Selection of Peptide Inhibitors for Double-Stranded RNA-Dependent Protein Kinase PKR
M.-J. Du1#, H.-K. Zhang2#, A.-J. He3, Y.-S. Chang1, Y. Yang1, Y. Wang1, C.-Z. Zhang1*, and Y. Cao1*
1Key Laboratory of Microbial Functional Genomics of the Ministry of Education, College of Life Sciences, Nankai University, 94 Weijin Road, Tianjin, 300071, P. R. China; fax: (8622) 2350-0808; E-mail: caoyj@nankai.edu.cn; dumjnk@126.com; changyunsong1026@126.com; yangy@mail.nankai.edu.cn; yuwang0805@126.com; czz912@yahoo.com.cn2Department of Molecular Biology, Scripps Research Institute, La Jolla, CA, 92037, USA; fax: (858) 784-2729; E-mail: hongkai@scripps.edu
3Tianjin Second Business College, Tianjin, 300193, P. R. China; E-mail: h_aj2005@126.com
# These authors contributed equally to the paper.
* To whom correspondence should be addressed.
Received April 25, 2013; Revision received June 28, 2013
Protein kinase inhibitors have been developed and applied as antitumor drugs. The majority of these inhibitors are derived from ATP analogs with limited specificity towards the kinase target. Here we present our proof-of-principle study on peptide inhibitors for kinases. Two peptides were selected by phage display against double-stranded RNA-dependent protein kinase (PKR). In vitro assay revealed that these peptides exhibit an inhibitory effect on PKR-catalyzed phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2α). The peptides also interrupt PKR activity in cells infected by viruses, as PKR activation is one of the hallmarks of host response to viral infection. Kinetic study revealed that one of the peptides, named P1, is a competitive inhibitor for PKR, while the other, named P2, exhibits a more complicated pattern of inhibition on PKR activity. Fragment-based docking of the PKR–peptide complex suggests that P1 occupies the substrate pocket of PKR and thus inhibits the binding between PKR and eIF2α, whereas P2 sits near the substrate pocket. The computational model of PKR–peptide complex agrees with their kinetic behavior. We surmise that peptide inhibitors for kinases have higher specificity than ATP analogs, and that they provide promising leads for the optimization of kinase inhibitors.
KEY WORDS: fragment-based docking, kinase inhibitors, PKR, phage display, peptidesDOI: 10.1134/S0006297913110059
Abbreviations: AD, Alzheimer’s disease; BSA, bovine serum albumin; eIF2α, alpha subunit of eukaryotic initiation factor 2; ELISA, enzyme-linked immunosorbent assay; HSV1, herpes simplex virus 1; PKR, double-stranded RNA-dependent protein kinase; PKRcat, PKR catalytic domain; PTD, permeable transduction domain; RMSD, root-mean-square deviation.
Among the kinases specific for the alpha subunit of eukaryotic
initiation factor 2 (eIF2α), double-stranded RNA-dependent
protein kinase (PKR, EC 2.7.11.1) is one of the best-characterized
kinases. Upon activation by autophosphorylation of Thr446 [1], PKR catalyzes the phosphorylation of eIF2α
on Ser51, and thus converts translation initiation factor eIF2α
into its inactive form [2]. Structurally, PKR has
two functional domains: an N-terminal double-stranded RNA
(dsRNA)-binding regulatory domain and a C-terminal kinase catalytic
domain [3]. In the catalytic domain (a.a. 251-551)
of PKR (named PKRcat), K296 acts as the binding site for ATP, and
mutation of K296 (K296R) has a dominant negative effect on the ability
of PKR to phosphorylate eIF2α [4].
PKR was originally discovered in the innate immune response, in which the expression and activation of PKR are induced by viral infection. The phosphorylation of eIF2α catalyzed by PKR blocks the initiation of protein synthesis, affecting viral replication and propagation [5]. Later, PKR was demonstrated to play an integral role in cell proliferation, cellular differentiation, and apoptosis [6-8]. For instance, PKR is reported to mediate apoptosis in neurodegenerative diseases [9-11]. The phosphorylation of eIF2α catalyzed by PKR has been observed in degenerating neurons from the brain of Alzheimer’s disease (AD) patients [12]. The primary neurons from pkr–/– knockout mice and neuroblastoma cells stably transfected with a dominant negative mutation of PKR are both less susceptible to β-amyloid peptide toxicity [3], one of the hallmarks in the pathogenesis of AD [13]. Thus, inhibiting PKR could be a therapeutic strategy in treatment of AD. Furthermore, higher expression of PKR is revealed in human cancer cells such as melanoma, colon cancer, and lung cancer cells compared to normal cells [14, 15]. The inhibition of PKR results in cancer cell death and increases chemosensitivity by mechanisms that block the activation of Akt-mediated survival [16]. The pathologic effect of PKR has sparked recent interest in developing inhibitors for this kinase.
