Modulation of Enzymatic Activity of Dengue Virus Nonstructural Protein NS3 Nucleoside Triphosphatase/Helicase by Poly(U)
M. Junaid1,2, C. Angsuthanasombat1, J. E. S. Wikberg3, N. Ali4, and G. Katzenmeier1*
1Laboratory of Molecular and Cellular Microbiology, Institute of Molecular Biosciences, Mahidol University, Salaya, 73170 Thailand; E-mail: katzenmeier.ger@mahidol.ac.th2Department of Pharmacy, University of Malakand, K. P. K, 18550 Pakistan; E-mail: juniphdr@gmail.com
3Department of Pharmaceutical Biosciences, Division of Pharmacology, Uppsala University, Uppsala, 75124 Sweden
4Department of Pharmacology, Institute of Basic Medical Sciences, Khyber Medical University, Peshawar, Khyber Pakhtunkhwa, 25000 Pakistan
* To whom correspondence should be addressed.
Received March 22, 2013; Revision received April 26, 2013
The nonstructural protein 3 (NS3) appears to be the most promising target for anti-flavivirus therapy because of its multiple enzymatic activities that are indispensable for virus replication. NS3 of dengue virus type 2 (DEN2) is composed of two domains, a serine protease in the N-terminal domain (NS3pro) and RNA-stimulated nucleoside triphosphatase (NTPase)/RNA helicase at the C-terminus (NS3h). NS3 plays an important role in viral replication and the coordinated regulation of all the catalytic activities in the full-length NS3 protein. In this study, a plasmid harboring the NS3 helicase domain (NS3h) was constructed by PCR. The 56.5 kDa NS3h protein was purified by metal-chelate affinity chromatography followed by renaturation, mediated by artificial chaperone-assisted refolding, which yielded the active helicase. NTPase activity was assayed with Malachite Green. The NTPase activity in the presence of poly(U) showed a higher turnover number (kcat) and a lower Km value than without poly(U). The activity increased approximately fourfold in the presence of polynucleotides. This indicates that NTPase activity of dengue NS3 can be stimulated by polynucleotides. A helicase assay based on internal fluorescence quenching was conducted using short internally quenched DNA oligonucleotides as substrates. Significant fluorescence signaling increase was observed in the absence of polynucleotides such as poly(U). No unwinding activity was observed with addition of poly(U). The approach we describe here is useful for the further characterization of substrate specificity and for the design of high-throughput assays aimed at discovery of inhibitors against NS3 NTPase/helicase activities.
KEY WORDS: Dengue virus, NS3 protein, NTPase, helicase, assay, substrate, polynucleotideDOI: 10.1134/S0006297913080105
Dengue fever and dengue hemorrhagic fever are the most rapidly spreading mosquito-borne diseases in the world today, with an observed 30-fold increase over the last 50 years [1]. World Health Organization estimates that globally 2.5 billion people (two-fifth of the world’s population) are at risk and annually 50-100 million people become infected, of which approximately 0.5 million require hospitalization [2].
The dengue virus particle is about 50 nm in diameter. The 10,723-nucleotide RNA genome of DEN serotype 2 (DEN2) encodes an uninterrupted open reading frame (ORF) directing the synthesis of a 3,391 amino acid residue polyprotein precursor in the order NH2-C-prM-E-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5-COOH, where C is the nucleocapsid, M the membrane-associated protein, E the envelope protein, and NS1 through NS5 are nonstructural proteins [3].
Flavivirus nonstructural protein 3 (NS3) bears multiple enzymatic activities essential for viral replication, and it therefore represents an attractive target for antiviral intervention. NS3 contains the viral serine protease at the N-terminus and ATPase, RTPase, and helicase activities at the C-terminus [4, 5]. The serine protease activity within ~180 residues at the N-terminus is vital for the cleavage of the polyprotein precursor. The combined activities of a polynucleotide-stimulated helicase/NTPase in the C-terminal domain are required for melting secondary structures prior to initiation of RNA synthesis and for the unwinding of RNA duplexes, either to separate dsRNA intermediates formed during viral RNA synthesis or as a translocase that can remove proteins bound to viral RNA [6]. Mutational analysis has shown that the ATPase/helicase and NTPase activities of DEN NS3 share a common active site [7, 8]. Dengue viruses with impaired helicase activity lose the ability to replicate, demonstrating the importance of the NS3 helicase domain in the viral life cycle [9] and that inhibitors or modulators for these enzymes are potentially of interest as therapeutic agents.
