES) have similar stability against the effect of elevated temperature and denaturants; however, the processes of GroEL denaturation and renaturation are substantially dependent on the presence of ligands, while for GroES such dependence is not marked.
Studies of GroEL denaturation and renaturation were started more than 20 years ago; they were substantially initiated by an article of N. M. Lisin et al. published in Nature in 1990 [52]. They showed that the complex tetradecameric particle of GroEL (Fig. 1) could be reconstructed in vitro from the urea-unfolded monomeric state in the presence of Mg-ATP. However, the assembly of the GroEL particle was more efficient both when the concentrations of the monomeric form increased and when native GroEL (self-chaperoning) or co-chaperone GroES was added.
Reconstruction began with the formation of a folded monomeric form GroELm, which folded after the removal of urea from the protein solution by gel filtration. The resulting monomeric form of GroEL was incapable of specific oligomerization in the absence of Mg-ATP, had a high content of secondary structure and compactness, but was less stable against the effects of temperature and urea than within the complete tetradecamer GroEL14 (see also [53, 54]). The folded monomeric form (GroELm) is also characterized by substantially higher content of hydrophobic clusters exposed to the solvent compared to the subunit within the oligomer (GroELp) [54, 55]. Our data have confirmed most of the previously published results. First, the study of full-size GroEL denaturation and renaturation from the urea-unfolded state in the absence of Mg-ATP by transverse urea gradient gel electrophoresis [56, 57] has shown two-stage variation in the hydrodynamic volume during unfolding of full-size GroEL: its decrease in the region of 2 M urea and then a noticeable increase. The first stage seems to be concerned with dissociation of oligomeric GroEL particle into monomers that are partially unfolded under dissociation [58]. This is in agreement with previous data showing that in this very interval of denaturant concentrations there is a drastic decrease in intensities of light [54, 58] and X-ray [59] scattering, as well as a decrease in the amplitude of circular dichroism in the region of absorption of peptide bonds [54, 58]. The second stage is associated with further unfolding of GroEL monomers and increase in their hydrodynamic volume. The hydrodynamic volume of unfolded monomeric form (electrophoretic mobility) is close to the hydrodynamic volume (electrophoretic mobility) of the fully native GroEL particle. However, the study of GroEL unfolding by small-angle diffuse X-ray scattering indicates that the radius of gyration of the unfolded monomeric state is 15 Å (~20%) greater than the radius of gyration of the native oligomer (~70 Å) [59, 60]. The decrease in the hydrodynamic volume during GroEL renaturation from the urea-unfolded state in the absence of Mg-ATP also has two stages. However, these stages reflect the change in the hydrodynamic volume during the folding of the GroEL monomer. The product of GroEL renaturation in the region of 0.5 M urea has much higher electrophoretic mobility than the full GroEL particle under the same conditions. The presence of two stages in the renaturation of the monomeric form of GroEL seems to be associated with sequential folding of domains of the GroEL subunit. It should be mentioned that electrophoretic mobility under denaturation (unfolding in urea) of the folded monomeric form of GroEL changes exactly as in case of renaturation. Two-stage unfolding of the monomeric form (subunit) of GroEL manifests itself also in the variation of parameters such as the amplitude of the circular dichroism spectrum at 220 nm and the intensity of fluorescence of tyrosine residues and hydrophobic probe ANS (data not shown). Since the monomeric form is unfolded generally under the conditions of oligomeric GroEL dissociation (~2 M urea), it can be supposed that the equatorial domain responsible for inter-subunit contacts is unfolded under these conditions, while the apical domain maintains some of the intramolecular interactions. This supposition is in agreement with data obtained by the authors of work [58] indicating that the apical domain of the GroEL subunit maintains residual structure in the region of 3 M urea.
The folded monomeric form of GroEL is destabilized and partially unfolded when temperature drops below 10°C [52-54, 61], which makes it incompetent for specific oligomerization at low temperatures even in the presence of magnesium ions and adenine nucleotides [53, 61].
The influence of ligands and external factors on stability of the oligomeric structure of the GroEL particle has become an object of research of some authors. It has been revealed that the oligomeric structure of GroEL can be destabilized by low concentrations of guanidine hydrochloride (<0.5 M) [62, 63], by the presence of Mg-ATP or Mg-ADP [55, 64], NaCl and KCl salts, and trivalent spermidine cation [63], as well as by high hydrostatic pressure [65-67] or low temperature [61]. Low concentrations of denaturants also result in destabilization of GroEL complexes with substrate proteins [68] and GroES [69]. However, Mg2+ ions alone and also the interaction with the substrate proteins and the co-chaperone GroES significantly stabilize the quaternary structure of GroEL [61, 64, 65, 67, 68, 70, 71]. Thus, the quaternary structure of GroEL is characterized by enhanced lability, while its stability is regulated both by various external factors and by ligands. Such property of GroEL possibly provides its interaction with substrate proteins of different amino acid composition, structure, and size [22].