In recent years, targeting protein kinases has become a widely used strategy in drug development. Various kinase inhibitors, such as Imatinib [17] (marketed as Gleevec by Novartis (Switzerland)), have been successfully applied in treatment of cancer. With the computational model, the ATP-binding pocket of a kinase provides a tremendous advantage for ATP analogs design, and it has presented challenges for developing highly selective kinase inhibitors. Meanwhile, with the strength of high specificity and low toxicity, peptide drugs are innovatively designed as peptide vaccines not only against viral and bacterial infection, but also against many other diseases, including AIDS, cancer, and AD [18].
Here we present the screening of peptide inhibitors for PKR using a phage display library of 12-mer peptides. Three peptides were selected, and two of them inhibit PKR activity towards eIF2α in biochemical and cellular assays. Kinetic analysis reveals that one of the selected peptides is a substrate competitive inhibitor. Molecular modeling predicts the interaction sites of PKR and the peptides to be the interface of eIF2α–PKR according to the crystal structure. The screening and remodeling of the PKR-specific peptides provide proof-of-concept modeling for peptide drug optimization, and it may provide a basis for the development of peptide inhibitors for PKR and other kinases.
MATERIALS AND METHODS
Cells and virus. HeLa cells were from the American Type Culture Collection, and they were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum (Ming Hai, GanSu, China). R3616, a mutation of herpes simplex virus 1 (HSV1) lacking the gene encoding ICP34.5, was kindly provided by Dr. Bin He (University of Illinois at Chicago) [19].
Protein production. The genes coding the PKR catalytic domain (PKRcat) and eIF2α were constructed into pET28 plasmid, respectively. A 6×His-tag was fused to the C-terminus of the two proteins. PKRcat and eIF2α were expressed in Escherichia coli (BL21) and purified with nickel-chelating resin (Qiagen, Germany) and desalted with Sephadex G25 (Amersham Pharmacia, Sweden) according to the manufacturers’ instructions. Protein concentrations were determined with the Bradford assay.
Screening for PKRcat-binding peptides by phage display. The phage display library of the 12-mer peptides was screened according to manufacturer’s instructions (New England Biolabs, UK). Briefly, the 96-well plates were first coated with purified PKRcat at 4°C overnight and blocked with bovine serum albumin (BSA). Then the phage library containing 1·1011 transducing units was incubated in wells at room temperature for 1 h, the wells were washed 10 times with TBST buffer (25 mM Tris-HCl, 150 mM NaCl, 2 mM KCl, pH 7.4, containing 0.5% (v/v) Tween 20), and bound phages were eluted with 0.1 M glycine-HCl, pH 2.2, containing 1 g/liter BSA and neutralized with 1 M Tris-HCl buffer, pH 9.0. Finally, the eluted phages were amplified by infecting E. coli strain K12 and precipitated using polyethylene glycol (Sigma, USA). Binding of the selected phage pools or individual phage clones to PKR was determined by Phage-ELISA.
Peptide synthesis. The peptides screened by phage display were named P1, P2, and P3 and synthesized by Hysbio Ltd. (China). All peptides were HPLC-purified to 95% (w/w). For intracellular experiments, the peptides were made cell permeable by coupling to a Tat permeable transduction domain (PTD) sequence (YGRKKRRQRRR) at the carboxyl termini [20]. For cellular localization analysis, the peptides were labeled with FITC at the N-termini.
In vitro biochemical assay of PKR activity. Purified eIF2α (500 nM) was incubated in kinase assay buffer (20 mM Tris-HCl, 40 mM KCl, 2 mM MgCl2, pH 7.5) with 25 nM active PKRcat and increasing amounts (0.01-50 µM) of different peptides to a final volume of 40 μl for 10 min at 30°C. The reactions were stopped with 6× loading buffer (0.3 M Tris-HCl, pH 6.8, 0.6 M dithiothreitol, 12% (w/v) SDS, 0.6% (w/v) Bromophenol Blue, 60% (w/v) glycerol) and subjected to 12% SDS-PAGE. The level of phosphorylated eIF2α (p-eIF2α) and total eIF2α were determined by Western blot with anti-phospho-Ser51-eIF2α (Biosource, USA) and anti-eIF2α (Santa Cruz Biotechnology, USA) antibodies, respectively, and the density of each band was quantitated using Quantity One software (Bio-Rad; version 4.4.0). The relative activity of PKR was represented by the ratio of p-eIF2α to total eIF2α.