Unlike the hepaciviruses, such as hepatitis C virus (HCV) whose NS3 helicases are extensively characterized [10-16], the helicases from flaviviruses are comparatively less studied mechanistically.
The crystal structure of the dengue virus NS3 helicase (residues 171-618) has been resolved at 2.4 Å resolution [17]. Since the functional domains of NS3 protease and helicase have been shown to overlap within a region of 20 amino acids between residues 160 and 180 of the DEN NS3 protein, an interaction between the protease and helicase domains was proposed to be involved in a coordinated regulation of both protease and helicase activities [18]. In HCV NS3 protein, such interdependence of different domains for their respective activities has been extensively characterized, and a strong interdependence in the enzymatic activities of the protease and helicase domains of HCV NS3 protein has been demonstrated. Previous studies have suggested a significant regulatory role of the protease domain on flavivirus NS3 helicase activity [17, 19-24]. In contrast to earlier reports, a recent study employing the DEN2 NS3 full-length protein (residues 1-618) and the NS3-helicase domain (residues 171-618) showed that the NS3 protease domain had no significant effect on the ATPase and helicase activities of DEN NS3 [5].
A role for polynucleotides as modulators of enzymatic activity on the HCV NS3 NTPase and helicase activities has been thoroughly investigated [25], and a comparison between full-length and truncated NS3 proteins demonstrated significant differences in pH optima and poly(U) concentration required for ATPase activity [26]. Binding of polynucleotides to the NS3 helicase domain not only stimulated NTPase activity, but it also modulated the helicase and protease activities of HCV NS3 [26]. In contrast, much less is known about the influence of polynucleotides on NS3 NTPase and helicase activities of DEN virus.
It is noteworthy that a comparison of crystal structures of the full-length NS3 from DEN and HCV revealed major differences in the relative orientations between the protease and helicase domains in the two proteins. Specifically, a β-strand in the HCV NS3 clamps the protease domain next to the helicase domain, thereby creating a compact conformational state that differs from the extended conformation observed in the DEN protein [10, 24]. Moreover, in contrast to the NS3 from HCV, the DEN NS3h displays low affinity and inefficient unwinding of double-stranded DNA molecules, while it appears to preferentially bind to single-stranded RNA [27]. This favors the notion that the NS3 proteins from different viruses should be studied individually to uncover their structural characteristics and evaluate their kinetic parameters, substrate preferences, and inhibition.
The objective of this study was to establish a simplified procedure for cloning, expression, and single-step purification of dengue NS3h protein, and to determine its enzymatic activities in vitro in the presence and absence of synthetic polynucleotides. We show that the DEN NS3 protein can be obtained in sufficient quantity and purity and describe a straightforward procedure for assay of both NTPase and helicase activities, which should be useful for the design of high-throughput assays aimed at antiviral inhibitor discovery.
MATERIALS AND METHODS
Construction of pTrcHisA/NS3h plasmid. The full-length gene sequence of DEN serotype 2 NS2B(H)-NS3 was obtained as a synthetic gene in pUC57-NS2B(H)-NS3FL plasmid (TOPGENE Technologies, Canada). The pUC57-NS2B(H)-NS3FL plasmid was digested with restriction enzymes BamHI and HindIII (Promega, USA). The desired 2,043 nucleotide NS2B(H)-NS3FL fragment was cloned into pTricHisA vector (Invitrogen, USA).
The catalytic domain of the dengue virus helicase (residues 160-618) (nucleotides 4999 to 6375 in DEN2 strain 16681, GenBank accession No. U87411) was amplified by PCR using pTH/NS2B-NS3FL as a template. The specific PCR primers used are NS3(h) 160_F: 5′-AGGGGATCCGCATATGTGAGTGCTATAG-3′ (the underlined sequence represents the BamHI restriction site) and NS3(h)_R: 5′-CGCAAGCTTCAGCTACTTTCTTCCGGC-3′ (the underlined sequence represents the HindIII restriction site, and the sequence in italic indicates the introduced stop codon). All oligonucleotides were purchased from Proligo Singapore Pte Ltd.
