REVIEW: High-Performance Anion-Exchange Chromatography with Pulsed Amperometric Detection for Carbohydrate Analysis of Glycoproteins
J. S. Rohrer*, L. Basumallick, and D. Hurum
Thermo Fisher Scientific, 1214 Oakmead Parkway, Sunnyvale, CA, USA; fax: 408-730-9403; E-mail: Jeff.Rohrer@thermofisher.com* To whom correspondence should be addressed.
Received January 28, 2013
High-performance anion-exchange chromatography with pulsed amperometric detection (HPAE-PAD) is an established technique for the carbohydrate analysis of glycoproteins. HPAE-PAD is routinely used for determinations of monosaccharide, sialic acid, mannose-6-phosphate (M-6-P), and oligosaccharide contents of a glycoprotein. This is true for both the initial investigation of a glycoprotein and routine assays of recombinant therapeutic glycoproteins. This contribution reviews the fundamentals of HPAE-PAD, recent technological improvements, and advances in the last ten years in its application to carbohydrate analysis of glycoproteins. The application areas reviewed include monosaccharide determinations, sialic acid determinations, M-6-P determinations, sugar alcohol determinations, analysis of polysialic acids, neutral and charged oligosaccharide analysis, following glycosidase and glycosyltransferase reactions, and coupling HPAE-PAD to mass spectrometry (MS).
KEY WORDS: monosaccharide, sialic acid, oligosaccharide, glycoprotein, carbohydrate, mannose-6-phosphate, HPAE-PADDOI: 10.1134/S000629791307002X
Abbreviations: DMB, 1,2-diamino-4,5-methylenedioxybenzene; DP, degree of polymerization; HPAE-PAD, high-performance anion-exchange chromatography with pulsed amperometric detection; IC, ion chromatography; LC, liquid chromatography; MALDI-TOF-MS, matrix-assisted laser desorption time-of-flight mass spectrometry; M-6-P, mannose-6-phosphate; MS, mass spectrometry; Neu5Ac, N-acetylneuraminic acid; Neu5Gc, N-glycolylneuraminic acid; PE, polyester; PEEK, polyether ether ketone polymer; PTFE, polytetrafluoroethylene.
HPAE-PAD couples high-resolution separations of carbohydrates, nearly
universally at high pH (>12), with sensitive amperometric detection
where only a small percentage (<5%) of the eluting carbohydrate is
oxidized for detection. Though HPAE-PAD was first described in 1983 [1], it was first applied to glycoprotein analysis in
the late 1980s [2, 3]. The
technique enjoyed rapid adoption for the analysis of protein
glycosylation including in the then emerging field of recombinant
glycoproteins for human therapeutic use [4]. This
success was due to the ability of HPAE-PAD to deliver high-resolution
separations and sensitive detection of a wide variety of carbohydrates
including monosaccharides, sialic acids, sugar phosphates, and
oligosaccharides, without analyte derivatization. HPAE-PAD was the
subject of early reviews [5-8]
and since then has become a well-established technique for glycoprotein
analysis as well as for other fields of carbohydrate analysis. A
comprehensive review, with emphasis on the application of HPAE-PAD to
glycoprotein analysis and on its application in the biopharmaceutical
industry, was published in 2007 [9]. Our review
updates the application of HPAE-PAD to glycoprotein analysis since the
writing of the 2007 review.
HPAE-PAD still enjoys wide acceptance in the biopharmaceutical industry for the characterization of glycoproteins as noted by Higgins in her 2010 review [10]. That review focused on the methods most commonly found in regulatory filings and lot-release testing of therapeutic glycoproteins. She cited the use of HPAE-PAD for the determination of monosaccharide composition, the determination of sialic acid composition, the determination of mannose-6-phosphate (M-6-P), and both asparagine-linked (N-linked) and serine/threonine (O-linked) oligosaccharide profiling. Another recent review cited the ongoing need for HPAE-PAD for analyzing glycosylation [11]. The authors concluded that after more than 20 years HPAE-PAD was still appropriate for the analysis of protein glycosylation, and also for the investigation of disease biomarkers.
This review begins with a section that briefly describes the fundamentals of HPAE-PAD. That is followed by discussion of the technological improvements to the technique that have been introduced in the past seven years. This contribution concludes with an update on each of the major HPAE-PAD applications for glycoprotein analysis and the coupling of HPAE-PAD to mass spectrometry (MS).