The I50 of peptides in the inhibition of PKR activity was calculated by plotting the relative activity of PKR versus log(1 + log[peptide]). The curves were fit linearly with Excel (Microsoft).
To determine the kinetics of peptide inhibition of PKR activity, increasing concentrations (215-945 µM) of eIF2α were incubated with 25 nM PKRcat in the presence of 1.6 µM peptide P1 or 16 µM P2 to a final volume of 40 μl for 10 min at 30°C. The reactions were stopped with loading buffer and subjected to 12% SDS-PAGE. The relative activity of PKR was represented as the ratio of p-eIF2α to total eIF2α. The kinetics of peptide inhibition of PKR activity was analyzed according to the Lineweaver–Burk plot [21, 22].
Cell-based in vivo assay for PKR activity and activation. The peptides were first fused with an HIV Tat PTD sequence. HeLa cells were infected with R3616 at multiplicity of infection (MOI) of 5 in the absence or presence of the peptides. Peptides P1 and P2 were added at concentrations of 50, 25, 12, and 6 µM, while the concentrations of the control peptide (Pscr) were 50 and 25 µM. The cells were harvested 8 h after infection, and the phosphorylated and total forms of eIF2α and PKR were detected by Western blot with antibodies against phospho-Thr446-PKR and anti-PKR antibodies (Santa Cruz Biotechnology). PKR activity was reflected by the changes in the phospho-eIF2α level, while its activation was represented by the changes in the phospho-PKR level in the absence or presence of peptides with viral infection.
Computational modeling. The PKR coordinates were obtained from http://www.rcsb.org (PDB ID: 2A1A). Starting conformations of fragments were built and optimized with the Hyperchem program package. Gasteiger charges, atom types, and solvation parameters were assigned to the proteins, and the torsions of ligands were set actively using AutoDock Tools 1.5.2 (http://autodock.scripps.edu/).
Blind docking was performed with Autodock4 (http://autodock.scripps.edu/). Tetramer fragments of the peptide were docked to the whole PKR protein surface. The Lamarckian genetic algorithm was used with population size of 250, 100 trials, and 50 million energy evaluations per trial. The docked conformations were clustered with root-mean-square deviation (RMSD) tolerance of 0.3 nm. The top five clusters of each fragment were overlapped on the PKR surface to determine the particular region that was preferred by the ligand.
In the second round of docking, overlapped hexamer fragment residues 1-6, residues 4-9, and residues 7-12 were docked to the binding site. Two and a half million energy evaluations and 200 trials were used. Conformations of two fragments that superimposed well in overlapped residues and showed no steric clash were assembled.
Statistical analysis. The results were representative of at least three independent experiments and were presented as the mean ± standard deviation (S.D.) of triplicate experiments. Statistics were performed using the unpaired student t-test. The statistical significance was set at P < 0.05 (*) and P < 0.01 (**).
RESULTS
Selection of PKR-binding peptides by phage display. Recombinant PKRcat was expressed in bacteria and purified by affinity chromatography for phage display screening. The phage-display library of the 12-mer peptides was used to screen the PKRcat-binding peptides. After five rounds of panning, the phages specific to PKRcat were enriched by 105-fold (Table 1). More than 100 clones were tested for their association towards PKRcat by phage-ELISA, and 11 clones with high binding index were subjected to DNA sequencing. Three sequences of high occurrence were obtained, and they are listed in Table 2 with designated names peptide P1, P2, and P3. Their frequencies of occurrence in the 11 clones implied these three peptide-phage clones were equally enriched with repeated panning (Table 2, right column). To verify the binding between the phages and PKRcat, we performed phage-ELISA with the three clones. All of the three clones showed nearly 3-fold higher absorption compared to the control (library phage) (Fig. 1a), this indicating the specificity of their interaction with PKRcat. BSA was used here to eliminate the absorption of the plate or nonspecific background.
Table 1. Enrichment of PKR binding
phages*

* The enrichment of the phages was conducted for five rounds with the
initial input phage of 1·1011 pfu.
** The fold of the enrichment was calculated by the value of the
Output/Input divided by the value of the Output/Input of the previous
round.
