Human Leptin Triggers Proliferation of A549 Cells via Blocking Endoplasmic Reticulum Stress-Related Apoptosis
Wei Wang1#, Haicheng Yan2#, Changwu Dou2*, and Youle Su2
1Guangzhou Institute of Respiratory Diseases, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou Medical University, Guangzhou, 510120, China2Neurosurgery, Affiliated Hospital of Inner Mongolia Medical University, Huhehaote, 010051, China; fax: +86 (0471) 663-7640; E-mail: changwudou@126.com
# These authors equally contributed to this work.
* To whom correspondence should be addressed.
Received May 4, 2013; Revision received September 13, 2013
Lung cancer is a disease characterized by uncontrolled cell growth in tissues of the lung. Leptin is a pleiotropic hormone with antiapoptotic and proliferative roles involved in several systems. However, there is no known antiapoptotic mechanism of leptin in non-small cell lung cancer (NSCLC). So, we investigated the antiapoptotic mechanism of leptin in NSCLC. Proliferation, apoptosis, and the specific mechanism of leptin-transfected cells were analyzed in this study. Leptin, p-Perk, IRE1, cleaved ATF6, spliced XBP1, eIF2-α, TRAF2, CHOP, and caspase 12 proteins were detected by Western blot, and endoplasmic reticulum (ER) stress-related mRNA was detected by semi-quantitative reverse transcription PCR (RT-PCR). Leptin in A549 and transfected cells inhibited cisplatin-activated ER stress-associated mRNA transcription and activation of proteins. ER stress unfolded protein response (UPR) proteins, PERK and ATF6, were involved in leptin-triggered apoptosis. XBP1 and TRAF2 were increased significantly when treated with cisplatin in A549-siLPT and non-transfected cells. CHOP expression was blocked in A549 and transfected cells (LPT-PeP and LPT-EX cells). In conclusion, leptin can promote the proliferation of A549 cells through blocking ER stress-mediated apoptosis. This blocking is mediated by the p-Perk and ATF6 pathway through blocking activation of CHOP.
KEY WORDS: apoptosis, ER stress, cell growth, leptin, TRAF2, XPB1DOI: 10.1134/S0006297913120031
Lung cancer is a disease characterized by uncontrolled cell growth in lung tissues. If left untreated, this growth can spread beyond the lung in the process called metastasis into nearby tissue or other parts of the body. According to its histological characteristics, non-small cell lung cancer (NSCLC) is divided into adenocarcinoma, squamous cell carcinoma, and large cell lung cancer [1]. NSCLC accounts for >80% of all lung cancers [2]. Most cancers that start in the lungs, known as primary lung cancers, are carcinomas that derive from epithelial cells. Presently, novel therapies are needed to reduce the increasing incidence of pulmonary neoplasms [3]. So investigating the targets and exploring the mechanism of NSCLC is an increasingly important field for lung cancer therapy.
There are mainly three apoptotic pathways including the mitochondrial pathway, the endoplasmic reticulum (ER) pathway, and the death receptor-mediated pathway [4]. Many studies have indicated that ER stress plays a crucial role in the regulation of apoptosis [5, 6]. In the process of apoptosis, ER stress can trigger several specific apoptotic signaling pathways including ER-associated protein degradation (ERAD) and unfolded protein response (UPR) [7]. The inositol-requiring protein 1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) are involved in the reaction of UPR in ER stress [8, 9]. Activation of PERK phosphorylates eucaryotic translation initiation factor-2α (eIF-2α), which suppresses protein synthesis. Activation of the RNase activity of IRE1 initiates splicing of X-box transcription factor-1 (XBP-1) into spliced variant XBP-1 mRNA, which is subsequently translated into a potent transcription factor. A combination of ATF6 and the spliced variant of XBP-1 positively regulate a wide variety of UPR target gene expression, including ER resident chaperones. CHOP is a proapoptotic transcription factor that suppresses the transcription of Bcl-2, which can be induced by a combination of the PERK/ATF4 and ATF6 pathways [9].
Leptin is a 16-kDa protein hormone that plays a key role in regulating energy intake and expenditure including appetite and hunger, metabolism, and behavior. Leptin is also an important hormone with proliferative and antiapoptotic functions involved in several cancers such as lung cancer, gastric cancer, and breast cancer [10-12]. Leptin functions by binding to the leptin receptor in normal human lung tissue. However, the antiapoptotic effect and mechanism of leptin in lung cancer remain unknown. The study reported here attempts to explore the antiapoptotic mechanism of leptin in lung cancer (especially NSCLC).