FUNDAMENTALS OF HPAE-PAD
At the time of its introduction, both the separation and detection components of HPAE-PAD were unique. We will first focus on separation and then turn our attention to detection. Most carbohydrates are not anionic at pH 7, so their separation as anions was not obvious. At high pH carbohydrates become oxyanions, and these can be separated by anion-exchange chromatography. For example, glucose has pKa of 12.28. Therefore, above pH 12, glucose will be in equilibrium with its oxyanion. Thus anion-exchange chromatography of glucose requires a stationary phase that can tolerate mobile phases of pH 12 and greater. Also, the structural similarity of glucose to other monosaccharides that would be present in many samples requires a stationary phase capable of high-resolution separations. At the time, most liquid chromatography (LC) stationary phases were prepared from silica, which is not stable in high pH mobile phases. To meet the two stationary phase requirements, Dionex Corporation (now Thermo Fisher Scientific) introduced the HPIC-AS6 column (later rebranded as the CarboPac PA1 column). The stationary phase in this column was composed of essentially non-porous (microporous) 10 µm polystyrene-divinylbenzene beads that had been chemically treated to create a cation-exchanger. These substrate beads were then treated (agglomerated) with smaller polystyrene-divinylbenzene beads (<1 µm, the latex) that had quaternary amine groups (a strong anion-exchanger) on their surface. The result of this latex agglomeration was the electrostatic binding of the latex to the large bead to create an anion-exchange resin that in chromatographic terms would have short diffusion path lengths and therefore be capable of high-resolution separations. The polystyrene-divinylbenzene beads are compatible with highly acidic and, more important to carbohydrate separation, highly basic mobile phase, so both requirements for chromatography of carbohydrates as anions were met. The column hardware was made out of the polymer polyether ether ketone (PEEK), which unlike the more commonly used stainless steel does not shed metal ions when exposed to alkaline solutions. Metals will contaminate the column and, as discussed below, also interfere with detection. There are now five other Thermo Fisher Dionex CarboPac columns (PA10, PA20, PA100, PA200, and SA10) with stationary phases constructed using this basic recipe with variations in substrate bead size, latex bead size, substrate bead cross-linking, latex bead cross-linking, porosity of the substrate bead, substrate composition (always pH 0-14 compatible), and the quaternary amine group on the latex. A seventh CarboPac column, the CarboPac MA1, is prepared in a different manner. For its preparation, a 7.5-µm vinylbenzyl chloride/divinylbenzene macroporous substrate bead is fully functionalized with an alkyl quaternary ammonium group to create a strong anion-exchange phase that has a much higher capacity than the other CarboPac columns, but unlike the other columns, has a maximum backpressure rating of only 2000 psi.
To separate a wide variety of carbohydrates only two types of mobile phases are required: sodium (or potassium) hydroxide and sodium acetate. While there are other mobile phases that have been used (e.g. sodium nitrate, which will be discussed later in this paper), nearly all HPAE-PAD carbohydrate applications use either a hydroxide mobile phase, or a mobile phase that is a combination of hydroxide and acetate. In general, hydroxide-only mobile phases are used to separate mono-, di-, tri-, and tetrasaccharides as well as the sugar alcohols of those saccharides. Acetate and hydroxide mobile phases are used to separate larger carbohydrates and carbohydrates that possess a negative charge at neutral pH such as the sialic acids, phosphorylated sugars (e.g. M-6-P), and sulfated sugars. For most separations of larger and charged carbohydrates related to glycoproteins the concentration of hydroxide is held constant at 100 mM and a gradient of sodium acetate is used with the maximum concentration usually no greater than 200 mM. The general separation conditions for each class of glycoprotein-related carbohydrates will be described in sections of this paper devoted to those carbohydrates.
After separation the carbohydrates are detected by PAD, which is a direct detection technique. When HPAE-PAD was introduced the only direct detection techniques available for LC of carbohydrates were UV at short wavelengths and refractive index (RI). Both UV and RI lacked the sensitivity required for analyzing mono- and oligosaccharides from glycoproteins. PAD solved this problem as it was able to routinely detect low picomolar amounts of carbohydrates. While it had been known for a long time that carbohydrates could be oxidized on gold working electrodes in highly alkaline solution to generate a current that could be measured (i.e. dc amperometry), it was not appropriate for LC detection because the oxidation products of the carbohydrates would foul the working electrode and cause a rapid loss of signal in subsequent analyses. The fouling problem was solved by application of a series of potentials (i.e. voltages) to the gold working electrode. The initial potential was for detection and subsequent potentials were for cleaning and restoring the working electrode. This series of potentials (a.k.a. the waveform) was executed in a second or less, therefore making it ideal for application to LC separations. It was also ideal for HPAE separations of carbohydrates because the highly alkaline mobile phases met the pH requirement of the detection.