Table 2. Selected peptide sequences
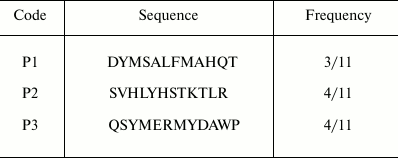
Fig. 1. Detection of binding between the synthesized peptides and PKRcat by phage-ELISA. a) Selected phage clones named P1, P2, and P3 were incubated with PKRcat, and binding was detected with phage-ELISA by measuring the absorbance at 450 nm. The amplified phage library was used as negative control. b) Competition of phage binding to PKRcat with the synthesized peptides. Increasing amounts of selected or scrambled peptides were added to PKRcat-coated plates before the addition of phage. The remaining bound phage was determined by ELISA, and the bound phage in the presence of scrambled peptides was set as 100%. The competition index is presented as percentage of bound phage (y-axis) against the concentration of peptides (x-axis). % Bound phage = (A450 in the presence of corresponding peptide/A450 in the presence of scrambled peptide) × 100%. All the data represent mean ± standard deviation (S.D.) of triplicate experiments.
We then chemically synthesized the three peptides and examined their association with PKRcat by competitive phage-ELISA. As shown in Fig. 1b, the detected amount of phage was reduced with increasing concentration of P1, P2, and P3, implying that all three peptides compete with the corresponding phage-peptide for PKRcat binding in a dose-dependent manner. Among the three peptides, P1 exhibited the most efficient binding with PKRcat (Fig. 1b).
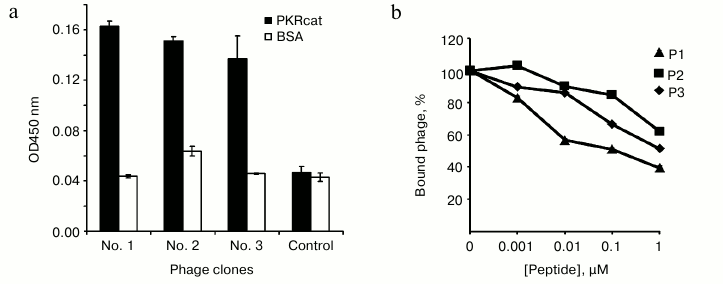
Inhibitory effect of peptides on PKR activity in vitro. We next examined whether the PKRcat-binding peptides affected the enzymatic activity of PKR, which was represented by its ability to phosphorylate eIF2α at Ser51. In vitro assays were performed to analyze the phosphorylated eIF2α catalyzed by PKRcat in the presence of peptides P1 (Fig. 2a) and P2 (Fig. 2b). Increasing concentrations (0-12.5 µM) of P1 or (0-50 µM) of P2 inhibited PKRcat activity with I50 of 0.57 μM for P1 and 1.65 μM for P2. Peptide P3 did not show any inhibition activity on PKRcat up to 50 μM concentration (data not shown). In correlation with the binding efficiency of the peptides shown above, P1 and P2 inhibited PKR activity in a concentration-dependent manner.
Fig. 2. In vitro assay for the inhibition of PKR activity. The eIF2α was phosphorylated by PKRcat in the presence of the indicated concentration of P1 (a) and P2 (b). The phosphorylation level of eIF2α was detected by Western blot with anti-phospho-eIF2α and anti-eIF2α antibodies, and the bands were quantitated using Quantity One software (Bio-Rad; version 4.4.0). PKR activity is represented by the ratio of p-eIF2α to total eIF2α, which was calculated and plotted as the relative activity (y-axis) versus peptide concentration (x-axis) in the lower panels. The result represents one of three experiments.
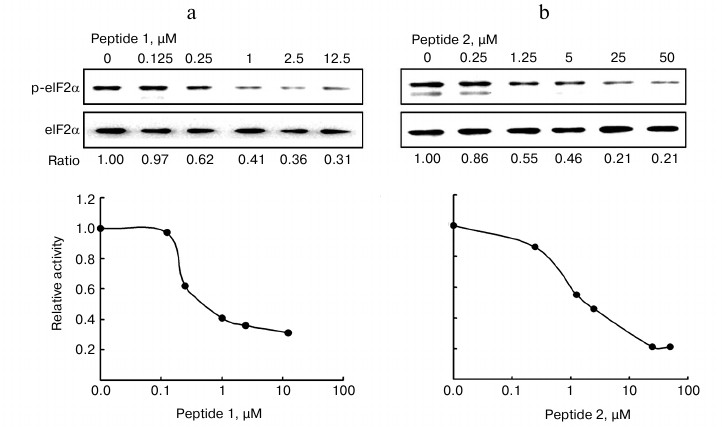
Kinetic characterization of peptide inhibition of PKR activity. We further investigated the kinetics of the two peptides in the inhibition of PKR activity. The analysis is based on the in vitro assay as described in the experimental procedures. The relative activity of PKR is expressed as the ratio of p-eIF2α to total eIF2α. The Vmax and Km were determined according to the Lineweaver–Burk plot (Fig. 3). The Km increased in the presence of P1 (triangles) in comparison with that of the control line (Fig. 3, open circles), while the Vmax remained unchanged. According to the Michaelis–Menten equation, this pattern indicated that P1 is a competitive inhibitor for PKR substrate eIF2α, implicating that P1 might occupy the binding site on PKR for eIF2α. On the other hand, in the presence of P2 an increased Km and a reduced Vmax were seen, suggesting a more complicated pattern of inhibition that is neither competitive nor noncompetitive inhibition.