In contrast to GroEL, denaturation by urea of its heptameric partner (co-chaperone GroES) is a fully reversible process that does not depend on any external factors. The hydrodynamic volume during unfolding of full-size GroES7 by urea has two well distinguishable stages, just as in case of GroEL14. In the region of 2.5 M urea, the oligomeric structure is decomposed with respective decrease in the hydrodynamic volume, followed by the final unfolding of monomers with increase in hydrodynamic volume. The succession of folding (renaturation) is the reverse: first, monomers reach a certain (maybe partially folded) conformational state competent for specific oligomerization. Then these monomeric states are oligomerized with the formation of the native heptamer GroES7.
This result is in full agreement with the literature data on denaturation and renaturation of co-chaperone GroES (Hsp10), indicating high reversibility of this process. Thus, the study of GroES stability by differential scanning calorimetry and circular dichroism with different solvent compositions [72] showed that the temperature-induced unfolding/folding of the protein was a spontaneous reversible process described by a highly cooperative transition between folded heptamers and unfolded monomers. During denaturation, the folded monomeric state is energetically disadvantageous and, therefore, does not accumulate in noticeable amounts. Stabilization of the structure of the monomeric GroES occurs mainly due to inter-subunit interactions [72]. These interactions make oligomerization both enthalpically and entropically favorable. In spite of the high density of charged residues, GroES stability actually does not depend on salt concentrations at pH 7. However, millimolar concentrations of Mg2+ noticeably stabilize the structure of GroES, probably due to their specific binding (approximately three Mg2+-binding centers per heptamer) [72]. Thus, GroES (Hsp10) is a good example of the role of quaternary structure in stabilization of small proteins. Each subunit within the heptameric GroES and its analogs contains a very mobile loop (residues 16 to 33) located at the lower end of the molecule (Fig. 1) and acquiring a stable β-hairpin conformation only during the interaction with GroEL [11, 12, 73, 74]. It was also shown that the stability (denaturing temperature) of GroES increased at higher protein concentrations [72]. This result indicates that the unfolding of GroES at higher temperatures leads to dissociation of its oligomeric structure. It has been shown that the oligomeric structure of GroES has a strong tendency to dissociate at protein concentrations below the micromolar level [75]. GroES denaturation was also shown to be highly cooperative and reversible in the case of protein unfolding by guanidine hydrochloride [76, 77] or urea [77, 78]. The possible existence of the folded monomeric form of GroES under nondenaturing conditions was demonstrated by mechanical unfolding of covalently bound GroES subunits in an atomic-force microscope [79].
The reversibility of GroES denaturation substantially depends on the duration of protein incubation in the denatured state [72], which may be associated with aggregation of the protein in this state [77, 80]. The important structural element of GroES subunits, which provides the formation of their heptameric quaternary structure, is seven C-terminal amino acid residues. The proteolytic (carboxy peptidase Y) cleavage of these residues prevents specific oligomerization of GroES subunits [81]. We confirmed the importance of these residues in the formation of a GroES subunit competent for oligomerization also in the case of limited trypsinolysis. Thus, based on the literature and our own data, we know that the denaturation (unfolding) of heptameric GroES molecule is a highly cooperative process. Dissociation of the oligomeric structure is accompanied by unfolding of the monomeric form, leading to the absence of noticeable accumulation of intermediate states in the region of the denaturation transition, which is well described by a two-state (native heptameric and unfolded monomeric) model. However, the data on denaturation of eukaryotic Hsp10 [77] showing that the unfolding of this protein by urea does not lead to dissociation of the heptamer, while denaturation by guanidine hydrochloride results in dissociation of the heptamer, do not fit well with this model. At the same time, some literature data [78] and our data indicate that dissociation of the heptameric structure of prokaryotic GroES during its unfolding by urea occurs in exactly the same way as during denaturation of the protein by guanidine hydrochloride [76, 77] or higher temperatures [72].