MATERIALS AND METHODS
Cell culture and transfection. Both the lung adenocarcinoma cell line A549 and the non-tumorigenic human bronchial epithelial cell line BEAS2B were purchased from the American Type Culture Collection (ATCC). The cells were cultured in complete culture medium (RPMI 1640 containing 10% FCS, 200 IU/ml penicillin, 100 µg/ml streptomycin), with condition of 37°C and 5% CO2. The BEAS2B cells were plated into 6- or 96-well plates (Falcon, Japan) 24 h before transfection. Different amounts of plasmids were transfected into the BEAS2B monolayer cells mediated with the Lipofectamine 2000 transfection reagent (Invitrogen, USA). The A549 and BEAS2B cells were harvested by trypsin/EDTA in PBS 24 h after transfection, pelleted by brief centrifugation, and suspended in lysis buffer according to the procedures of Wang et al. [13].
Plasmid construction. The human leptin gene was amplified from the cDNA of a human adipocyte cell isolated from a patient’s subcutaneous fat with the forward primer (5′-GGATCCGCGAATTCATGGTTCCAATCCAAAAAGTCCAAGAGG-3′, with the BamHI site underlined), and the reverse primer (5′-GCGGCCGCTATGGATCCTCAGCACCCAGGGCTGAGG-3′, with the NotI site underlined). The PCR product was ligated to the pcDNA3.1(+) vector yielding recombinant plasmid pcDNA3.1-LPT, which can express the human leptin protein. PCR was conducted as follows: 94°C for 1.5 min, followed with 94°C for 30 s, 63°C for 30 s, and 72°C for 30 s, totally 30 cycles, and a final extension at 72°C for 10 min.
In the present study, the patient who provided the subcutaneous fat signed a written informed consent. This study was also approved by the Ethics Committee.
Leptin peptide treatments. Human leptin peptide (100 nmol) (Asn-Val-Ile-Gln-Ile-Ser-Asn-Asp-Leu-Glu-Asn-Leu-Arg, LPT-PeP) (ChemicalBook, USA) was introduced into BEAS2B cells 4 h before treatment with cisplatin or without such treatment. Later, the ER stress-associated proteins were detected by Western blot or XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) analysis.
siRNA interference. siRNA interference agents against leptin protein (siLPT) were purchased from Invitrogen (USA). The cells were transiently transfected with Lipofectamine 2000 transfection reagent according to the manufacturer’s protocol. A549 cells were seeded on 6-well plates for RNA or protein preparation and 96-well plates for DNA fragmentation or cell growth assays (named A549-siLPT). After 24 h incubation, the media were replaced with serum-free RPMI 1640 containing siRNA (100 nM) and the transfection reagent. The cells were harvested for assays daily for three consecutive days after transfection with the siRNA duplexes.
Trial grouping. According to the above-described experimental methods, there were five groups involved in this study including A549, A549-siLPT, LPT-PeP, LPT-EX (transfected BEAS2B group), and not-transfected BEAS2B group.
Western blotting. SDS-PAGE with 15% polyacrylamide gel was used to separate the protein lysates, and the proteins were electrotransferred onto nitrocellulose membranes. The membranes were blocked with 5% defatted milk (in PBST) overnight at 4°C, and they were incubated with specific antibodies including 1 : 2000 leptin specific monoclonal antibody (mAb) (Santa Cruz, USA), 1 : 3000 mice mAb anti-human IRE1, 1 : 2000 mouse mAb anti-human p-Perk, 1 : 1000 goat pAb anti-human CHOP, 1 : 1000 polyclonal Ab anti-human caspase 12 (Santa Cruz), 1 : 1000 mAb anti-full length and spliced XBP1 (Stressgen, USA), 1 : 1000 mAb anti-full length and cleaved ATF6 (Santa Cruz), 1 : 1000 anti-eIF2-α (Santa Cruz), and 1 : 600 mAb anti-human β-actin (Santa Cruz) for 2 h at room temperature, and then incubated with 1 : 4000 horseradish peroxidase (HRP)-conjugated anti-mouse, 1 : 1000 anti-rabbit or anti-goat IgG (Santa Cruz). The reactive signals were visualized using an ECL kit (PE Applied Biosystems, USA).
Cisplatin treatment. BEAS2B cells (1·105) or A549 cells (1·105) were grown in 6-well plates and treated with cisplatin at the final concentration of 1 µM. Data were collected from three independent experiments in triplicate.