PCR was performed by using a thermal cycler GeneAmp PCR System Model 2400 (Perkin Elmer Cetus, USA) with pre-denaturing (94°C, 5 min), 25 cycles of denaturing (94°C, 60 s), annealing (59°C, 30 s), extension (72°C, 3 min), and final extension (72°C, 7 min). The resulting PCR products were separated by agarose gel electrophoresis and purified using a QIAquick gel extraction kit (Qiagen, Germany). The purified PCR product and pTrcHisA (Invitrogen) vector were digested with restriction enzymes BamHI and HindIII and purified using a QIAquick purification kit. Insert DNA and pTrcHisA vector were combined at 6 : 1 molar ratio in ligation reaction containing 1× ligation buffer (50 mM Tris-HCl, pH 7.6, 10 mM MgCl2, 1 mM ATP, 1 mM DTT, 25% (w/v) polyethylene glycol 8000) and 5 units of T4 DNA ligase (Gibco BRL, USA) in a final volume of 20 µl and incubated overnight at 14°C, resulting in NS2B(H)-NS3pro carrying an N-terminal (His)6 purification tag.
The sequence of the recombinant pTrcHis/NS3h construct was confirmed by DNA sequence analysis by using an ABI PRISM Dye Termination Cycle Sequencing Ready Reaction Kit (Perkin Elmer, USA). Sequencing reactions were run on ABI PRISM Model 377 DNA Sequencer (Perkin Elmer). The sequence of the construct was confirmed by nucleotide sequencing using the sequencing primers, pTrcHis-F: 5′-GAGGTATATATTAATGTATCG-3′ and pTrcHis-R: 5′-CTGAAAATCTTCTCTCATC-3′, and no mutations were found in the construct.
Expression and purification of NS3h. The pTrcHisA vector was used for expression of hexahistidine-tagged recombinant proteins. Cultures of the E. coli strain C41(DE3) transformed with the expression plasmid were grown in 1 liter of LB medium containing 100 µg/ml ampicillin, at 37°C, until A600 reached 0.5-0.6. Expression of the recombinant protein was induced by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) to final concentration 0.1 mM and incubated for an additional 8 h at 37°C. Cells were harvested by centrifugation (6000g, 4°C, 10 min).
The cell pellet was resuspended in 30 ml of cell lysis buffer (0.1 M Tris-HCl, pH 7.5, 300 mM NaCl, 250 µg/µl lysozyme, 10 µg/ml DNase, and 5 mM MgCl2) followed by sonication (10 bursts at power setting 3 for 15 s, on ice) using an XL Sonicator Ultrasonic Processor (Misonix Inc., USA). The cell lysate was clarified by centrifugation (15,000g, 4°C, 30 min). The soluble fraction was collected, and the insoluble fraction was washed two times with 20 ml cell wash buffer (0.1 M Tris-HCl, pH 7.5, 0.3 M NaCl) containing 1% Triton X-100, and then two times with 20 ml cell lysis buffer (without Triton X-100). The pellet was suspended in 30 ml of denaturing buffer (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, 8 M urea) followed by a 15 s brief sonication and centrifugation (15,000g, 4°C, 30 min), and by filtration through 0.22 µm filters (Pall Corporation, USA).
The resulting fusion protein was purified using a 5 ml HisTrap HP column (GE Healthcare, Sweden). The column was equilibrated with five column volumes (25 ml) of buffer A (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, 8 M urea). The filtered protein solution was loaded onto the equilibrated 5 ml HisTrap HP column with a flow rate of 1.0 ml/min using an FPLC pump (ÄKTA FPLC system; GE Healthcare) and the column was washed with ten column volumes (50 ml) of binding buffer containing 20 mM imidazole. Protein was eluted from the column with five column volumes (25 ml) of buffer B (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, 8 M urea, 100 mM imidazole), and fractions of 1 ml were collected and analyzed by SDS-PAGE and Western blotting.
Aliquots of 20 µl from each fraction were loaded onto a 12% SDS-PAGE gel, and electrophoresis was performed in Tris-glycine buffer (25 mM Tris-HCl, pH 8.3, 192 mM glycine and 0.1% SDS). The gel was stained with Coomassie Blue staining solution (0.1% Coomassie Brilliant Blue R250, 50% methanol, and 10% glacial acetic acid) with shaking at room temperature for 2 h, and then destained in destaining solution (10% methanol and 10% glacial acetic acid). Western blotting of the protein fractions was performed using anti-hexahistidine antiserum (Invitrogen) at 1 : 10,000 dilution with alkaline phosphatase color development.