Over the first two decades of the application of HPAE-PAD there were changes in the waveform to improve first baseline and noise performance, and later long term peak area response stability. In 1997 a four-potential waveform (also commonly referred to as either the “quadruple potential” waveform or Waveform A in Thermo Fisher Scientific literature), was introduced and it has become the standard for HPAE-PAD analyses [12, 13]. This waveform and all prior waveforms can be used for all types of carbohydrates. In other words, separate waveforms are not needed for monosaccharides, sialic acids, oligosaccharides, etc. The waveform is also rather selective for carbohydrates. While amines and non-fully oxidized sulfur-containing compounds (e.g. thiols) can be oxidized on gold working electrodes under the conditions used to detect carbohydrates, the sensitivity for these compounds under these conditions is poor. Therefore, unless these compounds are present in the sample at concentrations far exceeding the carbohydrate concentrations, they do not interfere with the analysis.
As discussed previously, nearly all HPAE-PAD separations require a highly caustic mobile phase and carbohydrates must be in a caustic mobile phase for PAD. Because a typical stainless steel HPLC system will corrode when exposed to caustic and leach metal ions that contaminate the column and the working electrode, another type of chromatography system was required for HPAE-PAD. The HPAE-PAD system is metal-free, with the pump heads and tubing fabricated out of PEEK. The autosampler needle is also coated with PEEK. Using systems that were not designed for HPAE-PAD invariably leads to separation and/or detection problems.
HPAE-PAD TECHNOLOGY IMPROVEMENTS
As a mature technology, recent HPAE-PAD technological advancements focus on improving the reproducibility, ruggedness, and ease-of-use of the technique. This is exemplified by the evolution of disposable gold working electrode technology. The recently introduced gold on polytetrafluoroethylene (PTFE) disposable working electrodes offer significant improvements over gold on polyester (PE) disposable working electrodes. Both types of disposable electrodes offer greater electrode-to-electrode reproducibility compared to conventional gold working electrodes, and they do not require electrode polishing. The gold on PE disposable working electrodes deliver a one to two week lifetime depending on the application, and they are limited to use with mobile phases with ≤100 mM sodium or potassium hydroxide. The gold on PTFE disposable working electrodes have lifetimes of at least four weeks and can be used with mobile phases with ≤750 mM sodium or potassium hydroxide, which covers the strongest commonly used HPAE-PAD mobile phases. The proper use and typical performance of gold on PTFE disposable working electrodes for a variety of typical HPAE-PAD carbohydrate applications are described in Thermo Fisher Scientific (formerly Dionex) Technical Note 110 [14].
In the decades since HPAE-PAD carbohydrate analysis was first described much has been learned about the preparation of mobile phases for successful analysis. HPAE-PAD mobile phases are not as widely used as mobile phases for other LC techniques and PAD has different mobile phase requirements compared to other detection techniques. This has resulted in a lower knowledge base compared to other LC mobile phases (e.g. acetonitrile/trifluoroacetic acid (TFA) for reversed-phase HPLC). While high quality deionized water and high purity hydroxide mobile phases are also needed for ion chromatography (IC), PAD places different demands on mobile phase quality compared to suppressed conductivity detection. For example, water from a deionized water system recently sterilized with hydrogen peroxide will only benefit an IC system (e.g. lower acetate and formate contamination) while it will temporarily cause a HPAE-PAD system to have a high background if the deionized water system has not been sufficiently rinsed (i.e. allowed to produce a sufficient quantity of deionized water to remove the residual peroxide). Sodium acetate mobile phases are not used for IC with suppressed conductivity detection so there is even less overall knowledge about their preparation, storage, and use. Poor preparation and maintenance of a sodium acetate mobile phase can cause significant problems in part because acetate can promote microbial growth. The proper preparation and maintenance of the mobile phases used for HPAE-PAD carbohydrate applications are described in Thermo Scientific (formerly Dionex) Technical Note 71 [15]. That document also describes how to differentiate mobile phase issues from other possible problems.