RENATURATION OF GroEL AND GroES. ROLE OF LIGANDS AND EXTERNAL
CONDITIONS
Renaturation of the monomeric form (subunit) of GroEL at moderate concentrations of the protein is highly efficient and needs no additional factors [52-55]. However, as noted in one work, simple dilution of GroEL solution at high protein concentrations in the presence of more than 3 M urea by native buffer down to the conditions of existence of the folded monomeric form (0.7 M urea) is accompanied by substantial aggregation [59]. Therefore, it is preferable to obtain the folded monomeric form GroELm by the methods proposed in work [52], using gel filtration to get rid of the high concentrations of urea. We confirmed that such methods (followed by concentration) make it possible to obtain the folded monomeric form of GroEL with high yield and minimum aggregation [54]. In addition, this approach allows immediate separation of the aggregates from the monomeric protein. It should be noted that the monomeric form of GroEL folds quickly enough (in the second time interval) in spite of its high molecular weight (~60 kDa) and domain organization (our unpublished data). The kinetics of tyrosine fluorescence recovery is well described by a single-exponential process with a rate constant of ~0.23 s–1 (t1/2 ~ 3 s). The secondary structure of the monomeric form during its renaturation also recovers quickly (in the second time interval), but this process has three stages well separated in time. The greater part of the secondary protein structure (~60%) is formed during the dead time of kinetic experiments (<10–2 s). The rest of the secondary structure is formed in the measured interval of times during two kinetic stages: (1) ~30% per characteristic time t1/2 ~ 0.7 s, and (2) ~10% per characteristic time t1/2 ~ 7 s. The complex character of formation of the secondary structure of the monomeric form of GroEL is typical of the folding of globular proteins, and it is associated with rapid formation of an intermediate state of the “molten globule” type and with different stability of secondary structure elements [82]. It is possible that the domain organization of GroEL subunit plays a certain role in this process. Under conditions of initiation of specific oligomerization of the monomeric form of GroEL (in the presence of Mg2+ and glycerol or Mg-ATP [52, 54, 59, 61]), the times of kinetic stages are practically invariable, but the amplitude of the slowest third stage noticeably (~2-fold) increases. This is evidence of stabilization (under conditions of oligomerization) of the accessory elements of subunit secondary structure that may be responsible for intersubunit interactions.
The addition of a few factors leads to initiation of oligomerization of the folded monomeric form (subunits) of GroEL and enhancement of its efficiency. The first work on the assembly of the GroEL particle from the folded monomeric state [52] showed that specific oligomerization of the protein occurs in the presence of Mg-ATP, and its efficiency increases upon addition of GroES. As noted in the same work, specific oligomerization does not occur in the presence of Mg-ADP, while the introduction of native GroEL in addition to Mg-ATP enhances the efficiency of GroEL particle assembly from the monomeric state (self-chaperoning). The presence of Mg-adenine nucleotides is necessary for the assembly of not only GroEL but also similar chaperones from various organisms [61]. Further studies of the assembly of the GroEL particle from the denatured state yielded ambiguous results. For example, in work [83] it was noted that the tetradecameric particle of GroEL could be assembled in vitro from monomers also in the absence of Mg-ATP. The authors of works [55, 59, 84] have shown that the assembly of the GroEL particle in the presence of ammonium sulfate can also be initiated by Mg-ADP [59, 84], and even without adenine nucleotides at high concentrations of ammonium sulfate (~1 M) [55]. The presence of native GroEL is not necessary for the assembly of subunits (i.e. the phenomenon of “self-chaperoning” is unnecessary for initiation of the assembly of a GroEL particle from the monomeric state) [59, 84]. Moreover, adenine nucleotides are not at all necessary if GroEL assembly occurs in the presence of 20% glycerol and Mg2+ ions [61].
Thus, analysis of literature data leads to the conclusion that the assembly of the quaternary structure of GroEL in vitro is determined both by its ligands and by external conditions (temperature and ionic strength). We have studied this process more thoroughly to elucidate the external and structural factors that influence the assembly of the GroEL particle from the monomeric form. Our data lead to the following conclusions: first, as has already been mentioned in the literature [52-55, 59, 61, 84], the assembly of the oligomeric GroEL particle does not occur in the absence of ligands or in the presence of each ligand separately and at a moderate ionic strength of the solution. Second, specific oligomerization of the monomeric form is initiated by the addition of a certain combination of GroEL ligands, which depends on the ionic strength of solution and the ionic composition. So, at low ionic strength (~20 mM Tris-HCl), noticeable oligomerization is observed only in the presence of Mg-ATP or Mg-ADP and twofold molar excess of native GroES. At moderate ionic strength (0.2 M NaCl), the presence of even high concentrations of Mg-ADP or Mg-ATP (up to 100 mM) does not lead to oligomerization of GroEL subunits without GroES. As mentioned in previous works [55, 59, 84], ammonium sulfate has the maximum stimulating effect on GroEL oligomerization. In the presence of 0.1 M ammonium sulfate, noticeable oligomerization is observed in the presence of either Mg-ATP alone or Mg-ADP alone. In addition, Mg-ATP is more effective for the assembly of the GroEL particle than Mg-ADP. However, the additional presence of twofold molar excess of GroES (GroEL14/GroES7 = 1 : 2) also substantially increases the efficiency of oligomerization in these cases.
