Improvement of the Efficiency of Transglycosylation Catalyzed by α-Galactosidase from Thermotoga maritima by Protein Engineering
K. S. Bobrov1#, A. S. Borisova1#, E. V. Eneyskaya1, D. R. Ivanen1, K. A. Shabalin1, A. A. Kulminskaya1, and G. N. Rychkov1,2*
1Petersburg Nuclear Physics Institute, Orlova Roscha, 188300 Gatchina, Leningrad Region, Russia; fax: (81371) 32014; E-mail: georgy-rychkov@yandex.ru; rychkov@omrb.pnpi.spb.ru2St. Petersburg State Polytechnical University, 29 Politekhnicheskaya st., 195251 St. Petersburg, Russia
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received March 26, 2013; Revision received June 24, 2013
At high concentrations of p-nitrophenyl-α-D-galactopyranoside (pNPGal) as a substrate, its hydrolysis catalyzed by α-galactosidase from Thermotoga maritima (TmGalA) is accompanied by transglycosylation resulting in production of a mixture of (α1,2)-, (α1,3)-, and (α1,6)-p-nitrophenyl (pNP)-digalactosides. Molecular modeling of the reaction stage preceding the formation of the pNP-digalactosides within the active site of the enzyme revealed amino acid residues which modification was expected to increase the efficiency of transglycosylation. Upon the site-directed mutagenesis to the predicted substitutions of the amino acid residues, genes encoding the wild type TmGalA and its mutants were expressed in E. coli, and the corresponding enzymes were isolated and tested for the presence of the transglycosylating activity in synthesis of different pNP-digalactosides. Three mutants, F328A, P402D, and G385L, were shown to markedly increase the total transglycosylation as compared to the wild type enzyme. Moreover, the F328A mutant displayed an ability to produce a regio-isomer with the (α1,2)-bond at yield 16-times higher than the wild type TmGalA.
KEY WORDS: α-D-galactosidase, transglycosylation, enzymatic synthesis of pNP-digalactosidesDOI: 10.1134/S0006297913100052
Carbohydrate structures (glycostructures), such as glycoconjugates or oligo- and polysaccharide fragments, play numerous and often not fully understood roles in cellular processes. Glycostructures are involved in inflammation, intercellular interactions and signaling, immune response, viral and bacterial infections, energy accumulation, and in various matrix processes [1-3]. Natural glycostructures are characterized by a great variety, but to study and understand their functions caused by the structural variability it is necessary to have significant amounts of the compound under study. Moreover, multiple biological activities of glycostructures determine the wide application of oligosaccharides with the desired composition in medicine and biotechnology [4-7]. Traditional chemical approaches for synthesizing glycostructures consist of many time-consuming stages including procedures of blocking and deblocking of reactive chemical groups. Such syntheses, in addition to the desired product, give ecologically harmful waste. But for chemical synthesis of various carbohydrate-containing molecules there is an alternative, namely, enzymatic synthesis using two classes of enzymes, glycosyltransferases and glycoside hydrolases. However, the high price of nucleotide-activated sugars used as substrates for the directed synthesis of oligosaccharides with glycosyltransferases makes these enzymes less attractive. In turn, glycoside hydrolases capable of transglycosylating (transferring a carbohydrate substrate (donor) residue bound to the active site onto the hydroxyl of another sugar or alcohol (acceptor)) are promising tools for producing oligosaccharides [8]. As a rule, these enzymes are stable, can be easily isolated, and their substrates are rather available. Glycoside hydrolases have already been used for synthesis of carbohydrate-containing structures for several dozens of years. Hundreds of papers concerning the synthesis of the glycosidic bond using glycoside hydrolases have been published (reviews [9-12]).
α-D-Galactosidases (EC 3.2.1.22) are glycoside hydrolases cleaving off the terminal residues of α-D-galactose from the non-reducing end of α-galactooligosaccharides [13] with retention or inversion of the anomeric configuration of the substrate C1 atom in the product (the so-called “retaining” or “inverting” glycoside hydrolases [14]). In some cases “retaining” α-D-galactosidases display a transglycosylating activity, i.e. the enzyme is able not only to hydrolyze but also to synthesize galactose-containing compounds [15] required in different fields of biotechnology, pharmaceutics, and medicine. More often, α-galactosidases have a low specificity in the +1 subsite of the active site, and that is why many “retaining” α-galactosidases can synthesize virtually the whole spectrum of galactosidic bonds [16-21].
The gene of α-D-galactosidase from Thermotoga maritima (TmGalA) used in our work was sequenced and cloned in E. coli [22]. Based on comparative analysis of the amino acid sequences, this enzyme was classified to family 36 of glycoside hydrolases (http://www.cazy.org/glycoside-hydrolases.html [23]). Due to biochemical features of TmGalA (the thermostability and transglycosylating activity in the presence of high concentrations of the acceptor), this enzyme seems promising for synthesis of α-galactose-containing structures.
The aim of this work was to establish the possibility of managing the transglycosylating activity of TmGalA. The paper presents theoretical calculations and results of experimental assay that have confirmed that the +1 subsite contains amino acid residues whose substitution can result in a significant increase in the transglycosylating activity of the α-galactosidase.
MATERIALS AND METHODS
Molecular modeling. Molecular modeling was performed using the Molsoft ICM Pro 3.6 program [24]. The known crystal structure of TmGalA (PDB code 1ZY9) was taken as an initial model for the rational design. The position of the α-D-galactopyranose (α-Galp) binding site in the catalytic center of TmGalA was determined by spatial superposition of this enzyme structure with structures of homologous α-galactosidases containing α-Galp in the active site and isolated from Oryza sativa (PDB code 1UAS; family 27 [25]) as well as from Lactobacillus acidophilus (PDB code 2XN2; family 36 [26]). Then the α-Galp orientation in the catalytic center of TmGalA was refined by flexible molecular docking (a flexible ligand and mobile side chains of the receptor amino acids) that allowed us to choose the sugar conformation in the active site with the position and orientation coinciding with α-Galp in two homologous enzymes. Then a galactosyl–enzymatic complex was created: the OH1 group of galactose was removed, and its anomeric atom C1 was artificially covalently bound to atom Oδ1 or Oδ2 of the side chain of the enzyme D327 residue acting as a nucleophile. Positions of water molecules obtained from crystal structure were taken into account only during structural optimization of the resulting intermediate (MMFF94 force field [27] was used). Mutant forms of this intermediate were modeled by replacement of target amino acids, followed by spatial optimization of the mutated side chain and its neighbors within 5 Å. The optimized models were used for flexible docking with p-nitrophenyl α-D-galactopyranoside (pNPGal) to evaluate possible mutual orientations of the intermediate (β-galactosyl–TmGalA) and the acceptor (pNPGal) located in subsites –1 and +1, respectively.