While the four-potential waveform first described in 1998 has become the standard waveform for HPAE-PAD carbohydrate analysis, two new waveforms have been recently reported. One waveform uses just two potentials [16]. This two-potential waveform was applied to clinical pediatric urine samples to measure both endogenous and administered carbohydrates [17]. The authors show equivalent performance to the four-potential waveform with respect to sensitivity and long-term performance. With only two potentials needed, the waveform can be executed at a 20% greater frequency compared to the four-potential waveform, thereby allowing a faster data collection rate. This enables more data points to be collected for early eluting narrow peaks ensuring good reproducibility. While extremely fast (1-2 min) separations are currently not used for HPAE-PAD, the current trend in LC is for faster separations, and a faster waveform will be needed if HPAE-PAD separations are to be faster. The authors suggest that with a change in the electronics of the commercial electrochemical detector it would be possible to execute the waveform faster and collect data at 50 Hz. A recently reported 4.5 min HPAE-PAD analysis of glycoprotein sialic acids that is discussed later in this review [18] spurred the development of a faster waveform at Thermo Fisher Scientific that allows data to be collected at a rate of 3 Hz compared to the 2 Hz of the four-potential waveform [19]. This waveform has been in use in our laboratory for over six months with good results and is scheduled to be included in the software controlling HPAE-PAD systems in 2013.
In 2011 a capillary IC system was introduced and this system could be purchased with an electrochemical cell. At present the products available for HPAE-PAD carbohydrate analysis are a capillary (0.4 × 250 mm) Thermo Scientific Dionex CarboPac PA20 column, capillary Thermo Scientific Dionex AminoTrap column, the aforementioned electrochemical cell that can be outfitted with the traditional Ag/AgCl reference electrode or a new palladium hydrogen reference electrode, and an eluent generator capable of producing up to 200 mM potassium hydroxide (a standard eluent generator has a maximum concentration of 100 mM potassium hydroxide). These components should allow all HPAE-PAD carbohydrate applications using mobile phases with ≤200 mM potassium hydroxide and a Dionex CarboPac PA20 column (i.e. most monosaccharide applications).
MONOSACCHARIDE ANALYSIS
The application of HPAE-PAD for the determination of the monosaccharide content of a glycoprotein was first reported by Hardy et al. in 1988 [2]. They showed that the method accurately determined the monosaccharide content of a glycoprotein and reported the following advantages over existing glycoprotein monosaccharide analysis techniques: good sensitivity without pre- or post-column labeling of the monosaccharide (i.e. direct detection), and no need for re-N-acetylation of the amino sugars. Twenty-five years later, these advantages persist.
Though there have been improvements to HPAE-PAD monosaccharide analysis over the past 25 years, the basic method outlined by Hardy et al. has not significantly changed. Briefly, the glycoprotein or other glycoconjugate is hydrolyzed in a volatile acid (e.g. TFA) to release the monosaccharides, the acid removed by evaporation, the sample reconstituted in deionized water, and then injected into the HPAE-PAD system for analysis. The monosaccharides are then separated on a Dionex CarboPac PA1 column with a 16 mM sodium hydroxide mobile phase, and then detected by PAD.
Over the years there have been advancements in column technology to improve monosaccharide resolution, shorten analysis time, and reduce mobile phase consumption. There have also been advancements in PAD to improve sensitivity, reproducibility, ruggedness, and ease-of-use. The principles of HPAE-PAD monosaccharide analysis, its evolution, and basic execution have recently been reviewed [20]. In the next paragraph we describe how the basic HPAE-PAD monosaccharide analysis of a glycoprotein, or other glycoconjugate, experiment is conducted today.
The preparation of the glycoprotein sample prior to injection on the HPAE-PAD system remains the same with the following typical conditions. Tens of micrograms of glycoprotein are hydrolyzed in 2 N TFA for 4 h at 100°C for neutral sugar analysis and 6 N HCl for 4 h at 100°C for amino sugar analysis. For example, for the lightly glycosylated human serum IgG, hydrolyzing 66 µg allows 20 analyses of 3.3 µg of acid-hydrolyzed IgG. Three point three micrograms are more than sufficient for easy monosaccharide peak quantification, so much less protein can be hydrolyzed and/or injected if needed, and more highly glycosylated proteins require even less sample. The acid-hydrolyzed sample is dried in a vacuum concentrator equipped with an acid trap and then reconstituted in a small volume of deionized water (e.g. 200 µl). With these conditions 10 µl of sample are injected onto the HPAE-PAD system, and the monosaccharides are separated using a Dionex CarboPac PA20 column (3 × 150 mm) preceded by a Dionex AminoTrap column (3 × 30 mm) with a 10 mM sodium hydroxide mobile phase flowing at 0.5 ml/min. The mobile phase is often potassium hydroxide produced by an eluent generator rather than manually prepared sodium hydroxide. If an eluent generator is used, the 13 min separation is followed by a 3 min column wash with 100 mM KOH, and then 8 min at 10 mM potassium hydroxide to equilibrate the column for the next injection. With manually prepared sodium hydroxide, a longer 200 mM sodium hydroxide column wash is required to remove carbonate from the column (the eluent generator produces essentially carbonate-free potassium hydroxide) and maintain stable retention times. The method with manually prepared sodium hydroxide is typically 6 min longer than when the method is executed with an eluent generator. The Dionex AminoTrap column functions to delay the elution of amino acids and small peptides from the glycoprotein acid hydrolysis, so that they do not interfere with monosaccharide quantification. After separation, the monosaccharides are detected on a gold working electrode, either a disposable Au on PTFE electrode or a conventional electrode, using the four-potential carbohydrate waveform. Compared to earlier Dionex CarboPac columns, the PA20 delivers higher monosaccharide resolution and greater mass sensitivity, reduces mobile phase consumption and waste generation, and requires less time per analysis. The eluent generator eliminates error associated with hydroxide mobile phase preparation (e.g. variable carbonate contamination and therefore variable retention times), and shortens analysis time.