Fig. 3. Kinetics of inhibition of PKR activity by the peptides. Increasing concentration (215-945 µM) of eIF2α was incubated with 25 nM active PKRcat and 1.6 µM peptide P1 or 16 µM P2. PKR activity is represented by the ratio of p-eIF2α to total eIF2α, which was calculated and plotted as the Lineweaver–Burk plot.
Peptide effects on PKR activity in cell-based in vivo assay. PKR was one of the important interferon response genes that were triggered by viral infection. We next investigated whether the two selected peptides are involved in changing the phosphorylation of eIF2α during viral infection.
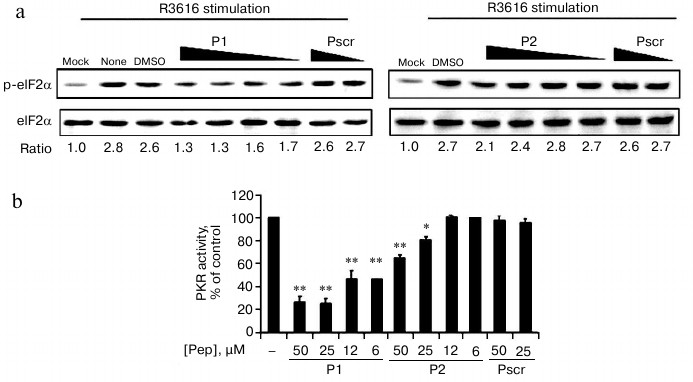
Cells were infected by R3616 strain of HSV1 in the absence or presence of the permeable fusion (FITC-labeled) peptides. The fusion peptides penetrated into cells in less than 10 min as observed by fluorescence microscopy, and cell viability was not affected at the maximum concentration of 50 μM (data not shown). Endogenous PKR activity was determined by the ratio of phospho-eIF2α to the total eIF2α resolved by Western blot. The ratio of mock-infected cells was set as 1 to indicate the basal level of phosphorylation of eIF2α in this assay (Fig. 4a, “mock”). After viral infection, the ratio of “no treatment” increased to 2.8-fold (Fig. 4a, “none”), which acted as the positive control for endogenous PKR activity. The addition of 50 μM of P1 and P2 reduced the activity to 1.3 and 2.1, respectively, demonstrating the viral-stimulated PKR activity was inhibited by the specific peptides P1 and P2, but not the peptide with scrambled sequence (Pscr). Furthermore, this inhibition effect was concentration dependent, as the relative level of the phosphorylated eIF2α was resumed as the amount of the peptides decreased (Fig. 4a). As indicated by the relative phosphorylated eIF2α, the PKR activity was reduced after treatment with P1 or P2, while the scrambled peptide had no effect (Fig. 4b). Dimethyl sulfoxide was used as a solvent control (Fig. 4a, “DMSO”). The results demonstrate that both P1 and P2 inhibit the enzymatic activity of PKR, and P1 is more efficient than P2.
Fig. 4. Cell-based in vivo assay for the inhibition of PKR activity. a) HeLa cells were infected with R3616 at multiplicity of infection (MOI) of 5 in the absence or presence of the peptides. Peptides P1 (left panel) and P2 (right panel) were added to concentrations of 50, 25, 12, and 6 µM, while the control peptide (Pscr) was at 50 and 25 µM. The cells were harvested 8 h after stimulation and analyzed by Western blot with anti-phospho-eIF2α and anti-eIF2α antibodies. The PKR activity is presented as the ratio of p-eIF2α to total eIF2α (under the gel panels), the ratio of mock infection being set as 1 to indicate the basal level of p-eIF2α. b) The full scale of PKR activity was calculated as the virus-stimulated activity in the absence of peptide (“DMSO”) minus that of “mock” infection, and it was set as 100%. The inhibition activity at each peptide concentration was calculated as percentage of control using the formula: %PKR activity of control = [(p-eIF2α/eIF2α in presence of peptide) – (p-eIF2α/eIF2α of mock-infected cells)]/[(p-eIF2α/eIF2α in absence of peptides) – (p-eIF2α/eIF2α of mock-infected cells)] × 100%. The data represent mean ± S.D. for three experiments. Statistics were performed using the unpaired student t-test. (*) and (**) indicate P < 0.05 and P < 0.01, respectively, versus the groups of DMSO treatment and Pscr treatment.