A third conclusion is that the effects of individual ligands can be replaced by certain external conditions (solvent composition). So, the effect of Mg2+ can be replaced by high (~2 M KCl or NaCl) ionic strength of solution. In the presence of 20% glycerol, the GroEL particle is assembled in the absence of adenine nucleotides, with only Mg2+ present in the solution [61], while 1 M ammonium sulfate initiates the assembly of full-size GroEL14 both in the absence of Mg2+ and adenine nucleotides and in the absence of GroES [55]. All the above suggests that some structural element of the subunit (supposedly an element of the secondary structure) must be stabilized to initiate the oligomerization of the GroEL particle, which occurs during the interaction of the monomeric form with Mg2+ and adenine nucleotides in the presence of 1 M ammonium sulfate or a combination of various factors.
The kinetics of GroEL oligomerization can be easily recorded by the increase in light scattering intensity, since the large oligomeric particle of GroEL more effectively scatters light compared to the monomeric form with much lower molecular weight [54, 63, 66]. We obtained the time dependences of enhancement of light scattering intensity (at wavelength 330 nm) during the oligomerization of the GroEL particle from the folded monomeric form, initiated by the appropriate concentrations of necessary factors (data not shown). It turned out that, both in case of oligomerization initiation by Mg2+ (1 mM) and glycerol (20%) and in the presence of ammonium sulfate (50 mM), ATP (0.05 mM), and Mg2+ (1 mM), the kinetics of formation of the full GroEL particle has two phases. The first phase is an order of magnitude faster than the second (final) phase. The two-phase nature of GroEL particle formation (assembly) may reflect the accumulation of an intermediate oligomeric state containing a large number of GroEL subunits. The intermediate oligomer can be observed during GroEL oligomerization on electrophoresis under nondenaturing conditions in the presence of Mg-ATP (data not shown). In this case, the observed enhanced fuzziness of the electrophoretic bands of the monomer and the intermediate oligomer may be evidence of a sufficiently rapid exchange between these states, while the electrophoretic band corresponding to the state of the full-size oligomer GroEL14 is more distinct, which is probably associated with its high stability.
The nature and structure of the intermediate oligomeric state on the pathway of GroEL particle assembly are the key moments for understanding this process. However, the available data are insufficient for characterization of the intermediate oligomer. Decomposition and formation of the oligomeric structure of GroEL in equilibrium processes of its denaturation and renaturation occur cooperatively, and there has been no noticeable accumulation of intermediate oligomeric structures [55, 83]. However, some data demonstrate the possibility of stabilization of the intermediate oligomeric state of GroEL (supposedly heptameric) in the presence of substrate protein at intermediate concentrations of urea [68]. We have also obtained indirect data indicating that such intermediate oligomer may be a heptameric ring capable of interacting with the full-size GroES. First, the linear dependences of the rates of two phases in the kinetics of GroEL oligomerization on concentration of the monomeric form, which reflect the biomolecular nature of these phases, have substantially different inclinations. The increase in the rate of the first (the faster) phase exceeds almost 7-fold the increase in the rate of the second (slower) phase, with the concentration of the monomeric form increasing from 0.2 up to 1.2 mg/ml. This fact demonstrates that molar concentrations of the molecules interacting at these kinetic stages are very different, being much higher at in first stage compared to the second one. Second, the rate of the first phase increases along with increase in the concentration of native co-chaperone GroES, while the rate of the second (slower) one is actually independent of GroES concentration. Third, at low concentrations of the monomeric form of GroEL (below 0.1 mg/ml) GroEL oligomerization occurs only in the presence of GroES, while at high concentrations (~1 mg/ml) the effect of GroES on the kinetics of GroEL oligomerization is much less (our unpublished data). All these data suggest that the formation of the full oligomeric GroEL14 particle occurs via the formation of an intermediate heptameric ring, which is unstable and is decomposed into monomers until it interacts with another heptameric ring with the formation of a tetradecameric structure. This tetradecameric structure is stable both at low protein concentrations and at low ionic strengths or in the absence of ligands. It can be supposed that stabilization of the heptameric intermediate oligomer during GroEL-particle assembly at low concentrations of the subunits or low ionic strengths occurs in the presence of Mg-ATP through the interaction with the heptameric co-chaperone GroES. This supposition allows us to understand why the GroES gene is located before the GroEL gene in the GroE operon of E. coli cells. Obviously, the full-size GroES is necessary for the effective assembly of GroEL at low concentrations of the chaperone at the initial stage of biosynthesis. Hence, it seems to be especially important that the folding and assembly of GroES occur in the absence of any accessory factors [72, 76, 78]. In addition, the rate of spontaneous self-organization of GroES is much higher than the rate of ligand-dependent self-organization of GroEL. Our kinetic experiments on renaturation of GroES from the urea-unfolded monomeric state for different protein concentrations have shown that the affinity of folding GroES to the hydrophobic probe ANS increases rather quickly (with a constant rate of ~2 s–1) independent of protein concentration. This process is supposedly intramolecular and seems to reflect the folding of GroES subunits to the oligomerization-competent conformational state. This conformational state of subunits is characterized by the high exposure of hydrophobic clusters to the solvent and seems to be not rigidly packed. At the same time, the kinetic constant of this process is in good agreement with the literature data on the rate of folding of GroES subunits [85]. The second process, manifesting itself in enhanced light scattering intensity, depends on protein concentrations and is noticeably accelerated at higher concentrations, which is evidence of its biomolecular nature. The rate of this process is much higher than the rate of GroEL oligomerization at the same molar protein concentrations. Thus, the rate of self-organization of the co-chaperone GroES is much higher than the rate of self-organization of the chaperone GroEL. Taking into consideration that the GroES gene is expressed prior to the GroEL gene, it can be supposed that the native full-size GroES is already present in the cell at the moment of GroEL oligomerization and may facilitate the assembly of the chaperone at low concentrations of the latter.