Conformations of pNPGal and galactose molecules were initially analyzed in the MM+ force field using a molecular mechanics approach. The conformations of molecules having the minimal free energy were subjected to geometry optimization in the PM3 force field using the Polak—Ribiere algorithm with the energy gradient vector accuracy of 0.01 kcal/mol. These calculations were performed using the HYPERCHEM 8.0 program. Partial charges resulting from the optimization procedure were assigned to the ligand atoms prior to the docking procedure. During the docking, the sugar ring in pNPGal retained the “chair” conformation. Three independent runs of molecular docking, starting from different initial spatial location of pNPGal in the vicinity of the enzyme active center, were applied.
Strains and plasmids. All chemicals were obtained from Sigma-Aldrich or Acros Organics unless otherwise noted. pNPGal was synthesized from D-galactose as described for a glucoside [28]. A plasmid with the wild type α-galactosidase gene was kindly donated by Prof. Robert Kelly (North Carolina State University, USA). The α-galactosidase gene (galA) was cloned from the thermophilic bacterium Thermotoga maritima MSB8 (ORF TM1192) [22]. The pET24D vector (Novagen, USA) containing the α-galactosidase gene was isolated using QIAGEN plasmid midi kit (cat. No. 12143). Site-directed mutagenesis was carried out and the sequence was confirmed by Evrogen (http://www.evrogen.com/), yielding seven plasmids with point mutations P402D, P402S, G385L, F328A, L195C, F194K, and W85Y. Native and mutant galA genes on these plasmids were expressed in E. coli BL-21(DE3) [29], and the recombinant enzymes were purified from cells of culture grown overnight as previously described [22].
Analysis of hydrolytic activity of the wild type TmGalA and its mutants. The kinetics of hydrolysis of pNPGal by the wild-type (wt) and mutant TmGalA were measured at 37°C in 50 mM sodium phosphate buffer, pH 5.0. One unit of the activity was defined as amount of the enzyme releasing 1 µmol of nitrophenol from pNPGal per minute. Reaction mixtures (total volume of 50 µl) were pre-equilibrated, and the reaction was initiated by the addition of 10 µl of enzyme solution (0.02-0.4 µg/µl in wt or mutant enzyme, depending on specific activity). After 10 min of incubation at 37°C, the reactions were stopped by addition of 10% Na2CO3 (2 ml) and the optical densities at 400 nm were measured. The constants Km and kcat were determined by non-linear least squares fitting of initial rates to the Michaelis–Menten equation by programs available in DYNAFIT [30].
Synthesis of pNP-digalactosides in the transglycosylation reaction catalyzed by the wild type TmGalA. The reaction mixture (3 ml) contained the substrate pNPGal (180 mg) in 50 mM sodium phosphate buffer (pH 7.0). The reaction was initiated by addition of 0.35 mg TmGalA and incubated at 50°C. The reaction was monitored by TLC as described in the section “Analysis of transglycosylation of TmGalA mutants”. To determine the substrate conversion, aliquots (5 µl) were taken and the released nitrophenol concentration was measured after alkalization as described above. Reaction was stopped by boiling for 10 min after reaching 80% conversion of the substrate. The transglycosylation products were separated by HPLC on a Discovery BIO Wide Pore C18 column (250 × 21.2 mm, 10 µm; Supelco, USA). The products were eluted using a linear gradient of MeCN (0-90%) in water (τ = 180 min; velution = 4 ml/min, λ = 303 nm), followed by lyophilization of the resulted fractions. NMR spectra for pNP-digalactosides were recorded on a DRX-400 NMR-spectrometer (Bruker, Germany) in D2O at ambient temperature with acetone as an internal standard (δH 2.225). 1H NMR 1D, 1H-13C HSQC and 1H-1H ROESY spectra of chromatography fractions identified as the (α1,2)-, (α1,3)-, and (α1,6)-pNP-digalactosides were in a good agreement with those previously reported [16, 31]. The percent ratio of synthesized digalactosides was calculated relatively to amount of the substrate used.
Analysis of transglycosylation of TmGalA mutants. The presence of enzymatic hydrolysis and transglycosylation products was assessed by TLC on Kieselgel 60 plates (Merck, Germany) with a solvent system of butane-1-ol–ethanol–water (at the ratio of 3 : 3 : 1 v/v) as the mobile phase. The reaction mixture (250 µl) containing 25 µmol pNPGal and the enzyme (28-178 µg) (the amount of the wild type or mutant enzyme was used in dependence of the specific activity of each specimen) in 50 mM potassium phosphate buffer (pH 7.0) was incubated at 50°C until substrate conversion to ~35-45%. The reaction was stopped by freezing in liquid nitrogen and immediate lyophilization for subsequent analysis of the products by NMR. The substrate conversion was evaluated by the amount of released nitrophenol as described above in the section “Analysis of hydrolytic activity of the wild type TmGalA and its mutants”. Concentrations of the glycosylation products were calculated by integrating areas of peaks of the characteristic non-overlapping proton signals for different components of the reaction mixture: δ 5.82 for Gal(α1,3)Gal-pNP (H1), δ 6.00 for Gal(α1,2)Gal-pNP (H1), δ 5.83 for Gal(α1,6)Gal-pNP, δ 5.26 and 4.58 for α- and β-D-galactopyranose (H1), δ 5.77 for pNPGal (sugar H1), and δ 6.91 (2H) for p-nitrophenol H2 and H6. The yield of each regio-isomer of Gal(α1,X)Gal-pNP was evaluated at the moment of its maximal production and calculated by integrating intensities I(product) according to the relationship: % Gal(α1,X)Gal-pNP = {2IGalα1,XGal-pNP/[IGal + IpNPG + 2Σij(IGalα1,XGal-pNP]}100.