Proliferation and apoptosis analysis. In the present study, the XTT assay was employed to measure cell proliferation using a cytotoxicity detection kit (Cayman, USA). The lactate dehydrogenase activity from each well was detected and terminated by adding 1 N HCl to dissolve the formazan product. The multi-well plates were read at 490 nm on an ELISA plate reader (Thermo, USA). Cell apoptosis was detected by flow cytometric analysis and TUNEL staining. A FACScan flow cytometer (Beckman, USA) was employed to monitor annexin V–FITC binding and propidium iodide (PI). TUNEL analysis was performed using an apoptosis detection kit (Chemicon, USA). Apoptosis and death bodies were stained brilliant blue. Every analysis was performed in at least six wells in duplicate.
Semi-quantitative RT-PCR. For the five groups, a series of semi-quantitative RT-PCR assays were performed to evaluate transcriptional situations of ER stress-associated factors including XBP1, eIF-2α, and TRAF2, for which primers were synthesized according to previous studies (table) [14-16]. In parallel, individual β-actin was selected as the internal control. With an RNAsimple Total RNA Kit (TIANGEN, China), total cellular RNA semi-quantitative RT-PCR were performed according to the procedures given elsewhere [13]. Relative transcriptional values of each factor in semi-quantitative RT-PCR are presented as a ratio of the signal value of the specific PCR product and that of the individual β-actin.
Sequences of primers for ER stress-related genes
Statistical analysis. Immunoblot images were quantitatively analyzed by computer using Image Total Tech software (Pharmacia, USA). Briefly, the image of the immunoblot was scanned with Typhoon (Pharmacia) and digitalized, then saved in TIF format. The values of each target blot were evaluated. All data are presented as mean ± SD. The data were statistically analyzed using the t-test. Probabilities of less than 0.05 were considered to be statistically significant.
RESULTS
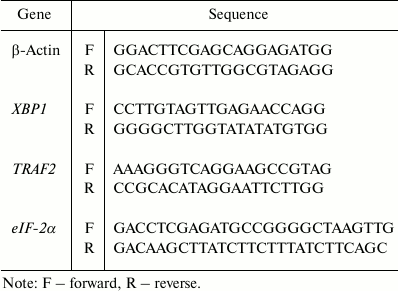
Detection of leptin expression in non-transfected BEAS2B, transfected BEAS2B, and A549 cells. In the preparations of A549 cells, high levels of leptin were intrinsically expressed 24 h after culturing. For the siRNA interfered control group (A549-siLPT), there were even no leptin bands detected (Fig. 1). The leptin-transfected cells (LPT-PeP and LPT-EX) revealed significant leptin expression. The non-transfected BEAS2B cells could not activate high levels of leptin protein 24 h after incubation. Furthermore, intrinsic expressed leptin in the A549 cells was higher than the leptin expressed in the LPT-PeP and LPT-EX group (p < 0.05).
Fig. 1. Detection of leptin (LPT) expression in non-transfected BEAS2B, transfected BEAS2B, and A549 cells with Western blot assay. a) Intrinsic or transfected leptin detected with leptin-specific monoclonal antibody. Various leptin proteins are indicated above the Western blot bands. b) Statistical analysis. The relative value of each preparation is calculated by each gray numerical value of specific product vs. that of β-actin. The averaged data of each preparation are evaluated based on three independent reactions and represented as mean ± S.D. Here and further statistical differences of the data of cisplatin-treated compared with that of untreated are illustrated as *p < 0.05, **p < 0.01 and p < 0.001, respectively.
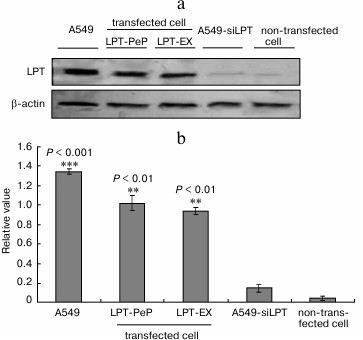
Leptin promotes proliferation of human BEAS2B cells. To show the link between leptin and proliferation, the proliferation curves for non-transfected BEAS2B cells and transfected cells (LPT-PeP and LPT-EX cells). The results indicated that the proliferation of the leptin-transfected cells was significantly higher compared with the non-transfected cells from 16 to 64 h (Fig. 2a). The cell proliferation of the leptin-transfected group remained at a high level until 64 h. The cell proliferation of the non-transfected group decreased significantly from 32 to 64 h compared with 24 h (Fig. 2a). XTT analysis results showed no difference in the proliferation viabilities among A549, LPT-PeP, and LPT-EX groups both cisplatin-treated and without cisplatin treatment. Also, there was no significant difference between A549-siLPT and non-transfected BEAS2B cells. However, the cell proliferation in the leptin-transfected BEAS2B cells was higher significantly compared with the non-transfected BEAS2B cells or the A549-siLPT cells (both p > 0.05). In the preparations of A549-siLPT and non-transfected BEAS2B group, when treated with cisplatin, the proliferation viabilities were decreased significantly (p < 0.001) 24 h after transfection (Fig. 2b) compared with the leptin-transfected BEAS2B cells.