Based on the literature and our own data on the processes of denaturation and renaturation of the molecular chaperone GroEL and its co-chaperone GroES, we propose the following scheme of self-organization of the GroEL/ES complex in vitro (Fig. 2). Let us note that the fundamentals of this scheme may be true also for the assembly of this complex in vivo. According to the scheme, the unfolded monomeric forms of GroEL and GroES (in vitro) or the newly synthesized subunits of these proteins (in vivo) acquire the oligomerization-competent conformational state rather quickly. However, the rate of this process for GroES is higher by an order of magnitude than that for GroEL. In addition, GroES subunits can undergo specific oligomerization (both in vitro and in vivo) in the absence of any specific factor, and the rate of this process depends only on subunit concentrations. In the case of GroEL, the initiation of subunit oligomerization needs the presence of magnesium ions and adenine nucleotides, as well as moderate ionic strength and full-size GroES. Note that all these factors in concentrations sufficient for GroEL oligomerization may be present also in vivo.
Fig. 2. Scheme of GroEL/ES chaperone folding in vitro.
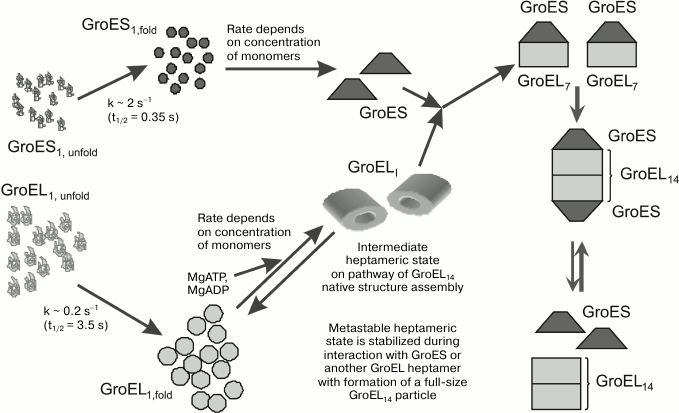
GroEL subunits are oligomerized into an intermediate ring heptamer, which seems to be unstable at low protein concentrations and physiological ionic strengths, and it is quickly decomposed to monomers until it interacts with the full-size GroES. The stable complex of the intermediate heptamer GroEL with heptameric GroES forms a two-ring GroEL structure, which is stable in the absence of both Mg-adenine nucleotides and GroES. Some aspects of this scheme (e.g. the existence of heptameric GroEL and its complex with heptameric GroES) need further experimental verification. Nevertheless, the proposed scheme (model) of the assembly of the molecular chaperone GroEL/ES does not contradict the available experimental data, and so it can be adopted for further experimental validation.
CONCLUSION
The analysis of literature data and our own experimental data on the equilibrium and kinetic processes of denaturation (unfolding) and folding (renaturation) of the molecular chaperone GroEL and its co-chaperone GroES leads to the following conclusions. First, in spite of the complex oligomeric structure, these proteins are able to acquire the native functionally active conformation from the denaturant-unfolded state in vitro. Second, the unfolding (denaturation) of both GroEL and GroES begins with dissociation of their oligomeric structure to the monomeric state, which is unfolded to a considerable extent under conditions of destruction of the quaternary protein structure. The folding (renaturation) of these oligomeric proteins begins with the folding of subunits to the conformational state competent for specific oligomerization. In the case of GroES, such state is formed spontaneously without the involvement of any accessory factors. In the case of GroEL, its ligands or certain external conditions (solvent composition) are necessary for the formation of the oligomerization-competent conformational state. We suppose that the oligomerization competence of GroEL subunits is ensured by stabilization of some secondary structure element important for intersubunit contacts, on one hand, and by suppression of electrostatic repulsion by the strong negatively charged monomeric form on the other hand. All the above is accomplished by GroEL ligands (Mg2+, adenine nucleotides, GroES) or solvent composition (ammonium sulfate, glycerol, and ionic strength). Third, it is necessary to mention the key role of the co-chaperone GroES in the assembly of the chaperone GroEL at its low concentrations or at the low or moderate ionic strengths of the solution, which may be important for understanding the mechanisms of GroEL self-organization in vivo. We suppose that this is due to the low stability (low probability of formation) under these conditions of the intermediate heptameric (one-ring) state, which is necessary for the assembly of the full-size tetradecameric GroEL particle. It seems that the intermediate heptameric state of GroEL interacts with the heptameric GroES, is stabilized, and the probability of formation of the full-size stable tetradecamer (two-ring) GroEL drastically increases. However, this needs further experimental verification. The analyzed results also indicate that GroEL ligands actively participate not only in its functioning as a molecular chaperone, but also in its self-organization.