Determination of pH optimum for reactions catalyzed by the wild type TmGalA and its mutants. The pH dependencies for hydrolytic reactions of wt and mutant TmGalA were measured in 50 mM sodium citrate-phosphate buffers, pH 3-8. Reaction mixtures (50 µl) containing 10 mM solution of pNPGal in buffer were pre-warmed to 37°C and the reaction was initiated by addition of 0.5-4 µg of the enzyme dissolved in buffer. After 10 min of incubation at 37°C, the reactions were stopped by alkalinization and concentration of the released nitrophenol was determined by standard method.
The pH-dependence of transglycosylation was determined using the same set of citrate-phosphate buffers as above. The reaction mixtures (250 µl) contained 100 mM pNPGal and 88 µg of TmGalA wt, 69 µg of F328A, or 41.2 µg of P402D. Reactions were incubated at 50°C until 45-50% substrate conversion, and then stopped by freezing and lyophilization. We have shown the reaction not to proceed further under these conditions (data not shown). For NMR spectroscopy analysis of the reaction mixture, each sample was dissolved in 650 µl of D2O and concentration of each regio-isomer was determined in each case.
RESULTS
Under the conditions when the initial concentration of pNPGal is above 50 mM, TmGalA(wt)-catalyzed hydrolysis of the substrate is accompanied by synthesis of different oligogalactosides. TLC analysis revealed the formation of two major and one minor products of transglycosylation, along with D-galactose and p-nitrophenol hydrolysis products. Then the pNP-digalactosides were synthesized in sufficient amounts for subsequent experiments, purified by column chromatography, and their structures were identified by NMR-spectroscopy. The following regio-isomers were prepared and identified: Gal(α1,3)Gal-pNP with the yield of 2.43% (3.5 mg); Gal(α1,6)Gal-pNP – of 1.9% (2.7 mg); Gal(α1,2)Gal-pNP – of 0.35% (0.5 mg). Because the transglycosylating ability of TmGalA is lower than this activity of other similar α-galactosidases [16, 17, 19], we attempted to modify properties of the enzyme using modern approaches of protein engineering including computer-aided design and site-directed mutagenesis. Our purpose was to identify certain amino acid residues located within the active center of TmGalA, changes in which will lead to an improvement of efficiency of transglycosylation reaction catalyzed by wild-type enzyme.
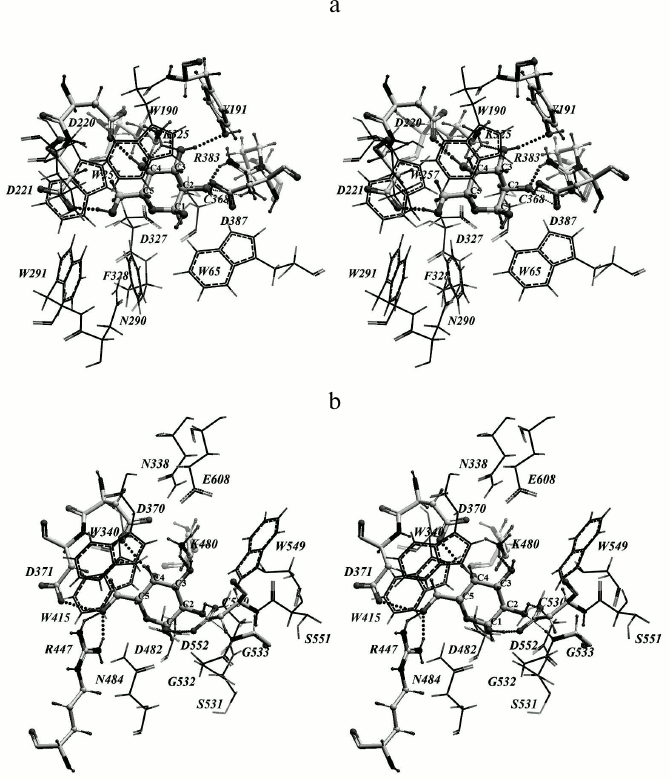
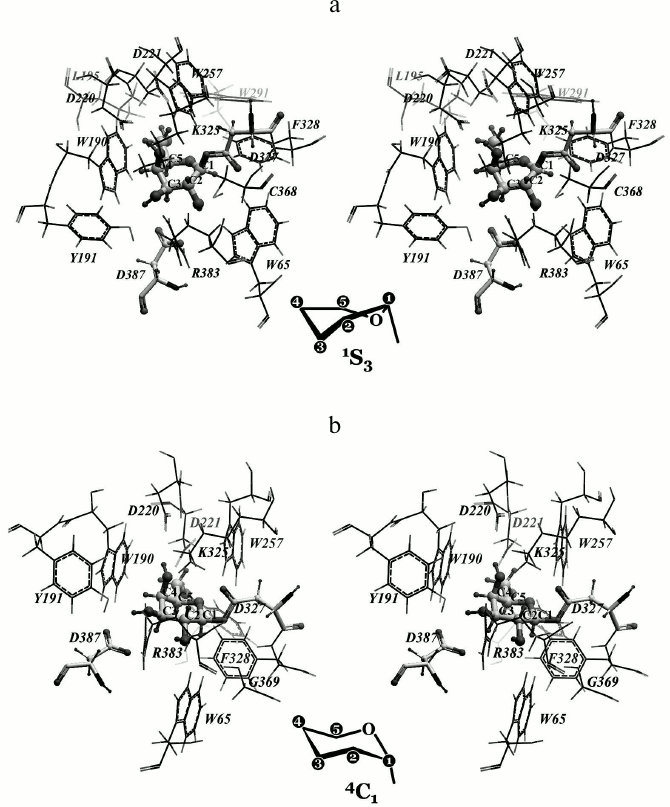
Rational designing of TmGalA mutants. We found that α-Galp is stabilized by nine possible hydrogen bonds in the active site of TmGalA (Fig. 1a). The OH2 group can form four hydrogen bonds with residues D387 and R383, OH3 forms two hydrogen bonds with residues Y191 and K325, OH4 forms two hydrogen bonds with residues K325 and D220, whereas OH6 can form a hydrogen bond with D221. The hydroxyl OH1 at the anomeric carbon atom sticks out of the active site and makes no detectable H-bonds with the enzyme. And the C1 atom is located at approximately the same distances of 3.7 and 3.6 Å from the nearest oxygen atoms of carboxyl groups of the catalytic residues D327 and D387, respectively. A similar location of α-Galp in the active site was found within the crystal structure of α-galactosidase from Lactobacillus acidophilus from the same family 36 (LaMel36, PDB code 2XN2) [26] (Fig. 1b). In the active sites of both enzymes there are six conserved residues: W190/W340, D220/D370, D221/D371, K325/K480, D327/D482, and D387/D552 (in TmGalA/LaMel36, respectively). The residues D220/D370, D221/D371, K325/K480 form the same hydrogen bonds with OH6, OH4, and OH3 of α-Galp. In addition to hydrogen bonds, the α-Galp position is stabilized by a tight hydrophobic contact of its atoms C3, C4, and C5 with the W190/W340 residue. Comparison of the active sites of two highly homologous enzymes has shown that the stable position of α-Galp within the active site of TmGalA obtained by docking is rather close to its position found experimentally within the crystal structure of LaMel36.