Renaturation of recombinant protein. The purified protein was renatured by artificial chaperone-assisted refolding, which utilized a detergent and cycloamylose to mediate the folding process. The purified protein (3-10 mg/ml) was diluted 50-60 times with 0.1 M Tris-HCl buffer, pH 8.0, containing 0.05% (v/v) cetyltrimethylammonium bromide (CTAB) and incubated at room temperature for 1 h. Subsequently, cycloamylose (Ezaki Glico Co., Japan; degree of polymerization from 22 to 50) was added to the protein–detergent complex solution, giving a 0.6% (w/v) final concentration, and the mixture was incubated at 4°C for 12-16 h.
The enzyme was concentrated and dialyzed in assay buffer (50 mM Tris-HCl, pH 7.5). The dialyzed NS3h was further concentrated to 1 mg/ml using a centrifugal filter device (Centricon 15 ml, 30 kDa MWCO; Millipore, USA) at 4°C. Protein concentrations were determined using a Bradford protein micro-assay [28] (Bio-Rad, USA) with bovine serum albumin (Sigma, USA) as the calibration standard. Samples were stored in 50 mM Tris-HCl, pH 9.0, 50% (v/v) glycerol, at –20°C.
Assay of enzyme activities. NTPase activity of NS3h was assayed as described in the literature [29, 30] by using Malachite Green reagent (Sigma-Aldrich, Singapore) in a 96-well plate. The assay was conducted in 20-µl volume reaction mixture containing 50 nM refolded enzyme, various ATP concentrations (0.125 to 4.0 mM), and 5 mM MgCl2 in assay buffer (25 mM Tris-HCl, pH 7.5), for 10 min at 37°C. The assay was performed in the presence and absence of 100 μM poly(U) (Invitrogen). The reaction was stopped by the addition of 50 mM EDTA, and the color was developed with 200 µl AM/MG reagent (0.034% Malachite Green, 1.05% ammonium molybdate and 0.04% (v/v) Tween 20 in 1 M hydrochloric acid). After 5 min, 30 µl of 34% (w/v) trisodium citrate was added, and absorbance was measured at 630 nm. The amount of ATP hydrolyzed was calculated from an inorganic phosphate standard curve. The kinetic parameters, Km (µM), kcat (s–1), and kcat/Km (M–1·s–1), were determined from measurements of the initial rates at different ATP concentrations using GraphPad Prism software.
Helicase activity of NS3h was tested by a fluorescence resonance energy transfer (FRET)-based assay in which the substrate used is prepared by annealing at 1 : 1.2 molar ratio a cyanine (Cy3)-labeled 36-mer (TAGTACCGCCACCCTCAGAACCTTTTTTTTTTTTTT) to a 3′-BHQ-2 (Black Hole Quencher-2)-labeled 22-mer (GGTTCTGAGGGTGGCGGTACTA) (METABION, Germany) in 20 mM Tris-HCl, pH 7.5, by brief heating to 90°C, followed by slow cooling to room temperature [31]. The standard helicase assay was performed in a 96-well microtiter plate in reaction volume of 100 µl, which contained 30 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 0.075% Triton X-100, 0.05% sodium azide, 20 nM substrate, 1 mM ATP, and 250 nM capture strand. The assay was performed in the presence and absence of 100 μM poly(U). The unwinding reaction was started by addition of enzyme (50 nM) and was carried out at 37°C for up to 45 min. The fluorescent signal was measured using an automatic microplate reader (Perkin Elmer) at excitation/emission wavelengths of 550/620 nm. Enzyme activity was calculated as the initial reaction velocity, expressed in relative fluorescence units (RFU) per unit time (minute) from the linear part of the progress curve.