This work was supported by the Russian Foundation for Basic Research (projects No. 09-04-00768-a and 12-04-31966-mol_a) and HHMI (55005607), by the programs “Molecular and Cell Biology” of the Presidium of the Russian Academy of Sciences and “Scientific and Scientific-Pedagogical Personnel of the Innovative Russia” of the Ministry of Education and Sciences of the Russian Federation (State contract No. 2.02.740.11.0295).
REFERENCES
1.Anfinsen, C. B. (1973) Science, 181,
223-230.
2.Seckler, R., and Jaenicke, R. (1992) FASEB
J., 6, 2545-2552.
3.Gething, M. J., and Sambrook, J. (1992)
Nature, 355, 33-45.
4.Freedman, R. B. (1992) in Protein Folding
(Creighton, T. E., ed.) WH Freeman, New York, pp. 457-541.
5.Ellis, J. (1987) Nature, 328,
378-379.
6.Lindquist, S., and Craig, E. A. (1988) Annu.
Rev. Genet., 22, 631-677.
7.Gething, M.-J. (ed.) (1997) Guidebook to
Molecular Chaperones and Protein-Folding Catalysts, Oxford
University Press, Oxford.
8.Ellis, J., and Hemmingsen, S. M. (1989) Trends
Biochem. Sci., 14, 339-342.
9.Saibil, H., and Wood, S. (1993) Curr. Opin.
Struct. Biol., 3, 207-213.
10.Braig, K., Otwinowski, Z., Hegde, R., Boisvert,
D. C., Joachimiak, A., Horwich, A. L., and Sigler, P. B. (1994)
Nature, 371, 578-586.
11.Hunt, J. F., Weaver, A. J., Landry, S. J.,
Gierasch, L., and Deisenhofer, J. (1996) Nature, 379,
37-45.
12.Xu, Z., Horwich, A. L., and Sigler, P. B. (1997)
Nature, 388, 741-750.
13.Georgopoulos, C. P., Hendrix, R. W., Casjens, S.
R., and Kaiser, A. D. (1973) J. Mol. Biol., 76,
45-60.
14.Sternberg, N. (1973) J. Mol. Biol.,
76, 25-44.
15.Herendeen, S. L., VanBogelen, R. A., and
Neidhardt, F. C. (1979) J. Bacteriol., 139, 185-194.
16.Hemmingsen, S. M., Woolford, C., van der
Vies, S. M., Tilly, K., Dennis, D. T., Georgopoulos, C. P.,
Hendrix, R. W., and Ellis, R. J. (1988) Nature, 333,
330-334.
17.Bochkareva, E. S., Lissin, N. M., and Girshovich,
A. S. (1988) Nature, 336, 254-257.
18.Horwich, A. L., Low, K. B., Fenton, W. A.,
Hirshfield, I. N., and Furtak, K. (1993) Cell, 74,
909-917.
19.Ewalt, K. L., Hendrick, J. P., Houry, W. A., and
Hartl, F. U. (1997) Cell, 90, 491-500.
20.Hartl, F. U., and Martin, J. (1995) Curr.
Opin. Struct. Biol., 5, 92-102.
21.Fayet, O., Ziegelhoffer, T., and Georgopulos, C.
(1989) J. Bacteriol., 171, 1379-1385.
22.Viitanen, P. V., Gatenby, A. A., and Lorimer, G.
H. (1992) Protein Sci., 1, 363-369.
23.Buchner, J., Schmidt, M., Fuchs, M., Jaenicke,
R., Rudolph, R., Schmid, F. X., and Kiefhaber, T. (1991)
Biochemistry, 30, 1586-1591.
24.Goloubinoff, P., Christeller, J. T., Gatenby, A.
A., and Lorimer, G. H. (1989) Nature, 342, 884-889.
25.Braig, K., Adams, P. D., and Brunger, A. T.
(1995) Nat. Struct. Biol., 2, 1083-1094.