Fig. 1. Location of α-Galp in the active sites of α-galactosidases from two organisms: Thermotoga maritima (a) and Lactobacillus acidophilus (b). Here and further in Figs. 2 and 4, stereo images are presented. Amino acid residues forming hydrogen bonds with α-Galp (colored by atom types) and catalytic residues of aspartic acid (colored dark gray) are in backbone representation. Hydrogen bonds are presented as small spheres the size of which depends on the strength of the bond produced. In TmGalA the residues K325 and D387 can form two alternative hydrogen bonds with α-Galp.
At the first stage of rational design of the enzyme, it was necessary to establish what amino acid residues are involved in the binding of the galactosidic bond acceptor pNPGal, and therefore it was docked with the galactosyl–enzymatic complex (its creation has been described in “Materials and Methods”). Because the initial state of the galactosyl ring was “chair”, the produced covalent bond within galactosyl–enzymatic complex was improbably long. Galactosyl within the covalent intermediate of the highly homologous human α-galactosidase has the “skew (or twisted) boat” 1S3 conformation [32]. Therefore, just this conformation of galactosyl was chosen after the optimization of the galactosyl–enzymatic complex geometry for further modeling of probable variants of its attack by a pNPGal molecule.
Four galactosidic bonds (α1,2-, α1,3-, α1,4-, and α1,6-) could be potentially formed depending on which of the attacking pNPGal hydroxyls would spatially approach the anomeric carbon atom C1 of the galactosyl covalently bound with the catalytic residue D327 of the enzyme. The flexible docking procedure revealed a set of conformations with the galactose residue of pNPGal located in the +1 subsite of the enzyme. Among the conformations revealed, those were chosen in which the O2, O3, O4, and O6 atoms of pNPGal could attack the anomeric atom C1 of the galactosyl. To determine such possibility, we used two criterions: the first – the distance between the indicated oxygen atoms of pNPGal and the C1 atom of the galactosyl anomeric center should be minimal providing a tight van der Waals contact of the atoms (3.0-3.5 Å); and the other – the distance between the indicated oxygen atoms of pNPGal and one of the oxygen atoms Oδ of the catalytic residue D387 should also be minimal for providing the formation of a hydrogen bond (2.5-3.0 Å). Thus, the sum of the described distances should also be minimal. Based on these characteristic distances, the value of 6.5 Å was chosen as the threshold for their sum.
Target amino acid residues promising for increasing the transglycosylating activity of TmGalA were chosen based on two ideas: first, an enlargement of the entrance into the active site can promote the approach of the attacking oxygen atom of pNPGal (the acceptor) to the carbon atom of galactosyl (the donor) within the galactosyl–enzymatic complex; second: the substitution of amino acid residues interacting with pNP or galactose within pNPGal can influence the orientation of this acceptor towards the galactosidic bond donor that may affect the decrease in the above-mentioned interatomic distances. Among potential targets for enlargement of the entrance into the active site, W190 is a conserved residue that stabilizes the galactose position within the active site due to hydrophobic interactions. The W291 residue is a structural one: it forms an H-bond with the side chain of D221 and orients it within the active site to form an H-bond with galactose in the —1 subsite. The D387 and D327 are catalytic residues [22]. The W65 and N290 residues, in their turn, stabilize the acceptor position within the +1 subsite due to hydrophobic interactions and H-bonds, respectively. Thus, on the entrance into the active site of the enzyme only one amino acid residue, F328, remains that can be substituted. Results of flexible docking showed that the F194, L195, W85, and P402 residues can interact with pNP within pNPGal depending on its location in vicinity of the active site. Note also that W85 displayed a pronounced mobility on binding pNPGal.
Moreover, to increase the transglycosylating activity, it was reasonable to strengthen the acidic properties of the amino acid residue, which acted as an acidic–base catalyst (in TmGalA this role is played by the D387 residue). The R383 residue is conserved for active sites of some glycoside hydrolases of family 36 and acts as an “activator” of the acidic–alkaline residue D387 by lowering the value of its pKa. The location and configuration of the R383 residue is very strictly determined by chains of hydrogen bonds OD1 (D387) — NH2 (R383) — O (G385) and Nε (R383) — H2O565 — N (Gly385), which is confirmed by values of B-factors in the crystal structure of TmGalA. It could be expected that destruction of these chains closing on N and O atoms of G385 should weaken or make vacant the bonds holding the NH2 and Nε atoms of amino acid residue R383 and cause its displacement towards the hydroxyl of the D387 residue.