Fig. 2. Cell proliferation of the five groups. a) Proliferation curves for non-transfected BEAS2B cells and transfected cells. b) A549, transfected cells (LPT-PeP and LPT-EX group), A549-siLPT group cells, and non-transfected cells were incubated with cisplatin, respectively. The cell proliferations were measured by XTT analysis.
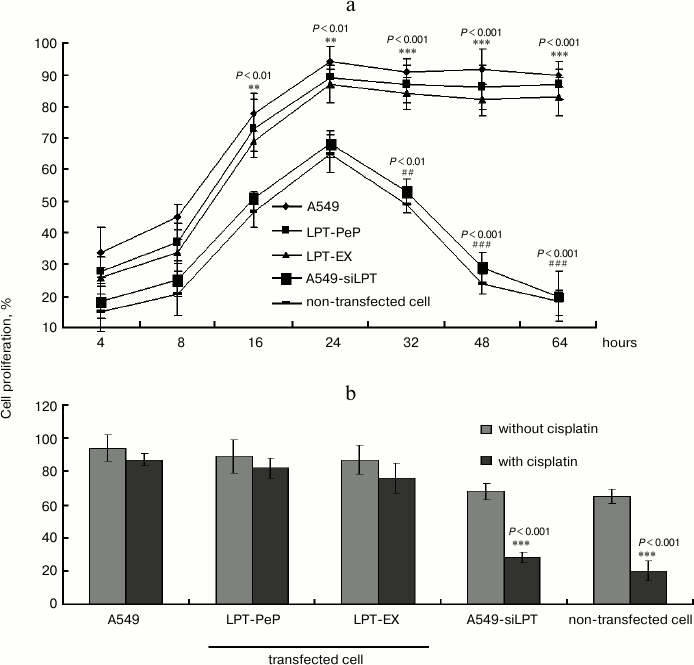
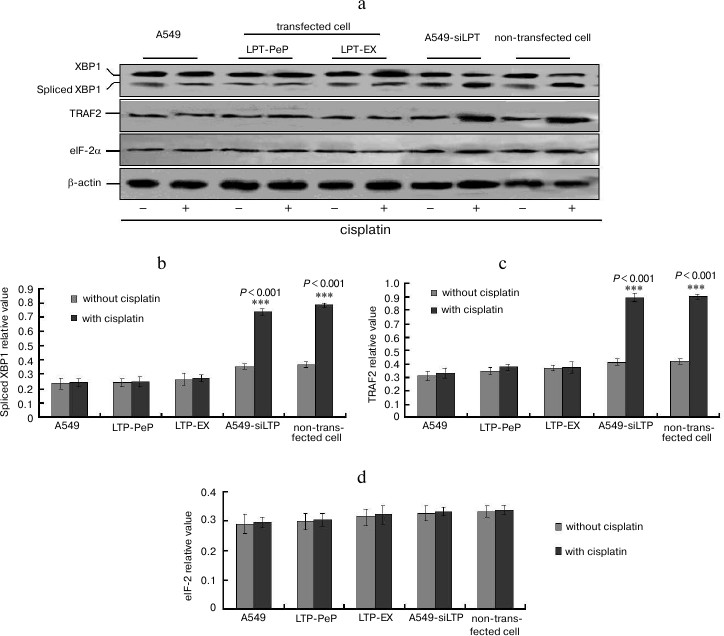
Apoptosis was inhibited in lung cells expressing leptin. TUNEL analysis indicated that cisplatin treatment could not affect cell death and apoptosis of A549 and leptin-transfected cells (LPT-PeP and LPT-EX group cells), which expressed leptin protein (Fig. 3; see color insert). The results also indicate that cisplatin can induce cell death and apoptosis in A549-siLPT cells and non-transfected BEAS2B cells, which had shortage or blockage of leptin expression. Flow cytometric analysis was employed to detect the early and late apoptosis of the five groups. For the A549 and leptin-transfected BEAS2B group (LPT-PeP, LPT-EX), there were no significant differences of early or late apoptosis between with cisplatin treatment and without treatment (p > 0.05). However, when treated with cisplatin, the early or late apoptosis of A549-siLPT cells and non-transfected BEAS2B group were significantly increased compared with no cisplatin treatment (Fig. 4) (p < 0.001).