RESULTS AND DISCUSSION
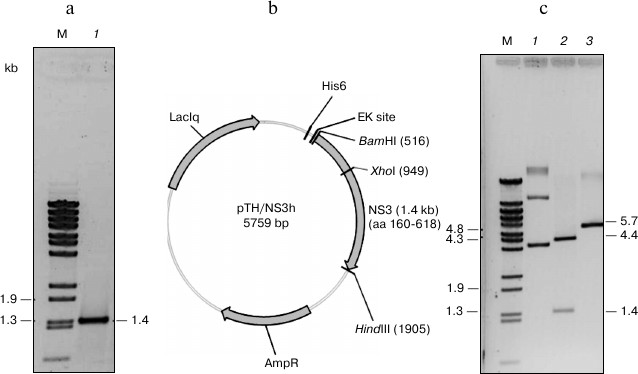
Cloning, expression, purification, and renaturation of DEN2 NS3h. The recombinant plasmid pTrcHisA/NS3h was constructed by using pTrcHisA/NS2B(H)-NS3FL as a template that encodes the full-length NS2B(H)-NS3 protein of dengue virus serotype 2 (DEN2). The DNA fragment encoding the 459 residues of the NS3 helicase domain (residues 160-170 of NS3 protease domain and residues 171-618 of the NS3 helicase domain) was amplified by PCR utilizing specific primer set (see “Materials and Methods”). Under the conditions used, the desired 1.4 kb fragment of NS3h was generated and cloned downstream of an N-terminal hexahistidine purification tag into expression vector pTrcHisA, resulting in a 5.7 kb pTH/NS3h expression plasmid. Recombinant plasmid DNA was transformed into E. coli DH5α followed by rapid size screening and restriction digestion analysis (Fig. 1). The complete sequence of cloned NS3h was analyzed by automated DNA sequencing, and resulting sequences were compared to the nucleotide sequence of the DEN2 D2V16681 sequence. The construct showed 100% identity to the template gene, and no premature stop codons or amino acid substitutions were introduced into the recombinant sequence.
Fig. 1. Physical map of pTH/NS3h. a) The 0.9% agarose gel electrophoresis (ethidium bromide stained) of the NS3h product. Lanes: M) λ-BstEII digested DNA markers; 1) PCR product containing the NS3 helicase domain). b) Restriction map of pTH/NS3h showing the positions of BamHI, HindIII and XhoI restriction endonuclease recognition sites. c) BamHI, HindIII and XhoI digestion pattern of the recombinant plasmid pTH/NS3h. Lanes: M) λBStEII digested DNA marker; 1) pTH/NS3h uncut; 2) pTH/NS3h/BamHI/HindIII double cut; 3) pTH/NS3h/XhoI cut.
Expression conditions were optimized by expression at varying temperatures, incubation times, and IPTG concentrations. Expression of NS3 helicase was performed under the optimized conditions. High yield expression was found at 0.1 mM IPTG concentration after 8-h incubation at 37°C. Western blot analysis of NS3 helicase expressed in E. coli with anti-(His)6 antibody showed that this protein migrates with an apparent molecular weight of approximately 56.5 kDa. This observation corresponds to the predicted molecular weight of NS3h carrying a hexahistidine tag at the N-terminus. The results from Western blotting of protein in the presence of 8 M urea showed that NS3h was predominantly expressed as inclusion bodies.
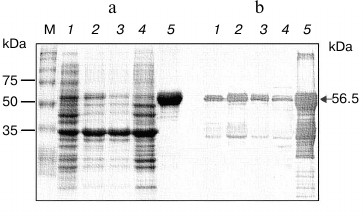
Therefore, purification and refolding from the insoluble lysate fraction was attempted. One step Ni2+-affinity chromatography yielded approximately 1.9 mg/ml of NS3h protein at purity >95% as determined from band intensities on SDS-PAGE gels and Western immunoblotting analysis (Fig. 2).
SDS-PAGE analysis revealed the presence of a single major band of 56.5 kDa molecular mass. Western blot analysis with anti-His tag antiserum demonstrated the presence of a single major band of 56.5 kDa molecular weight. Minor bands of molecular masses lower than 37 kDa were also detected; these might be degradation products of NS3 helicase protein produced during expression and purification (Fig. 2).
Fig. 2. SDS-PAGE profile (Coomassie staining) (a) and Western blot (b) of the purified NS3h protein fraction obtained after IMAC. Lanes: M) broad range standards (BioRad); 1) NS3h crude lysate; 2) NS3h pellet fraction; 3) flow through; 4) wash with 10 column volumes 20 mM imidazole; 5) elution fraction with 100 mM imidazole.
Purified NS3h was first refolded by an artificial chaperone-assisted procedure analogous to the procedures described for renaturation of HCV full-length NS3 protein, i.e. by successive dialysis [30]. However, we found in preliminary experiments that single-step dialysis, stepwise dialysis, and dialysis with additives, e.g. detergents and L-arginine, failed to efficiently reconstitute NS3 helicase activity. Instead, the protein was prone to the formation of aggregates. Thus, we set up the so-called “artificial chaperone technique”, to improve folding of the dengue protease. The procedure sequentially uses detergents and cycloamylose to mimic the bacterial GroEL/GroES protein folding system in vitro [32].