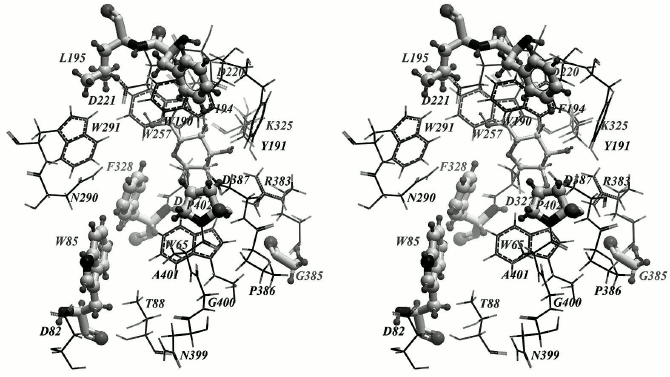
Finally, to introduce point substitutions that we believed to be capable of influencing the transglycosylating activity, we chose the following amino acid residues: F328, P402, W85, F194, L195, and G385 (Fig. 2). The F328 substitution by a smaller hydrophobic residue can enlarge the entrance into the active site and provide tighter contact between the donor and acceptor of the galactosidic bond. The P402 mutation to a polar residue can additionally stabilize pNPGal due to formation of a hydrogen bond with a galactose residue located in the +1 subsite. Substitution of the F194 and L195 residues can affect hydrophobic contacts of the nitrophenolic ring of pNPGal. Mutation of the mobile residue W85 can influence the pNPGal binding in total. And, finally, it can be expected that substitution of the small residue G385 by a large hydrophobic residue (leucine or isoleucine) can change its angle φ due to strengthening of hydrophobic interactions with the residue W65. This may lead to approaching of R383 with catalytic D387 residue and to a decrease in the pKa value of the latter.
Fig. 2. Key amino acid residues possibly capable of influencing the transglycosylating activity of TmGalA are in “ball and stick” representation; α-Galp is located within the —1 subsite of the enzyme.
Prior to experimental testing of definite amino acid substitutions, some point mutations were constructed in silico and their influence on the pNPGal binding with the galactosyl–enzymatic complex was tested by flexible docking. It was recommended to test experimentally two types of mutations: 1) those that promote the spatial approaching of the pNPGal attacking oxygen atom to the anomeric carbon C1 atom of the galactosyl covalently complexed with the enzyme; 2) those that lead to increase in the total number of active conformations (or in the number of visits of definite conformations) chosen based on the criterion of the sum of distances as described earlier. In the latter case, the conformational variety of pNPGal increases near the galactosyl–enzymatic complex, and, consequently, an increase can be expected in the probability of galactosyl transfer onto a pNPGal molecule. Based on molecular modeling results, it was recommended to study features of enzymes that included seven separate point mutations: W85Y, F194K, L195C, F328A, P402D, P402S, and also G385L.
Characteristics of TmGalA mutants. The wild type TmGalA enzyme and its mutants W85Y, F194K, L195C, F328A, P402D, P402S, and G385L were isolated using two chromatographic stages as described in [22]. At the final stage of purification, the resulting enzymes were homogenous as shown by electrophoretic data. The purified TmGalA mutants migrated on SDS-PAGE as a single band with a molecular mass of 60 kDa, identical with that of the wild-type enzyme (data not shown).
Kinetic parameters Km, kcat, and kcat/Km determined for pNPGal hydrolysis are presented in Table 1. No significant changes were observed in the kinetic parameters of hydrolysis for mutants P402S and W85Y, whereas the kcat/Km values for P402D, F328A, and L195C were more than twofold lower than for the wild type TmGalA. The mutation G385L resulted in a twofold decrease in the specific activity of the enzyme during its purification. The kinetic parameters were not determined for the mutant G385L due to its instability.
Table 1. Kinetic characteristics of wild
type TmGalA and its mutants in hydrolysis of pNPGal
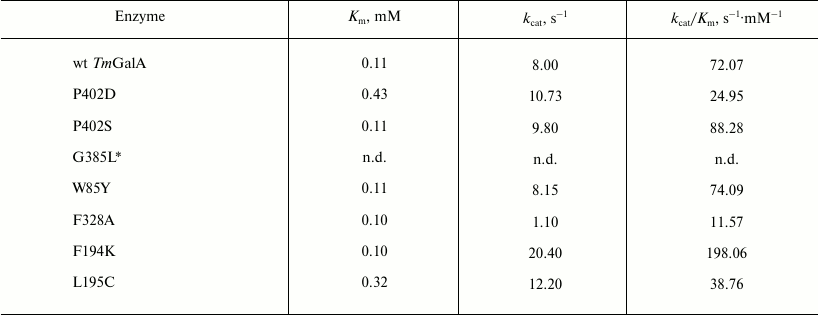
Note: The experimental error calculated as the average value for no less
than three measurements is ±3% of the presented values; n.d.,
not determined.
* Kinetic parameters of hydrolysis were not determined for G385L due to
instability of the purified enzyme.
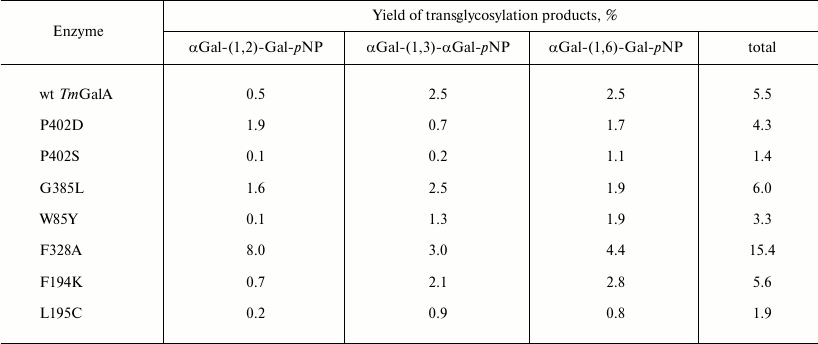
Transglycosylation reactions catalyzed by TmGalA mutants. To assess changes in the transglycosylating activity of the TmGalA, kinetic parameters of transglycosylation reactions were obtained for purified preparations of the enzymes, except for a crude preparation of mutant G385L taken for analysis after the stage of thermal treatment. The results were compared with the data obtained under the same conditions for transglycosylation catalyzed by the wild type TmGalA. The transglycosylating properties of three mutants were noticeably improved. In particular, the mutants P402D and F328A gave, respectively, 4- and 16-fold higher yields of products with the (α1,2)-bond as compared to those produced by the wild type enzyme (Table 2). This was associated with a general decrease in the yield of products with the (α1,3)-/(α1,6)-bonds relatively to the yield of products with the (α1,2)-bond. The crude preparation G385L also gave an increased yield of transglycosylation products, but it was not further considered because of its instability upon purification.