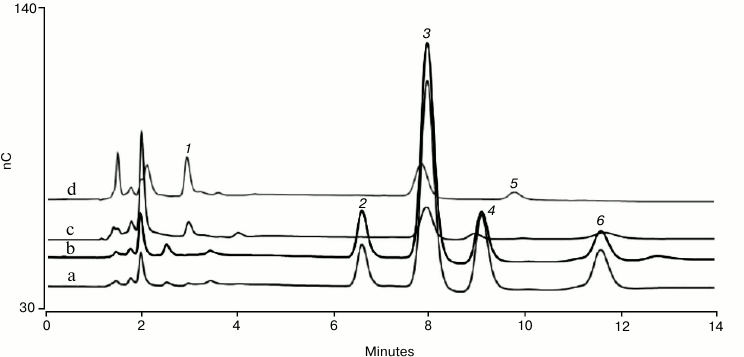
Figure 1 shows monosaccharide analysis of human polyclonal IgG and bovine fetuin using the conditions described above. Both the TFA and HCl hydrolysates of each glycoprotein are shown with the peaks identified by injecting a mixed monosaccharide standard containing fucose, galactosamine, glucosamine, galactose, glucose, and mannose. A Thermo Fisher Scientific technical note provides more information on glycoprotein monosaccharide analysis using an eluent generator [21]. That technical note shows that the monosaccharide compositions determined for human polyclonal IgG and bovine fetuin are consistent with values derived from the scientific literature, and that the method is reproducible.
Fig. 1. HPAE-PAD monosaccharide composition analysis of bovine fetuin (a, b) and human serum IgG (c, d). The monosaccharides are: 1) L-fucose; 2) D-galactosamine; 3) D-glucosamine; 4) D-galactose; 5) D-glucose; 6) D-mannose. Protein hydrolysate samples were separated on a Dionex CarboPac PA20 analytical column preceded by a Dionex AminoTrap column. The chromatographic parameters were: flow rate 0.5 ml/min, injection volume 10 µl, column temperature 30°C, electrolytically generated mobile phase, 10 mM KOH for 13 min, and 3 min regeneration at 100 mM KOH, and then a return to the starting conditions. The total run time was 24 min to allow full column re-equilibration. PAD was with the four-potential waveform and a gold on PTFE disposable working electrode.