The ability of detergents Tween-40, Tween-60, and CTAB in combination with cycloamylose to accommodate folding was examined. Recovery of NTPase activity was found after refolding with CTAB and cycloamylose, while complete loss of activity was observed by using Tween-40 and Tween-60.
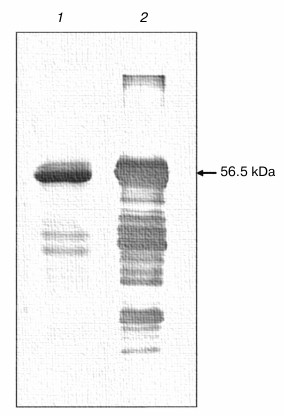
The refolded (His)6-NS3 helicase was analyzed on SDS-PAGE and Western blotting, demonstrating the presence of a major band of the NS3 helicase with molecular mass of 56.5 kDa. Some minor bands of molecular masses lower than 37 kDa might represent degradation products of the NS3 helicase protein (Fig. 3). A marked loss of approximately 35% of the NS3h protein was observed after refolding, probably due to the precipitation of protein aggregates formed during incubation.
Fig. 3. NS3h after renaturation. The figure shows the SDS-PAGE profile (Coomassie staining) (1) and Western blotting (2) of the IMAC-purified and refolded fraction.
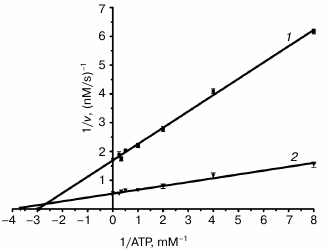
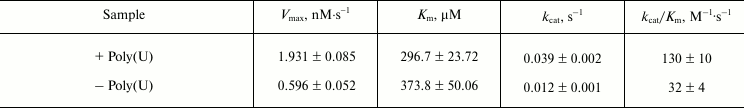
Assays of enzyme activities of purified NS3h. NTPase activity was assayed based on a colorimetric method using Malachite Green reagent, which detects free inorganic phosphate released from the NTP or NDP substrates [17, 29, 30]. In analogy to a previously published study using HCV, poly(U) was chosen because it is an efficient activator of the NS3 ATPase [33]. Kinetic parameters, kcat (turnover number) and Km (Michaelis constant), were determined from Lineweaver–Burk plots of hydrolysis activity for different concentrations of the substrate (Fig. 4). The kcat/Km value was calculated as a measure of the overall activity (catalytic efficiency) of the enzyme for substrate (table). The NTPase activity of NS3 helicase in the presence of poly(U) showed an increased turnover number and a lower Km value than the reaction without poly(U), whereby the overall catalytic efficiency (kcat/Km) was increased approximately fourfold.
Fig. 4. A double-reciprocal plot for DEN2 NS3h-catalyzed ATP substrate hydrolysis rates at different substrate concentrations. The graph shows reaction velocities of DEN2 NS3h for hydrolysis of ATP in the absence (1) and presence (2) of 100 µM poly(U). Assays were performed at 37°C in 25 mM Tris-HCl, pH 7.5, 5 mM MgCl2. Data are reported as the mean of three experiments ± standard error (SEM).
DEN2 NS3h kinetic parameters for ATP hydrolysis (ATPase activity)
Several studies have demonstrated that NS3h of HCV requires polynucleotide binding to turn on the NTPase activity [34-36]. In contrast, the response of the helicases to polynucleotides appears to be species-dependent. ATPase activity of the HCV enzyme is stimulated in the following order of efficiency: poly(U) = poly(dU) >> poly(A) > poly(dT) = poly(C) > poly(dI) > poly(I) > poly(dA) > poly(dC) > poly(G) [25, 36]. It was recently proposed that binding of poly(U) triggers a conformational rearrangement of the catalytic core of the NS3h enzyme resulting in an activation of NTP hydrolysis [27].