26.Langer, T., Pfeifer, G., Martin, J., Baumeister,
W., and Hartl, F. U. (1992) EMBO J., 11, 4757-4765.
27.Braig, K., Simon, M., Furuya, F., Hainfeld, J.
F., and Horwich, A. L. (1993) Proc. Natl. Acad. Sci. USA,
90, 3978-3982.
28.Chen, S., Roseman, A. M., Hunter, A. S., Wood, S.
P., Burston, S. G., Ranson, N. A., Clarke, A. R., and Saibil, H. R.
(1994) Nature, 371, 261-264.
29.Ishii, N., Taguchi, H., Sasabe, H., and Yoshida,
M. (1994) J. Mol. Biol., 236, 691-696.
30.Fenton, W. A., Kashi, Y., Furtak, K., and
Horwich, A. L. (1994) Nature, 371, 614-619.
31.Roseman, A. M., Chen, S., White, H., Braig, K.,
and Saibil, H. R. (1996) Cell, 87, 241-251.
32.Cheng, M. Y., Hartl, F. U., Martin, J., Pollock,
R. A., Kalousek, F., Neupert, W., Hallberg, E. M., Hallberg, R. L., and
Horwich, A. L. (1989) Nature, 337, 620-625.
33.Buckle, A. M., Zahn, R., and Fersht, A. R. (1997)
Proc. Natl. Acad. Sci. USA, 94, 3571-3575.
34.Chen, L., and Sigler, P. B. (1999) Cell,
99, 757-768.
35.Houry, W. A., Frishman, D., Eckerskorn, C.,
Lottspeich, F., and Hartl, F. U. (1999) Nature, 402,
147-154.
36.Chaudhuri, T. K., Farr, G. W., Fenton, W. A.,
Rospert, S., and Horwich, A. L. (2001) Cell, 107,
235-246.
37.Katsumata, K., Okazaki, A., Tsurupa, G. P., and
Kuwajima, K. (1996) J. Mol. Biol., 264, 643-649.
38.Martin, J., Langer, T., Boteva, R., Schramel, A.,
Horwich, A. L., and Hartl, F. U. (1991) Nature, 352,
36-42.
39.Lin, Z., Schwartz, F. P., and Eisenstein, E.
(1995) J. Biol. Chem., 270, 1011-1014.
40.Hayer-Hartl, M. K., Ewbank, J. J., Creighton, T.
E., and Hartl, F. U. (1994) EMBO J., 13, 3192-3202.
41.Perrett, S., Zahn, R., Stenberg, G., and Fersht,
A. R. (1997) J. Mol. Biol., 269, 892-901.
42.Aoki, K., Taguchi, H., Shindo, Y., Yoshida, M.,
Ogasahara, K., Yutani, K., and Tanaka, N. (1997) J. Biol. Chem.,
272, 32158-32162.
43.Katsumata, K., Okazaki, A., and Kuwajima, K.
(1996) J. Mol. Biol., 258, 827-838.
44.Marchenko, N. Yu., Marchenkov, V. V., Kaysheva,
A. L., Kashparov, I. A., Kotova, N. V., Kaliman, P. A., and Semisotnov,
G. V. (2006) Biochemistry (Moscow), 71, 1357-1364.
45.Viitanen, P. V., Lubben, T. H., Reed, J.,
Goloubinoff, P., O’Keefe, D. P., and Lorimer, G. H. (1990)
Biochemistry, 29, 5665-5671.
46.Schmidt, M., Rutkat, K., Rachel, R., Pfeifer, G.,
Jaenicke, R., Viitanen, P., Lorimer, G., and Buchner, J. (1994)
Science, 265, 656-659.
47.Jackson, G. S., Staniforth, R. A., Halsall, D.
J., Atkinson, T., Holbrook, J. J., Clarke, A. R., and Burston, S. G.
(1993) Biochemistry, 32, 2554-2563.
48.Burston, S. G., Ranson, N. A., and Clarke, A. R.
(1995) J. Mol. Biol., 249, 138-152.
49.Bochkareva, E. S., Lissin, N. M., Flynn, G. C.,
Rothman, J. E., and Girshovich, A. S. (1992) J. Biol. Chem.,
267, 6796-6800.
50.Todd, M. J., Viitanen, P. V., and Lorimer, G. H.
(1994) Science, 265, 659-666.
51.Llorca, O., Carrascosa, J. L., and Valpuesta, J.
M. (1996) J. Biol. Chem., 271, 68-76.
52.Lissin, N. M., Venyaminov, S. Yu., and
Girshovich, A. S. (1990) Nature, 348, 339-342.
53.Lissin, N. M., and Hemmingsen, S. M. (1993)
FEBS Lett., 324, 41-49.