Table 2. Percentage transglycosylation of
the wild type enzyme and its mutants
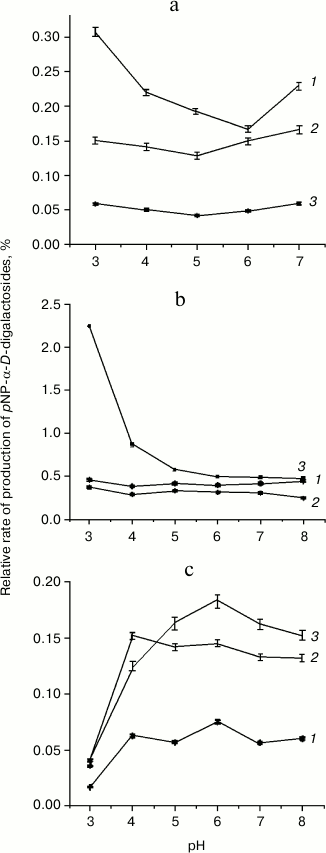
In order to find optimized reaction conditions for transglycosylation with F328A and P402D mutants, the pH variation of hydrolysis and transglycosylation was determined. The pH-optima of hydrolytic reactions did not differ significantly from those for TmGalA wt (data not shown) and were at pH 4.5-5.5. For transglycosylation, different behavior was observed for each enzyme. Yields of the (α1,2)-galactosidic linkage with the F328A mutant depended on pH (Fig. 3b), so that at pH 3.0-3.5 the rate of pNP1,2diGal formation was 4-5-fold higher than that for the other two isomers. Above pH 6.0, yields for all three isomers did not change. The behavior of the P402D variant was different (Fig. 3c). Overall yields of transglycosylation products were one order of magnitude lower than for F328A, and the pH-optimum for (1,2)-isomer formation was only just detectable, with a slight increase at pH 6.0. At pH > 6.0, the P402D mutant was less active. There was no clear dependence of Gal(α1,2)Gal-pNP and Gal(α1,6)Gal-pNP formation on pH with wt TmGalA (Fig. 3a), but Gal(α1,3)Gal-pNP was maximal at acidic pH.
Fig. 3. Dependencies of transglycosylation product formation by wt TmGalA (a), mutants F328A (b) and P402D (c) on pH conditions: 1) pNP-(α1,3)-digalactoside; 2) pNP-(α1,6)-digalactoside; 3) pNP-(α1,2)-digalactoside. Curves for the pH-dependence of regio-isomer synthesis by wt and mutant enzymes are plotted on the basis of values for transglycosylation/hydrolysis ratios.
DISCUSSION
All potential mutants were designed using molecular modeling approaches and then prepared by site-directed mutagenesis. Recently, crystal structure of α-galactosidase from Lactobacillus acidophilus (GHF 36) with α-D-galactose in the active center appeared (PDB-code 2XN2) [26] that gave a possibility to compare our data obtained by modeling with published experimental crystallographic ones. The interesting feature of galactosyl–TmGalA covalent complex formation obtained by molecular modeling is that the conformation of galactosyl depends on oxygen atom of D327 side chain to which it is covalently bonded. Upon the optimization of the galactosyl–enzymatic complex, the galactose residue bound to Oδ1 of D327 retained the “chair” 4C1 conformation. However, recently published experimental data have shown that in human α-galactosidase, highly homologous to TmGalA, galactose within the galactosyl–enzymatic complex has a “skew boat” 1S3 conformation [32]. Because the side chain of residue D327 has two equivalent oxygen atoms, we tested whether there is a possibility of formation of a covalent bond between the atom C1 of galactose and the atom Oδ2 of D327. The optimization of this structure showed that the covalent bond formation with the Oδ2 atom results in a conformational transition of galactosyl from 4C1 into 1S3 (Fig. 4).
Fig. 4. Model of the covalent galactosyl—enzymatic complex. Alternative conformations of galactosyl are presented: a) “skew boat” 1S3 where the atom C1 of galactosyl is covalently bound with atom Oδ2 of D327; b) “chair” 4C1 where the same carbon atom is bound to the Oδ1 atom of D327.
Based on results of the in silico experiment, some mutations in the enzyme were supposed to be capable of changing the transglycosylating activity of TmGalA. However, only three of seven proposed mutations really displayed a noticeable increase in the ability of TmGalA to form new galactosidic bonds. The most promising substitution F328A increased 16-fold the yield of (α1,2)-digalactoside. Such an increase seems to be caused by replacement of phenyl ring of F328 preventing an approach of OH2 of galactose to the anomeric center C1 of the galactosyl within the covalent complex. The substitution of this amino acid residue by a smaller one enlarges the entrance into the enzyme active site near to the anomeric carbon of the stable intermediate. This mutant corresponds to the above-described requirements: the minimal sum of two distances sought for is 5.64 Å (the distance from oxygen in the hydroxyl group of pNPGal to the anomeric carbon atom C1 in the intermediate galactosyl is 3.03 Å, i.e. the closest contact is provided between the attacking hydroxyl and the anomeric carbon; the distance to Oδ1 of D327 is 2.61 Å) (Fig. 5, c and d). The number of detected “active” conformations (with the sum of the indicated distances not higher than 6.5 Å) is 14. In the case of the wt TmGalA galactosyl–enzymatic intermediate the minimal sum of the same distances is 5.88 Å (3.20 Å + 2.68 Å) and the total number of “active” conformations is 10 (Fig. 5, a and b). Thus, in the F328A mutant oxygen atoms O2 and O6 of the attacking hydroxyl group of the acceptor (pNPGal) not only can have the closer contact with the anomeric carbon atom C1 of the donor galactosyl (the distance less by 0.11 and 0.17 Å, respectively), but the number of “active” conformations in the mutant increases nearly 1.5-fold compared to the data for the wild type enzyme. Moreover, experimental data for the mutant F328A revealed an increase in transglycosylating activity at acidic pH values. Possibly, protonation of both catalytic amino acid residues within the active site results in a decrease in the hydrolytic activity of the enzyme and in an increase in the yield of the αGal-(1,2)-Gal-pNP product.