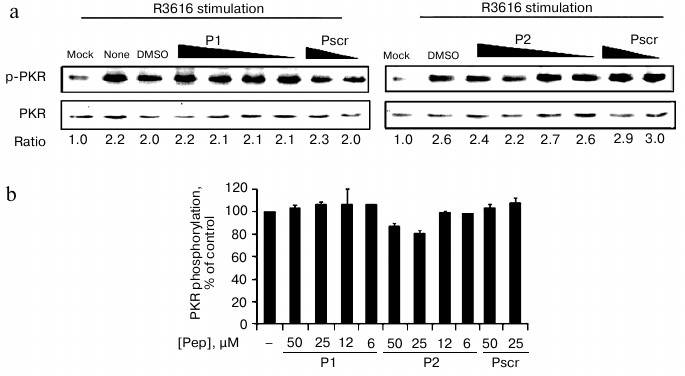
Effect of peptides on activation of PKR in cell-based in vivo assay. PKR can be activated by autophosphorylation on Thr446. We therefore tested whether the peptides inhibited the activation of PKR. As the purified recombinant PKR was always partially activated as represented by the phosphorylation at Thr446 of PKR (data not shown), we analyzed the endogenous PKR activation in mammalian cells with viral infection in the presence of the specific peptides or the control peptide (Fig. 5). PKR activation was determined by the ratio of phospho-PKR (p-PKR) to the total PKR resolved by Western blot. The ratio of the mock-infected cells without treatment was set as 1 to indicate the basal level of the p-PKR in this assay (Fig. 5a, “mock”). After infection with virus R3616, the ratio increased by 2.2-fold (Fig. 5a, “none”), which represented virally stimulated PKR activation as the positive control. As shown in Figs. 5a and 5b, PKR autophosphorylation levels were comparably elevated by viral infection even when P1 or P2 was added. These data indicate that the peptides exert their inhibitory effect solely on PKR activity towards eIF2a, rather than the activation of PKR.
Fig. 5. Effect of peptides on activation of PKR in cell-based in vivo assay. a) HeLa cells were infected with R3616 (MOI = 5) in the absence or presence of the peptides. Peptides P1 (left panel) and P2 (right panel) were added to concentrations of 50, 25, 12, and 6 µM, while the control peptide (Pscr) was at 50 and 25 µM. The cells were harvested 8 h after stimulation and analyzed by Western blot with anti-phospho-PKR and anti-PKR antibodies. The activation of PKR is presented as the ratio of p-PKR to total PKR (under gel panels), the ratio of mock infection being converted to 1 to indicate the basal level of p-PKR. b) Full scale of PKR activation calculated as the virus-stimulated activity in the absence of peptide (“DMSO”) minus that of “mock” infection, and it was set as 100%. The inhibited activation at each peptide concentration was calculated as percentage of control using the formula: %PKR phosphorylation of control = [(p-PKR/PKR in presence of peptide) – (p-PKR/PKR of mock-infected cells)]/[(p-PKR/PKR in absence of peptides) – (p-PKR/PKR of mock-infected cells)] × 100%. The data represent mean ± S.D. for three experiments.
Computational model of PKR–peptide complex. To test the different kinetics of the peptides, we used a computer-aided approach to observe the association of the peptides with PKR. Molecular docking has successfully reproduced binding models of small molecules [23-25], and we herein present a fragment-based docking to build a binding model for the peptides and PKR.
Referring to the previous work of docking fragments of heptapeptide to its antibody, most tetrapeptides and all larger peptides had considerably more favorable interaction energies when docked to the correct position than when docked to other positions [26]. This finding suggested that a tetrapeptide or larger peptide required rank of interaction energy to find the correct position with most interaction energy. To reveal the probable binding pockets for complete peptides, first the docking positions of the tetrapeptide fragments were statistically analyzed. Then the complete peptide was cleaved into longer overlapped fragments, which were independently docked to the defined protein-binding site, and fragment conformations with well-superposed residues were combined together to reproduce the complete peptide conformation. We tested the method with a set of protein–peptide complex structures and found that the predicted fragment conformations were highly accordant with their X-ray positions (data not shown).