54.Surin, A. K., Kotova, N. V., Marchenkova, S. Yu.,
Marchenkov, V. V., and Semisotnov, G. V. (1999) Bioorg. Khim.,
25, 358-364.
55.Ybarra, J., and Horowitz, P. M. (1995) J.
Biol. Chem., 270, 22962-22967.
56.Creighton, T. E. (1979) J. Mol. Biol.,
129, 235-264.
57.Goldenberg, D. P., and Creighton, T. E. (1984)
Anal. Biochem., 138, 1-18.
58.Gorovits, B. M., Seale, J. W., and Horowitz, P.
M. (1995) Biochemistry, 34, 13928-13933.
59.Arai, M., Inobe, T., Maki, K., Ikura, T., Kihara,
H., Amemiya, Y., and Kuwajima, K. (2003) Protein Sci.,
12, 672-680.
60.Hiragi, Yu., Seki, Ya., Ichimura, K., and Soda,
K. (2002) J. Appl. Cryst., 35, 1-7.
61.Lissin, N. M. (1995) FEBS Lett.,
361, 55-60.
62.Mizobata, T., and Kawata, Ya. (1994) Biochim.
Biophys. Acta, 1209, 83-88.
63.Horowitz, P. M., Hua, Su., and Gibbons, D. L.
(1995) J. Biol. Chem., 270, 1535-1542.
64.Gorovits, B. M., and Horowitz, P. M. (1995) J.
Biol. Chem., 270, 28551-28556.
65.Panda, M., Ybarra, J., and Horowitz, P. M. (2001)
J. Biol. Chem., 276, 5253-6259.
66.Panda, M., Ybarra, J., and Horowitz, P. M. (2002)
Biochemistry, 41, 12843-12849.
67.Panda, M., and Horowitz, P. M. (2002)
Biochemistry, 41, 1869-1876.
68.Mendosa, J. A., Demeler, B., and Horowitz, P. M.
(1994) J. Biol. Chem., 269, 2447-2451.
69.Todd, M. J., and Lorimer, G. H. (1995) J.
Biol. Chem., 270, 5388-5394.
70.Surin, A. K., Kotova, N. V., Kashparov, I. A.,
Marchenkov, V. V., and Semisotnov, G. V. (1997) FEBS Lett.,
405, 260-262.
71.Mendoza, J. A., and Horowitz, P. M. (1994) J.
Biol. Chem., 269, 25963-25965.
72.Boudker, O., Todd, M. J., and Freire, E. (1997)
J. Mol. Biol., 272, 770-779.
73.Landry, S. J., Zellstra-Ryalls, J., Fayet, O.,
Georgopoulos, C., and Gierasch, L. M. (1993) Nature, 364,
255-258.
74.Landry, S. J., Taher, A., Georgopoulos, C., and
Van Der Vies, S. M. (1996) Proc. Natl. Acad. Sci. USA,
93, 11622-11637.
75.Zondlo, J., Fisher, K. E., Lin, Zh., Ducote, K.
R., and Eisenstein, E. (1995) Biochemistry, 34,
10334-10339.
76.Higurashi, T., Nosaka, K., Mizobata, T., Nagai,
J., and Kawata, Ya. (1999) J. Mol. Biol., 291,
703-713.
77.Guidry, J. J., Moczygemba, Ch. K., Steede, N. K.,
Landry, S. J., and Wittung-Stafshede, P. (2000) Protein Sci.,
9, 2109-2117.
78.Seale, J. W., Gorovitz, B. M., Ybarra, J., and
Horowitz, P. M. (1996) Biochemistry, 35, 4079-4083.
79.Sakane, I., Hongo, K., Mizobata, T., and Kawata,
Ya. (2009) Protein Sci., 18, 252-257.
80.Iwasa, H., Meshitsuka, Sh., Hongo, K., Mizobata,
T., and Kawata, Ya. (2011) J. Biol. Chem., 286,
21796-21805.
81.Seale, J. W., and Horowitz, P. M. (1995) J.
Biol. Chem., 270, 30268-30270.
82.Kuwajima, K., Semisotnov, G. V., Finkelstein, A.
V., Sugai, S., and Ptitsyn, O. B. (1993) FEBS Lett., 334,
265-268.
83.Mendoza, J. A., Martinez, J. L., and Horowitz, P.
M. (1995) Biochim. Biophys. Acta, 1247, 209-214.
84.Ybarra, J., and Horowitz, P. M. (1995) J.
Biol. Chem., 270, 2213-2215.
85.Bascos, N., Guidry, J., and Wittung-Stafshede, P.
(2004) Protein Sci., 13, 1317-1321.
86.Boisvert, D. C., Wang, J., Otwinowski, Z.,
Horwich, A. L., and Sigler, P. B. (1996) Nat. Struct. Biol.,
3, 170-177.