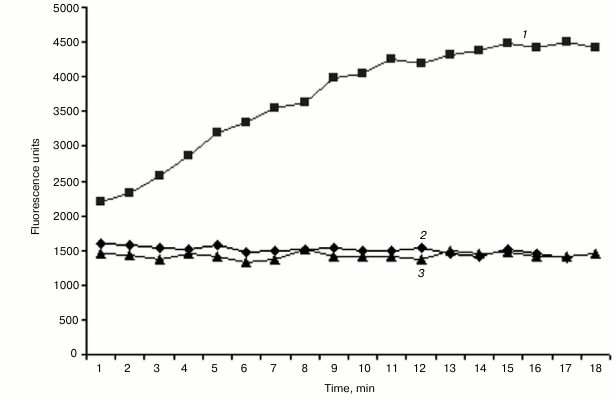
Helicase activity assay of purified NS3h. The RNA substrate unwinding activity of NS3 helicase was monitored with a quenched duplex DNA substrate. Fluorescence emission produced by the cyanine dye (Cy3 fluorophore), in the absence and presence of 100 μM poly(U), was recorded, and the time-course of the helicase reaction was plotted as shown in Fig. 5. A linear dependence of the unwinding activity as a function of incubation time was observed up to 10 min at a reaction velocity of 200 RFU/min. In the absence of helicase, no fluorescence signal was observed. Helicase activity in the presence of poly(U) was negligible, a finding in agreement with an earlier report by Morgenstern et al. [26] showing that addition of poly(U) results in a suppression of NS3 helicase activity. Related studies have suggested a similar inhibitory effect of added polynucleotides on NS3 helicase activity by a competition mechanism between the DNA/RNA substrates for the active site.
Fig. 5. Kinetics of the NS3 helicase reaction. Fluorescence release was measured in the absence (1) or presence (2) of 100 µM poly(U) compared to a control reaction without enzyme (3).
Inhibition of NS3h unwinding activity by polynucleotides can be mimicked by synthetic macromolecules acting as potential competitive inhibitors. A new selective and potent competitive inhibitor of the HCV helicase, QU663 (Ki = 0.75 μM), was recently discovered by Maga and coworkers [37]; it specifically inhibits the strand-displacement (unwinding) activity by decreasing the affinity of the enzyme for the substrate without affecting the ATP-hydrolyzing ability of NS3 helicase.
The cost- and time-efficient strategy described herein can be used to obtain NS3h in sufficient amounts and purity for the characterization of substrate specificity and for the design of high-throughput assays aimed at antiviral inhibitor discovery. The inhibitory activity of polynucleotides could be exploited to design competitive inhibitors that could specifically target the NS3 helicase activity of pathogenic flaviviruses.
The authors would like to thank Somsri Sakdee for excellent technical work and Anchalee Nirachanon for excellent secretarial assistance.
This work was supported by grant BRG4980008 from the Thailand Research Fund (TRF) to GK.
REFERENCES
1.World Health Organization (2009) Geneva: World
Health Organization and the Special Programme for Research and Training
in Tropical Diseases (TDR), p. 147.
2.World Health Organization (2012) Dengue and
Severe Dengue: Fact sheet number 117; available at
http://www.who.int/mediacentre/factsheets/fs117/en/index.html (accessed
February 22, 2013).
3.Chambers, T. J., Hahn, C. S., Galler, R., and Rice,
C. M. (1990) Annu. Rev. Microbiol., 44, 649-688.
4.Lescara, J., Luo, D., Xub, T., Sampath, A., Lim, S.
P., Canard, B., and Vasudeva, S. G. (2008) Antiviral Res.,
80, 94-101.
5.Gebhard, L. G., Kaufman, S. B., and Gamarnik, A. V.
(2012) PLoS ONE, 7, e36244.
6.Sampath, A., and Padmanabhan, R. (2009)
Antiviral Res., 81, 6-15.
7.Benarroch, D., Selisko, B., Locatelli, G. A., Maga,
G., Romette, J. L., and Canarad, B. (2004) Virology, 328,
208-218.
8.Bartelma, G., and Padmanabhan, R. (2002)
Virology, 299, 122-132.
9.Matusan, A. E., Pryor, M. J., Davidson, A. D., and
Wright, P. J. (2001) J. Virol., 75, 9633-9643.
10.Ding, S. C., Kohlway, A. S., and Pyle, A. M.
(2011) J. Virol., 85, 4343-4353.
11.Frick, D. N. (2006) in HCV Helicase:
Structure, Function, and Inhibition (Tan, S. L., ed.)
Chap. 6, Horizon Bioscience, Norfolk (UK) (http://www.ncbi.nlm.nih.gov/books/NBK1614/).
12.Sikora, B., Chen, Y., Lichti, C. F., Harrison, M.
K., Jennings, T. A., Tang, Y., Tackett, A. J., Jordan, J. B., Sakon,
J., Cameron, C. E., and Raney, K. D. (2008) J. Biol. Chem.,
283, 11516-11525.