Fig. 3. TUNEL analysis of leptin-expressing cells with or without cisplatin treatment. The bright blue staining cells represent the TUNEL positive cells.
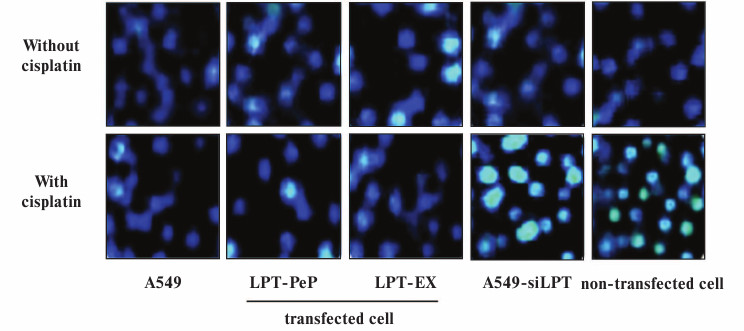
Fig. 4. Early and late apoptosis phenomena were observed in cisplatin-treated and untreated groups. a) Annexin V/PI double staining assays of cells incubated with or without cisplatin. b) Statistical analysis. The Y-axis indicates the numbers of PI stained cells. The X-axis indicates the numbers of annexin V–FITC stained cells. Results for three independent experiments are shown.
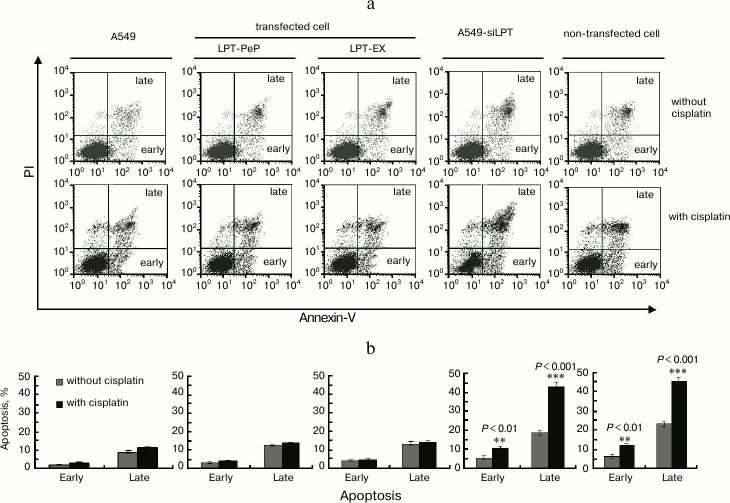
PERK and ATF6 pathway involved in leptin-caused apoptosis inhibition. The expression of leptin blocked the phosphorylation of PERK protein kinase and cleavage of ATF6 (activating transcription factor 6) protein (Fig. 5). So the amounts of p-PERK and cleaved ATF6 were not triggered by adding of cisplatin in the A549 and leptin-transfected BEAS2B group (PLT-PeP and PLT-EX group) (Fig. 5). However, in the A549-siPLT and non-transfected BEAS2B group the phosphorylation of PERK and cleavage of ATF6 were significantly increased when treated with cisplatin (Fig. 5).
Fig. 5. PERK and ATF6 pathway are involved in leptin-caused apoptosis inhibition. a) Observation of IRE1, p-PERK, and cleaved ATF6. b-d) Statistical analysis. The average gray value of each preparation was calculated by the gray numerical value of each blot vs. that of β-actin.