A sampling of recent publications using HPAE-PAD monosaccharide analysis show three Dionex CarboPac columns commonly employed for monosaccharide analysis, the PA1 [22-24], PA10 [25-27], and PA20 [28]. One publication using the PA1 column touted complete monosaccharide analysis in which after the neutral and amino sugars are eluted with a 15 mM sodium hydroxide mobile phase, the acidic sugars, including the sialic acids and uronic acids, are eluted with a hydroxide/acetate mobile phase [22]. They used this method to determine the composition of aliginate, fucoidan, and some glycosaminoglycans. This approach is similar to an approach published 21 years earlier for the compositional analysis of bacterial extracellular polysaccharides [29]. A second publication used acetate in the mobile phase to determine uronic acids after neutral and amino sugars were eluted with a hydroxide-only mobile phase to determine the carbohydrate composition of a proteoglycan [23]. HPAE-PAD monosaccharide analysis with a PA1 column was used in a study of the metabolic fate of N-glycolylneuraminic acid (Neu5Gc) in humans [24]. They separated N-acetylglucosamine (GlcNAc) and N-glycolylglucosamine from glycolic acid (not detected by PAD). When the sugars and/or sugar acid were radiolabeled, fractions were collected for subsequent scintillation counting. One publication that used the PA10 column for monosaccharide analysis was studying the glycosylation of salivary and buccal cell proteins to determine their role in protection against fungal infection [25]. The other two PA10 column publications determined the monosaccharide contents of recombinant glycoproteins with both reporting that they used the Dionex AminoTrap column [26, 27]. The authors of reference [26] used their knowledge of the glycoprotein oligosaccharide composition to predict the monosaccharide composition. They concluded that HPAE-PAD monosaccharide analysis can be considered accurate because the measured monosaccharide composition agreed with the predicted. Yu et al. used a PA20 column for their monosaccharide analysis of wild type and recombinant human lactoferrin, which was expressed in the milk of transgenic cloned cattle [28]. After TFA hydrolysis and subsequent drying, they washed the hydrolysate with methanol and dried three times to remove residual TFA. No reason was provided for why residual TFA removal was required prior to HPAE-PAD monosaccharide analysis. As detailed above, this is not the typical protocol for an HPAE-PAD glycoprotein monosaccharide analysis experiment.
Not included in the above summary of recent publications is a publication that described a comparative study of monosaccharide methods to determine the monosaccharide composition of biopharmaceutical glycoproteins [30]. Ten labs participated in that study with eight labs using pre-column monosaccharide derivatization methods and two labs using HPAE-PAD. The eight labs were divided into four groups of two with each of the four using a different pre-column derivatization method. One of the labs using HPAE-PAD used a PA1 column set and the other lab used a PA20 column preceded by a Dionex AminoTrap column. Each lab measured the monosaccharide composition of three biopharmaceutical glycoproteins, epoetin α, epoetin β, and alteplase. The study looked at the advantages and disadvantages of each method. For HPAE-PAD the authors concluded that as it did not require derivatization, and neutral and amino sugars could be separated in one injection, it had a shorter total analysis time compared to the other methods. The authors cited disadvantages as the need for specialized equipment to handle to high-pH mobile phases, and no method flexibility to resolve an interfering peak, if observed, because the separation has already been optimized. No HPAE-PAD monosaccharide interfering peaks were discussed in the manuscript, but the lab using the PA1 column did not use a Dionex AminoTrap column, making it more likely for that lab to observe interfering peaks. The alteplase preparation had a large amount of arginine, and the authors believe that the arginine may lead to low values for fucose by suppressing its response. Fucose is the first monosaccharide to elute after arginine.
SIALIC ACID ANALYSIS
The determination of sialic acids in glycoproteins, including those intended as human therapeutics, is performed not only to quantify the total sialic acid content, but also to determine the relative amounts of N-acetylneuraminic acid (Neu5Ac) and Neu5Gc. Due to the potential immunogenicity of Neu5Gc, it is considered undesirable in therapeutic proteins [31]. Furthermore, changes in the biopharmaceutical production conditions or changes in cell lines can lead to undesirable or unexpected sialylation [32, 33]. The application of HPAE-PAD to the determination of the sialic content of a glycoprotein was first reported by Manzi et al. [34]. They showed that the method accurately determined the Neu5Ac and Neu5Gc contents of a glycoprotein and as with monosaccharide analysis, had the advantage of good sensitivity without pre- or post-column labeling. HPAE-PAD sialic acid analysis was later reviewed and more recently updated [8, 9].
To determine the Neu5Ac and Neu5Gc contents of a glycoprotein, the sample is first acid hydrolyzed (e.g. 0.1 N HCl at 80°C for 1 h) or treated with neuraminidase to release the sialic acids. This can require as little as a few micrograms of glycoprotein. Neu5Ac and Neu5Gc are anionic at pH 7, so their elution from a Dionex CarboPac column requires sodium acetate in the mobile phase in addition to sodium hydroxide. Typical HPAE-PAD sialic acid methods use 100 mM sodium hydroxide with a gradient of sodium acetate to separate Neu5Ac and Neu5Gc using either a Dionex CarboPac PA1, PA10, or PA20 column. While there are other sialic acids besides Neu5Ac and Neu5Gc, many are base-labile and therefore are not easily determined by HPAE-PAD. Manzi et al. showed that O-acetylated sialic acids, which are base-labile and converted to their parent neuraminic acid in base, could be determined by removing sodium hydroxide from the mobile phase and separating with just an acetate mobile phase while adding sodium hydroxide post-column for detection [34]. Poor resolution of the O-acetylated sialic acids and the extra equipment required has resulted in most analysts using pre-column labeling with 1,2-diamino-4,5-methylenedioxybenzene (DMB) followed by reversed-phase HPLC and fluorescence detection for O-acetylated sialic acid analysis [35, 36].