13.Dumont, S., Cheng, W., Serebrov, V., Beran, R.
K., Tinoco, I., Jr., Pyle, A. M., and Bustamante, C. (2006)
Nature, 439, 105-108.
14.Serebrov, V., and Pyle, A. M. (2004)
Nature, 430, 476-480.
15.Serebrov, V., Beran, R. K., and Pyle, A. M.
(2009) J. Biol. Chem., 284, 2512-2521.
16.Cheng, W., Dumont, S., Tinoco, I., Jr., and
Bustamante, C. (2007) Proc. Natl. Acad. Sci. USA,
104, 13954-13959.
17.Xu, T. A., Sampath, A., Chao, D., Wen, M., Nanao,
P., Chene, Vasudevan, S. G., and Lescar, J. (2005) J. Virol.,
79, 10278-10288.
18.Li, H., Clum, S., You, S., Ebner, K. E., and
Padmanabhan, R. (1999) J. Virol., 73, 3108-3116.
19.Beran, R. K., Serebrov, V., and Pyle, A. M.
(2007) J. Biol. Chem., 282, 34913-34920.
20.Beran, R. K., and Pyle, A. M. (2008) J. Biol.
Chem., 283, 29929-29937.
21.Rajagopal, V., Gurjar, M., Levin, M. K., and
Patel, S. S. (2010) J. Biol. Chem., 285, 17821-17832.
22.Frick, D. N., Rypma, R. S., Lam, A. M., and Gu,
B. (2004) J. Biol. Chem., 279, 1269-1280.
23.Mastrangelo, E., Milani, M., Bollati, M.,
Selisko, B., Peyrane, F., Pandini, V., Sorrentino, G., Canard, B.,
Konarev, P. V., Svergun, D. I., de Lamballerie, X., Coutard, B.,
Khromykh, A. A., and Bolognesi, M. (2007) J. Mol. Biol.,
372, 444-455.
24.Lou, D., Xu, T., Hunke, C., Gruber, G.,
Vasudevan, S. G., and Lescar, J. (2008) J. Virol., 82,
173-183.
25.Banerjee, R., and Dasgupta, A. (2001) J.
Virol., 75, 1708-1721.
26.Morgenstern, K. A., Lando, J. A., Hsiao, K., Lin,
C., Gu, Y., Su, M. S., and Thomson, J. A. (1997) J. Virol.,
71, 3767-3775.
27.Wang, C. C., Huang, Z. S., Chiang, P. L., Chen,
C. T., and Wu, H. N. (2009) FEBS Lett., 583, 691-696.
28.Bradford, M. M. (1996) Anal. Biochem.,
72, 248-254.
29.Lanzetta, P. A., Alvarez, L. J., Reinach, P. S.,
and Candia, O. A. (1979) Anal. Biochem., 100, 95-97.
30.Wardell, A. D., Errington, W., Ciaramella, G.,
Merson, J., and McGarvey, M. J. (1999) J. Gen. Virol.,
80, 701-709.
31.Anna, M. B., Krawczyk, M., Stankiewicz, A.,
Gozdek, A., Haenni, A., and Strokovskaya, L. (2004) FEBS,
567, 253-258.
32.Machida, S., Ogawa, S., Xiaohua, S., Takaha, T.,
Fujii, K., and Hayashi, K. (2000) FEBS Lett., 486,
131-124.
33.Belon, C. A., and Frick, D. N. (2009) J. Mol.
Biol., 388, 851-864.
34.Yon, C., Teramoto, T., Mueller, N., Phelan, J.,
Ganesh, V. K., Murthy, K. H. M., and Padmanabhan, R. (2005) J. Biol.
Chem., 280, 27412-27419.
35.Tai, C. L., Chi, W. K., Chen, D. S., and Hwang,
L. H. (1996) J. Virol., 70, 8477-8484.
36.Suzich, J. A., Tamura, J. K., Palmer-Hill, F.,
Warrener, P., Grakoui, A., Rice, C. M., Feinstone, S. M., and Collett,
M. S. (1993) J. Virol., 67, 6152-6158.
37.Maga, G., Gemma, S., Fattorusso, C., Locatelle,
G. A., Butini, S., Persico, M., Kukreja, G., Romano, M. P.,
Chiasserini, L., Savini, L., Novellino, E., Nacci, V., Spadari, S., and
Campiani, G. (2005) Biochemistry, 44, 9637-9644.