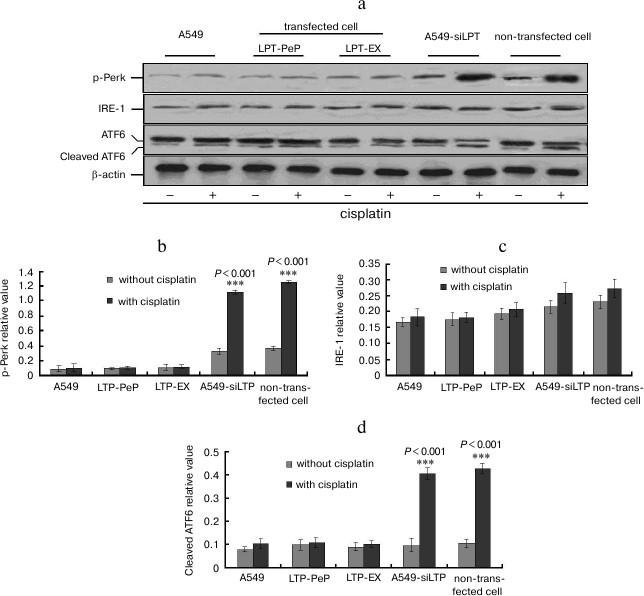
UPR downstream factor expression was highly blocked in leptin expressing cells. When treated with cisplatin, XBP1 and TRAF2 mRNA were increased significantly in A549-siLPT and non-transfected BEAS2B group compared with no cisplatin treatment (Fig. 6) (p < 0.001). Additionally, eIF-2α mRNA remained comparable among all of the groups (Fig. 6). There were no significant changes of spliced XBP1 and TRAF2 mRNA between treatment with and without cisplatin in the A549 and leptin-transfected group (LPT-PeP and LPT-EX) (Fig. 6). The above downstream ER stress-associated proteins (spliced XBP1, TRAF2, and eIF-2α proteins) were also detected. In the A549-siLPT and non-transfected BEAS2B group, spliced XBP1 and TRAF2 expression were increased significantly when treated with cisplatin (Fig. 7) (p < 0.001). But cisplatin could not affect the levels of XBP1 and TRAF2 proteins in the A549 and leptin-transfected group (LPT-PeP and LPT-EX).
Fig. 6. Analysis of mRNA levels of ER stress-associated genes (UPR). a) Analysis of spliced XBP1 mRNA. b) Analysis of TRAF2 mRNA. c) Analysis of eIF-2α mRNA. The relative value of spliced XBP1, TRAF2, and eIF-2α were calculated by the gray numerical value of each specific product vs. that of β-actin.
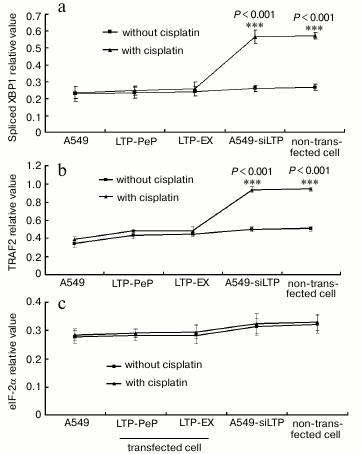
Fig. 7. a) Analysis of protein levels of the ER stress-associated genes (UPR). b-d) Statistical analysis of spliced XBP1, TRAF2, and eIF-2α protein, respectively. The relative value of spliced XBP1, TRAF2, and eIF-2α proteins were calculated by the gray numerical value of each specific product vs. that of β-actin.
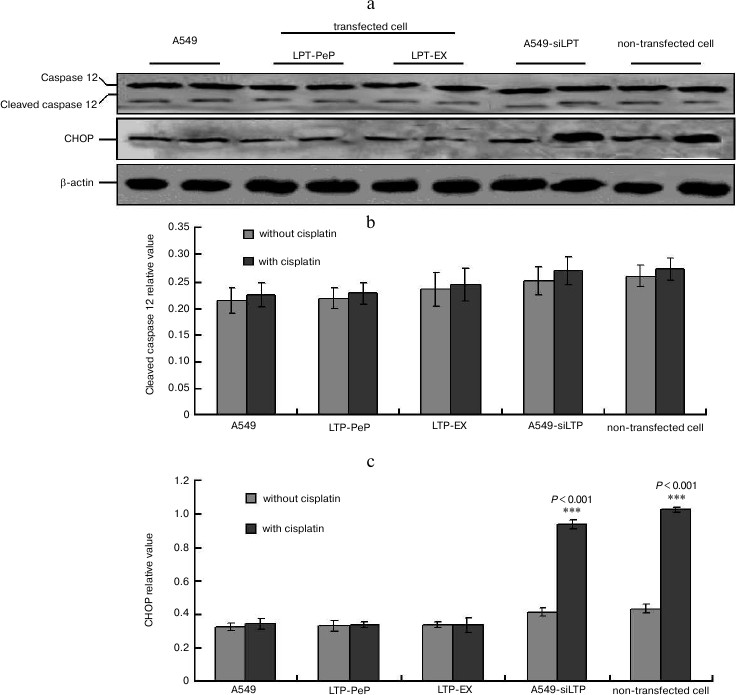
CHOP-triggered apoptosis was blocked in leptin-expressing cells. When there was no cisplatin treatment, there were no changes for caspase 12 and CHOP in every group (Fig. 8a). There were no significant differences of cleaved caspase 12 in the cisplatin-treated cells compared with no untreated cells in all of the groups (Fig. 8b). When treated with cisplatin, the levels CHOP in the preparations of A549-siLPT and non-transfected BEAS2B group were significantly higher compared with that of untreated cells (Fig. 8c). But no changes of CHOP were found either in cisplatin-treated or untreated cells of A549 and leptin-transfected group (LPT-PeP and LPT-EX) (Fig. 8c). These results indicate that CHOP in the A549-siLPT cells and non-transfected BEAS2B cells were activated, and this triggered apoptosis.