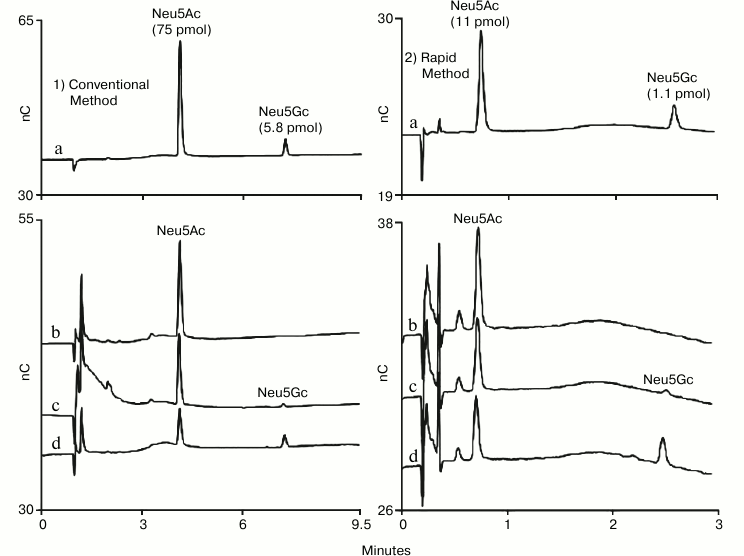
High-throughput methods are needed to meet the analysis needs of expression experiments to evaluate cell lines for therapeutic glycoproteins as well as optimize and monitor production methods. This led to the development of a rapid HPAE-PAD method for determination of sialic acids in glycoprotein hydrolysates using a short-format anion-exchange column (Dionex CarboPac PA20 Fast Sialic Acid Analysis Column) [18]. The resulting 4.5 min method was shown to provide data comparable to data collected with other HPAE-PAD methods (Fig. 2). This method was also compared to other methods, most notably the DMB method, and was found to produce comparable results without the labor of pre-column sample labeling.
Fig. 2. HPAE-PAD sialic acid determination by two methods. A conventional method (1) is presented on the left-hand column and a published rapid method (2) is presented on the right-hand column. Samples: a) Neu5Ac and Neu5Gc standards; b) acid hydrolysis of human α1-acid glycoprotein; c) acid hydrolysis of bovine fetuin; d) acid hydrolysis of ovine α1-acid glycoprotein. The rapid method used a CarboPac PA20 Fast Sialic Acid Analysis column (3 × 30 mm) with 100 mM NaOH and a 2.5 min gradient of 70-300 mM sodium acetate followed by 0.4 min at 300 mM, a return to 70 mM over 0.1 min, and 1.5 min at starting conditions prior to the next injection. The conventional method used a CarboPac PA20 column (3 × 150 mm) and its guard (3 × 30 mm) with 100 mM NaOH and a 7.5 min gradient of 70-300 mM sodium acetate followed by 1.5 min at 300 mM, a return to 70 mM over 0.5 min, and 7 min at starting conditions prior to the next injection. The two methods shared the following chromatographic parameters: flow rate 0.5 ml/min, injection volume 4.5 µl, column temperature 30°C, and the four-potential waveform and a gold on PTFE disposable working electrode for PAD.
HPAE-PAD methods for determining the amounts of individual sialic acids in proteins have been compared with other chromatographic methods requiring labeling of the sialic acids, showing comparable results [30, 37]. One study was a collaborative study that evaluated monosaccharide analysis methods for glycoprotein biopharmaceuticals [30]. It used the same samples to evaluate sialic acid analysis methods. Five labs used the DMB method and four labs used HPAE-PAD. Three of the HPAE-PAD labs used a Dionex CarboPac PA1 column set and the remaining lab used a Dionex CarboPac PA20 column set. The other study also compared the DMB method to HPAE-PAD (PA20 column) [37]. Both studies concluded that the two methods yielded similar results. The authors of the collaborative study also concluded that while the DMB method did provide good sensitivity and selectivity, the yield of derivatives can be compromised by the composition of the sample solution. Depending on sample analysis needs, the resolution of Neu5Ac and Neu5Gc achieved by HPAE-PAD is greater than by HPLC, HPAE-PAD sensitivity is within an order of magnitude of fluorescence detection methods, and the HPAE-PAD method is robust with no need for sample derivatization and subsequent cleanup.