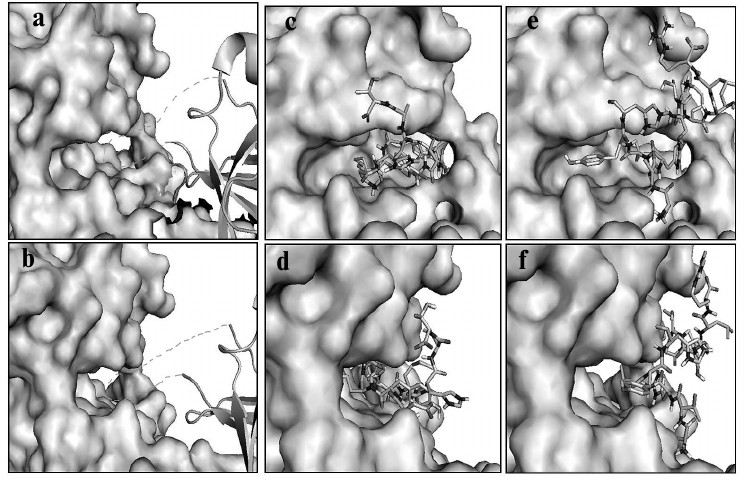
Based on the trial tests described above, we docked the selected peptides to PKR with this method as described in the experimental section. The helix insert of eIF2α (drawn as loops and sheets) made the Ser51 site fully accessible to the phosphoacceptor-binding site of PKR (shown as space-filling models) in the substrate pocket (Fig. 6, a and b) [21]. Peptide P1 fit in the same substrate pocket (Fig. 6, c and d), which inhibited the binding of eIF2α with PKR, and P2 located outside the substrate pocket and presumably affected the PKR activity by allosteric effect (Fig. 6, e and f). The predicted binding model of P1 to PKR substrate pocket rendered the kinetic results (Fig. 3), and this indicated that P1 is a competitive inhibitor. As P2 locates to the side of the pocket, both Vmax and Km could be affected. The molecular docking and predictions were well supported by the kinetic studies shown earlier (Fig. 3), and thereby the model would potentially be applied to the optimization of the PKR inhibitors.
Fig. 6. Computational model of PKR–peptide complex by molecular docking. a, b) Model for the association of eIF2α (drawn as loops and sheets) with PKR (shown as space-filling models). The Ser51 acceptor site within the helix insert of eIF2α is docked to the substrate pocket of PKR. (The helix insert of eIF2α is shown as dashed lines.) c, d) Model for peptide P1 (stick model) and PKR built by a fragment-based docking method. P1 fits in the substrate pocket of PKR. e, f) Model for peptide P2 (stick model) and PKR. P2 situates outside the substrate pocket of PKR. a, c, e) Frontal view; b, d, f) lateral view.
DISCUSSION
Recent studies show that PKR activation is implicated in Alzheimer’s disease and cancer in addition to its interferon responses. The screening and development for PKR inhibitory drugs have been drawing increasing interest. Jammi et al. discovered a small molecule inhibitor of PKR by screening a library of 26 ATP-binding-site-directed inhibitors in an in vitro phosphorylation assay [27]. The assay takes advantage of the reticulocyte lysate rich in integral PKR compounds to screen the PKR inhibitor; however, this method is not suitable for high-throughput screening with large-capacity libraries. The primary affinity based selection against the target used in our research can reduce the number of compounds to be screened. The screening with the phage display library offers several advantages: (1) the selection is based on binding; (2) linking between the binding phenotype and genotype facilitates peptide identification; and (3) it allows screening with a large capacity and diversity of libraries. In our study, we screen PKR-binding peptides from 1·109 complexity phage library and identify one potent inhibitory peptide (peptide P1) by biochemical and cellular assays. Kinetic analysis and molecular modeling elucidate that the peptide is a competitive inhibitor of eIF2α, and it explores the interaction specific to the PKR substrate pocket. All kinases possess a conserved kinase domain to bind ATP that compromises the selectivity of ATP analogs [28]. Peptide P1 exhibits specific inhibition on PKR activity compared with ATP analogs and thus endows selectivity to the peptide inhibitor for the particular enzyme.
In addition to its well-established role as a translational regulator, PKR is involved in signal transduction [1]. The first indication came from the observation that the chemical inhibition of PKR by the nucleotide analog 2-aminopurine interferes with gene induction triggered by interferon [29]. However, 2-aminopurine is not a generic inhibitor of a variety of kinases, thus the interpretation for the effect on cell signaling requires a higher level of specificity of the kinase inhibitor. Actually, PKR influences signals that involve not only eIF2α and protein synthesis, but also various factors such as STAT (signal transducers and activators of transcription), interferon regulatory factor 1 (IRF-1), p53, Jun N-terminal protein kinase (JNK), and p38 MAP (mitogen-activated protein kinase), as well as the NF-κB pathway [6, 30, 31]. Thus selective inhibitors for the catalytic activity can also serve as a tool to study PKR signaling.
The authors would like to thank Dr. Bin He (University of Illinois at Chicago, USA) for providing reagents for enzyme assays and Dr. Jianping Lin (Nankai University, China) for critical comments on the manuscript.