Fig. 8. CHOP-triggered apoptosis was blocked in leptin-expressing cells. a) Levels of CHOP and caspase 12 in A549, transfected cells (LPT-EX and LPT-PeP cell), A549-siLPT, and non-transfected cells were evaluated by individual Western blots. b, c) Statistical analysis for caspase 12 and CHOP, respectively. The average gray value of each preparation was calculated by the gray numerical value of each blot vs. that of β-actin.
DISCUSSION
The present study is the first to explore the antiapoptotic function of leptin protein in lung cancer. The results indicate that leptin inhibits cisplatin-triggered apoptosis of human non-small cell lung cancer cell line A549. However, BEAS2B cells could resist the cisplatin-induced apoptotic effects. Our study shows that leptin inhibits apoptosis through blocking ER stress through the PERK and ATF6 pathway, and this blocks apoptosis by blocking CHOP protein expression.
For leptin detection, the result indicated that all of the groups can express leptin protein, but the levels in the A549 cells and leptin-transfected cells were significantly higher compared with those in the A549-siLPT and non-transfected cells (Fig. 1). This result shows that malignant transformation of cancer requires the cancer cells to continue growth. Such a leptin expression pattern implies that the expression of leptin might inhibit the death and apoptosis of cancer cells.
Cell proliferation of A549, transfected cells (LPT-PeP and LPT-EX cells), A549-siLPT, and non-transfected cells was detected. We hypothesized that the higher proliferation in the A549 and leptin-transfected cells was triggered by the blocking of apoptosis. In the present study, the cells were treated with cisplatin to induce ER stress, and cell viability changes were observed. When adding the cisplatin to the cells, cell viability in leptin-transfected cells indicated no significant changes between cisplatin-treated and untreated cells. In the cisplatin-treated A549-siLPT cells and non-transfected cells, cell viabilities were obviously lower compared with that of untreated cells. This indicated that expressing of leptin inhibited cisplatin-mediated apoptosis and cell death.
To explore the specific mechanism of cell proliferation in leptin-transfected cells, ER stress-associated (UPR pathway) proteins, including p-PERK, IRE1 and cleaved ATF6 level, were detected. These results indicate that p-PERK and cleaved ATF6 proteins are activated in the A549-siLPT and non-transfected cells under treatment with cisplatin. Thus, the p-PERK and ATF6 UPR pathway may be involved in leptin blocked apoptosis. The leptin-induced inhibition of apoptosis may help to further illuminate the role of leptin in cancer progression. Moreover, the UPR pathway downstream factors, including spliced XBP1, TRAF2, and eIF-2α, were also detected. The activation of PERK phosphorylates eIF-2α. The data strongly indicate the emergence of an ER stress (or UPR pathway) after treatment with cisplatin in A549-siLPT and non-transfected cells. What is most important is that the expression of leptin inhibits the cisplatin-caused ER stress in transfected cells (LPT-PeP and LPT-EX cells) and A549 cells. In human non-small cell lung cancer cell lines, leptin may participate in the pathogenic process of lung cancer, especially for NSCLC.