Fig. 5. Results of pNPGal docking in the +1 subsite of the wild type galactosyl–enzymatic complex (a, b) and of its two mutants: F328A (c, d) and P402D (e, f). pNPGal (in the +1 subsite), galactosyl (in the —1 subsite), catalytic residues D327 and D387, and also mutant residues A328 and D402 are presented as backbone models. Dotted lines are shown distances from the atom C1 of the galactosyl anomeric center and from the oxygen atom of the D387 side chain to the oxygen atom of the attacking hydroxyl of pNPGal: OH6 (a, c, e) and OH2 (b, d, f). In the block (e) the hydrogen bond is presented formed by OH2 of pNPGal and the oxygen atom of the mutant residue D402 (dotted line).
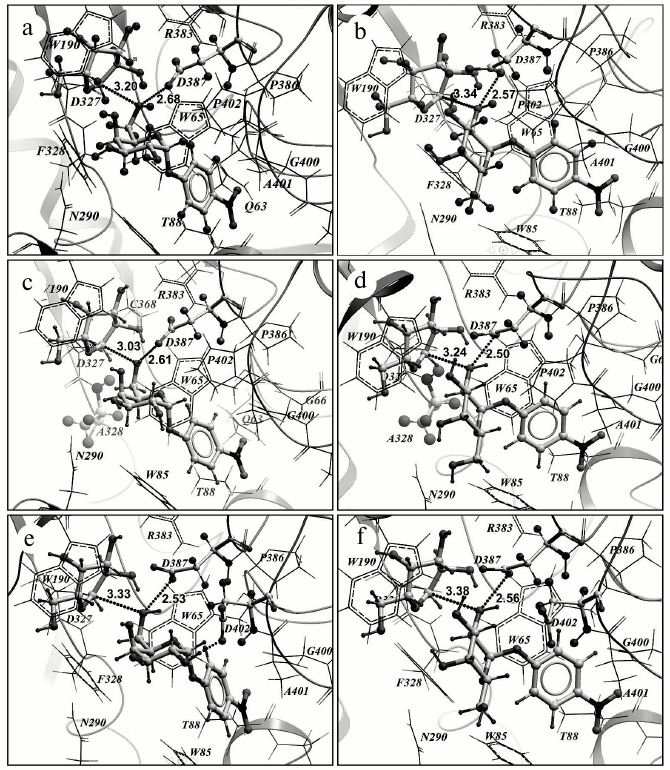
In the case of the P402D substitution, the minimal sum of distances is virtually unchanged (5.86 Å) compared to the wild type (Fig. 5, e and f), but the number of “active” conformations significantly increases (up to 19). Interactions of the OH2 and OH3 groups of pNPGal with D402 carboxylate stabilize the acceptor positions favorable for production of the (α1,6)-galactosidic bond, whereas similar interactions of OH6 with the carboxylate prevent a close contact of the OH2 and OH3 groups with the anomeric atom C1 of galactosyl. It should be noted that for the mutants F328A and P402D the frequency of visits favorable for formation of the (α1,2)- and (α1,6)-galactosidic bonds increases, and that for F328A additionally appears a possibility of formation of the (α1,4)-galactosidic bond; however, the minimal sum of distances in this case is somewhat higher at 6.05 Å.
It was hoped to weaken the linkages between the main-chain atoms of residue G385 and R383 NH2/Nε and thereby cause the guanidine group to bind more tightly to D387. This idea was derived from the approach demonstrated for α-L-fucosidase from Thermotoga maritima, which was transformed into a transglycosidase by directed evolution [33]. There, several amino acids located in the second amino acid shell of the enzyme active site influenced transglycosylating abilities for Th. maritima α-L-fucosidase by reorientation of the amino acids that are in a direct contact with substrates. We superimposed both structures (data not shown) and found an amino acid that potentially may cause the same effect in the case of TmGalA. However, the G385L mutation did not lead to significant results. Total transglycosylation output was only slightly changed. The G385L substitution did indeed lead to a 4-fold increase of the specific activity in cell lysate, but also to overall instability of the purified enzyme. Possibly, such a mutation causes multiple configuration changes within the protein structure that requires more precise methods of modeling including long-duration molecular dynamics simulations or separate structural research.
α-Galactosidases are known to be potentially useful in industrial and biomedical applications. The recent excellent review of Weignerova et al. [15] covers their application to various fields and ways to modify them. Several attempts have been made to change the regioselectivity of α-galactosidases to produce α-galacto-conjugates with preferably one type of linkage and good yields. Among the approaches that different investigators have been used is site-directed mutagenesis of α-galactosidase from Bifidobacterium adolescentis, which resulted in 16% increase of transglycosylation activity and the production of an array of (1,6)-linked di- and trisaccharides [19].
The main result of the present work is the detection of TmGalA mutations that result in the predicted increase in the transglycosylating efficiency of α-galactosidase. Note that rather high yields of transglycosylation products have been achieved for the (α1,2)-bond, which was synthesized in minor quantities by the wild type enzyme. The enzyme with the F328A mutation manifested a pronounced shift in the regioselectivity of the transglycosylation, and this confirms the possibility of managing features of the enzyme based on theoretical calculations and molecular modeling methods. The data of the present work on amino acid residues involved in the transglycosylation of TmGalA together with the literature data on the synthase activity of this enzyme [34] allows researchers to create effective tools for directed synthesis of α-galactose-containing compounds with high yields.