This work was supported by grants from the National Natural Science Foundation of China (81171556) and the Ministry of Science and Technology of China (2012CB917204) to Y.C. This work is also under the support of the Program of Introducing Talents of Discipline to Universities (B08011).
REFERENCES
1.Garcia, M. A., Gil, J., Ventoso, I., Guerra, S.,
Domingo, E., Rivas, C., and Esteban, M. (2006) Microbiol. Mol. Biol.
Rev., 70, 1032-1060.
2.Dever, T. E. (2002) Cell, 108,
545-556.
3.Chang, R. C., Suen, K. C., Ma, C. H., Elyaman, W.,
Ng, H. K., and Hugon, J. (2002) J. Neurochem., 83,
1215-1225.
4.Gil, J., Rullas, J., Garcia, M. A., Alcami, J., and
Esteban, M. (2001) Oncogene, 20, 385-394.
5.Barber, G. N. (2001) Cell Death Differ.,
8, 113-126.
6.Williams, B. R. (2001) Sci. STKE,
2001, re2.
7.Jagus, R., Joshi, B., and Barber, G. N. (1999)
Int. J. Biochem. Cell Biol., 31, 123-138.
8.Gil, J., and Esteban, M. (2000) Apoptosis,
5, 107-114.
9.Page, G., Rioux Bilan, A., Ingrand, S.,
Lafay-Chebassier, C., Pain, S., Perault Pochat, M. C., Bouras, C.,
Bayer, T., and Hugon, J. (2006) Neuroscience, 139,
1343-1354.
10.Peel, A. L. (2004) J. Neuropathol. Exp.
Neurol., 63, 97-105.
11.Suen, K. C., Yu, M. S., So, K. F., Chang, R. C.,
and Hugon, J. (2003) J. Biol. Chem., 278,
49819-49827.
12.Chang, R. C., Wong, A. K., Ng, H. K., and Hugon,
J. (2002) Neuroreport, 13, 2429-2432.
13.Ray, W. J., Ashall, F., and Goate, A. M. (1998)
Mol. Med. Today, 4, 151-157.
14.Kim, S. H., Gunnery, S., Choe, J. K., and
Mathews, M. B. (2002) Oncogene, 21, 8741-8748.
15.Roh, M. S., Kwak, J. Y., Kim, S. J., Lee, H. W.,
Kwon, H. C., Hwang, T. H., Choi, P. J., and Hong, Y. S. (2005)
Pathol. Int., 55, 688-693.
16.Pataer, A., Swisher, S. G., Roth, J. A.,
Logothetis, C. J., and Corn, P. G. (2009) Cancer Biol. Ther.,
8, 245-252.
17.Klein, S., and Levitzki, A. (2007) Adv. Cancer
Res., 97, 295-319.
18.Otvos, L., Jr. (2008) Methods Mol. Biol.,
494, 1-8.
19.Chou, J., Kern, E. R., Whitley, R. J., and
Roizman, B. (1990) Science, 250, 1262-1266.
20.Ho, A., Schwarze, S. R., Mermelstein, S. J.,
Waksman, G., and Dowdy, S. F. (2001) Cancer Res., 61,
474-477.
21.Dey, M., Cao, C., Dar, A. C., Tamura, T., Ozato,
K., Sicheri, F., and Dever, T. E. (2005) Cell, 122,
901-913.
22.Blum, G., Gazit, A., and Levitzki, A. (2003)
J. Biol. Chem., 278, 40442-40454.
23.Hetenyi, C., and van der Spoel, D. (2006) FEBS
Lett., 580, 1447-1450.
24.Rao, M. S., and Olson, A. J. (1999)
Proteins, 34, 173-183.
25.Hetenyi, C., and van der Spoel, D. (2002)
Protein Sci., 11, 1729-1737.
26.Friedman, A. R., Roberts, V. A., and Tainer, J.
A. (1994) Proteins, 20, 15-24.
27.Jammi, N. V., Whitby, L. R., and Beal, P. A.
(2003) Biochem. Biophys. Res. Commun., 308, 50-57.
28.Noble, M. E., Endicott, J. A., and Johnson, L. N.
(2004) Science, 303, 1800-1805.
29.Sekellick, M. J., Carra, S. A., Bowman, A.,
Hopkins, D. A., and Marcus, P. I. (2000) J. Interferon Cytokine
Res., 20, 963-970.
30.Verma, I. M., Stevenson, J. K., Schwarz, E. M.,
Van Antwerp, D., and Miyamoto, S. (1995) Genes Dev., 9,
2723-2735.
31.Goh, K. C., deVeer, M. J., and Williams, B. R.
(2000) EMBO J., 19, 4292-4297.