ER stress-associated factors (cleaved caspase 12 and CHOP protein) were also detected to identify the apoptotic factors. According to a study of Wang et al. [17], cleaved caspase 12 can activate caspase 3 and trigger apoptosis, and CHOP can directly induce ER stress-associated apoptosis. In the present study there were no significant changes of cleaved caspase 12 found (activated) in all of the five groups (p > 0.05) when treated with cisplatin. Interestingly, when treated with cisplatin, the CHOP levels in A549-siLPT cells and non-transfected cells were significantly increased compared with no cisplatin treatment (p < 0.05), but there were no changes in the A549 and transfected cells (LPT-PeP and LPT-EX cells). So the expression or the existence of leptin in the lung cells could indirectly inhibit CHOP protein-mediated apoptosis. Actually, the induction of the transcription factor CHOP/GADD153 can kill cells by an apoptotic mechanism [18, 19]. So we obtained the novel result that leptin can block the CHOP-induced ER stress in NSCLC cell lines.
In conclusion, leptin can promote the proliferation of A549 cells through blocking ER stress-mediated apoptosis. This blocking is mediated by the p-PERK and ATF6 pathway through blocking of CHOP activation.
REFERENCES
1.Petersen, I., and Petersen, S. (2001) Anal Cell
Pathol., 22, 111-121.
2.Molina, J. R., Yang, P., Cassivi, S. D., Schild, S.
E., and Adjei, A. A. (2008) Mayo Clin. Proc., 83,
584-594.
3.Al Husaini, H., Wheatley-Price, P., Clemons, M.,
and Shepherd, F. A. (2009) J. Thorac. Oncol., 4,
251-259.
4.Duan, H. Y., Cao, J. X., Qi, J. J., Wu, G. S., Li,
S. Y., An, G. S., Jia, H. T., Cai, W. W., and Ni, J. H. (2012)
Biochemistry (Moscow), 77, 261-269.
5.Moenner, M., Pluquet, O., Bouchecareilh, M., and
Chevet, E. (2007) Cancer Res., 67,
10631-10634.
6.Feldman, D. E., Chauhan, V., and Koong, A. C.
(2005) Mol Cancer Res., 3, 597-605.
7.Li, X. Q., Zhang, L. T., Ke, X. Z., and Wang, Y. M.
(2013) Biochemistry (Moscow), 78, 102-110.
8.Ben, S. B., Wang, Q. Y., Xia, L., Xia, J. Z., Cui,
J., Wang, J., Yang, F., Bai, H., Shim, M. S., Lee, B. J., Sun, L. G.,
and Chen, C. L. (2011) Biochemistry (Moscow), 76,
1030-1036.
9.Hung, J. Y., Hsu, Y. L., Ni, W. C., Tsai, Y. M.,
Yang, C. J., and Kuo, P. L. (2010) Lung Cancer, 68,
355-365.
10.Terzidis, A., Sergentanis, T. N., and
Antonopoulos, G. (2009) Oncology, 76, 19-25.
11.Caldefie-Chezet, F., Damez, M., de Latour, M.,
Konska, G., Mishellani, F., and Fusillier, C. (2005) Biochem.
Biophys. Res. Commun., 334, 737-741.
12.Geng, Y. T., Wang, J., Wang, R., Wang, K., Xu, Y.
J., Song, G. X., Wu, C., and Yin, Y. (2012) Biomed.
Pharmacother., 66, 419-424.
13.Wang, X., Dong, C. F., Shi, Q., Shi, S., Wang, G.
R., Lei, Y. J., Xu, K., An, R., Chen, J. M., Jiang, H. Y., Tian, C.,
Gao, C., Zhao, Y. J., Han, J., and Dong, X. P. (2009) BMB Rep.,
42, 444-449.
14.Xuan, B., Qian, Z., Toriqoi, E., and Yu, D.
(2009) J. Virol., 83, 3463-3476.
15.Wang, Q., He, Z., Zhang, J., Wang, T., Tong, S.,
Wang, L., Wang, S., and Chen, Y. (2005) Cancer Detect. Prev.,
29, 544-551.
16.Cherasse, Y., Maurin, A. C., Chaveroux, C.,
Jousse, C., Carraro, V., Parry, L., Deval, C., Chambon, C.,
Fafounoux, P., and Bruhat, A. (2007) Nucleic Acids Res.,
35, 5954-5965.
17.Wang, X., Shi, Q., Xu, K., Gao, C., Chen, C., Li,
X. L., Wang, G. R., Tian, C., Han, J., and Dong, X. P. (2011) PLoS
ONE, 6, e14602.
18.Nemukhin, A. V., Shadrina, M. S., Grigorenko, B.
L., and Du, X. (2009) Biochemistry (Moscow), 74,
1044-1048.
19.Turpaev, K. T. (2002) Biochemistry
(Moscow), 67, 281-292.