The authors are deeply grateful to Prof. Michael Sinnott and to senior researcher of the St. Petersburg Nuclear Physics Institute A. M. Golubev for their valuable remarks and analysis during discussion of the findings.
This work was supported by the Russian Foundation for Basic Research (project No. 12-08-00813-a), by the Committee on Science and Higher Schools of the St. Petersburg Government, by Government of the Leningrad Region, and by the Kurchatov Research Center.
REFERENCES
1.Varki, A. (2006) Cell, 126,
841-845.
2.Hricovini, M. (2004) Curr. Med. Chem.,
11, 2565-2583.
3.Mahal, L. K. (2008) Anticancer Agents Med.
Chem., 8, 37-51.
4.Shukla, R. K., and Tiwari, A. (2011) Crit. Rev.
Ther. Drug Carrier Syst., 28, 255-292.
5.Gabius, H. J., Andre, S., Jimenez-Barbero, J.,
Romero, A., and Solis, D. (2011) Trends Biochem. Sci.,
36, 298-313.
6.Ghazarian, H., Idoni, B., and Oppenheimer, S. B.
(2011) Acta Histochem., 113, 236-247.
7.Oppenheimer, S. B., Alvarez, M., and Nnoli, J.
(2008) Acta Histochem., 110, 6-13.
8.Wang, L. X. (2008) Carbohydr. Res.,
343, 1509-1522.
9.Shaikh, F. A., and Withers, S. G. (2008)
Biochem. Cell Biol.-Biochim., 86,
169-177.
10.Murata, T., and Usui, T. (1997) Biosci.
Biotechnol. Biochem., 61, 1059-1066.
11.Wang, L. X., and Huang, W. (2009) Curr.
Opin. Chem. Biol., 13, 592-600.
12.Teze, D., Dion, M., Daligault,
F., Tran, V., Andre-Miral, C., and Tellier, C. (2013)
Bioorg. Med. Chem. Lett., 23, 448-451.
13.Meier, H., and Reid, J. S. G. (1982) in
Encyclopedia of Plant Physiology, Vol. 13A (Loewus, F. A., and
Tanner, W., eds.) Springer Verlag, Berlin, pp. 418-471.
14.Davies, G., and Henrissat, B. (1995)
Structure, 3, 853-859.
15.Weignerova, L., Simerska, P., and Kren, V. (2009)
Biocatal. Biotrans., 27, 79-89.
16.Weignerova, L., Hunkova, Z., Kuzma, M., and Kren,
V. (2001) J. Mol. Catal. B: Enzym., 11, 219-224.
17.Nakai, H., Baumann, M. J., Petersen, B. O.,
Westphal, Y., Hachem, M. A., Dilokpimol, A., Duus, J. O., Schols, H.
A., and Svensson, B. (2010) FEBS J., 277, 3538-3551.
18.Zhao, H., Lu, L., Xiao, M., Wang, Q., Lu, Y.,
Liu, C., Wang, P., Kumagai, H., and Yamamoto, K. (2008) FEMS
Microbiol. Lett., 285, 278-283.
19.Hinz, S. W., Doeswijk-Voragen, C. H., Schipperus,
R., van den Broek, L. A., Vincken, J. P., and Voragen, A. G. (2006)
Biotechnol. Bioeng., 93, 122-131.
20.Simerska, P., Kuzma, M., Pisvejcova, A.,
Weignerova, L., Mackova, M., Riva, S., and Kren, V. (2003) Folia
Microbiol. (Praha), 48, 329-337.
21.Ashida, H., Yamamoto, K., and Kumagai, H. (2001)
Carbohydr. Res., 330, 487-493.
22.Comfort, D. A., Bobrov, K. S., Ivanen, D. R.,
Shabalin, K. A., Harris, J. M., Kulminskaya, A. A., Brumer, H., and
Kelly, R. M. (2007) Biochemistry, 46, 3319-3330.
23.Henrissat, B., and Davies, G. J. (1997) Curr.
Opin. Struct. Biol., 7, 637-644.
24.Abagyan, R. A., Totrov, M. M., and Kuznetsov, D.
A. (1994) J. Comp. Chem., 15, 488-506.
25.Fujimoto, Z., Kaneko, S., Momma, M., Kobayashi,
H., and Mizuno, H. (2003) J. Biol. Chem., 278,
20313-20318.
26.Fredslund, F., Hachem, M. A., Larsen, R. J.,
Sorensen, P. G., Coutinho, P. M., Lo Leggio, L., and Svensson, B.
(2011) J. Mol. Biol., 412, 466-480.
27.Halgren, T. A. (1996) J. Comp. Chem.,
17, 490-519.
28.Montgomery, E. M., Richtmyer, N. K., and Hudson,
C. S. (1942) J. Am. Chem. Soc., 64, 690-694.
29.Sambrook, J., and Russell, D. W. (2001)
Molecular Cloning. A Laboratory Manual, 3rd Edn., Cold Spring
Harbor Laboratory Press, N. Y.
30.Kuzmic, P. (1996) Anal.
Biochem., 237, 260-273.
31.Dion, M., Osanjo, G., Andre, C., Spangenberg, P.,
Rabiller, C., and Tellier, C. (2001) Glycoconj. J., 18,
457-464.
32.Guce, A. I., Clark, N. E., Salgado, E. N.,
Ivanen, D. R., Kulminskaya, A. A., Brumer III, H., and
Garman, S. C. (2010) J. Biol. Chem., 6, 3625-3632.
33.Osanjo, G., Dion, M., Drone, J., Solleux, C.,
Tran, V., Rabiller, C., and Tellier, C. (2007) Biochemistry,
46, 1022-1033.
34.Cobucci-Ponzano, B., Zorzetti, C., Strazzulli,
A., Carillo, S., Bedini, E., Corsaro, M. M., Comfort, D. A., Kelly, R.
M., Rossi, M., and Moracci, M. (2011) Glycobiology,
21, 448-456.