There have been other recent reports of using HPAE-PAD to determine the Neu5Ac and Neu5Gc contents of glycoproteins during the discovery process [25, 38, 39]. The publication previously discussed in the monosaccharide section used the Dionex CarboPac PA10 column to determine sialic acids. Another study that used a PA10 column determined the sialic acid content of the cancer-derived heat shock protein gp96 at different metabolic time points [38]. The third publication used the Dionex CarboPac PA20 to determine the sialic acid content (and monosaccharide content) of gull egg white glycoproteins [39]. The latter two studies used a neuraminidase for sialic acid release.
POLYSIALIC ACID ANALYSIS
Researchers continually seek to improve sensitivity for polysialic acid analysis, especially for detecting higher degrees of polymerization (DP) [40-44]. The DP information is very useful in the study of biosynthesis, degradation, and structure–function correlations for polysialic acids. Analysis of polysialic acids involves (i) separating them on an anion-exchange column (e.g. the Dionex CarboPac PA1, Dionex CarboPac PA100, Dionex DNAPac PA-100 and Mono Q columns) and (ii) detection, either by PAD [40], or by fluorescence (after labeling the polysialic acids with a fluorogenic reagent) [41-44]. A typical separation using a gradient of sodium acetate in the presence of 100 mM sodium hydroxide on a Dionex CarboPac PA100 column has a DP resolving power of up to 60 (with PAD) [40].
The use of sodium nitrate in place of sodium acetate has also been reported for polysialic acid separations [40-43] (nitrate earlier had been used for amylopectin and maltodextrin separations [45]). Nitrate being a stronger mobile phase allows elution of the higher DPs. Using sodium nitrate for polysialic acids, the maximum DP resolved is about 20 greater than with acetate [40-43].
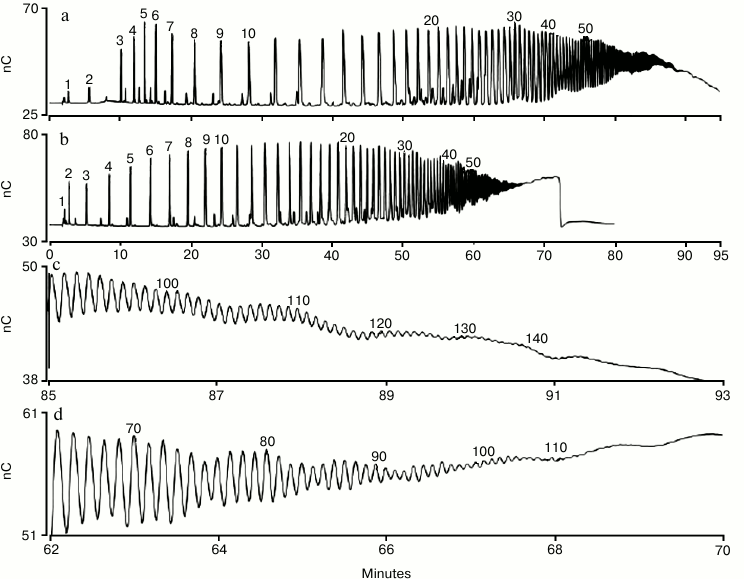
Using a column that has a smaller particle size (Dionex CarboPac PA200) provides higher resolution separations. The Dionex CarboPac PA200 resolves polysialic acid homologs up to DP 100 using a sodium acetate gradient (Fig. 3, b and d) and up to 140 using a sodium nitrate gradient (Fig. 3, a and c) [46]. This is an improvement over the maximum DP of about 60 for acetate and 80 for nitrate that could be achieved using other anion-exchange columns [40-44]. When using a nitrate gradient, we found that it was necessary to use a Thermo Scientific Dionex IonPac MFC-1 trap column (installed in the eluent line prior to the injection valve) to prevent significant loss of analyte peak area upon repeated sample injections [46]. The MFC-1 trap column removes trace transition metal contaminants from high-pH mobile phases, and we believe that metal contamination in the sodium nitrate (vendor specification for iron is 3 ppm on ACS grade sodium nitrate) led to electrode fouling (i.e. loss of analyte peak area). With a MFC-1 trap column installed we found that peak areas were higher, and for over 35 consecutive injections there was no peak area loss.
Fig. 3. HPAE-PAD profile of a commercial sample of colominic acid. Fifty micrograms were injected on a Dionex CarboPac PA200 column and eluted with nitrate (a) and acetate (b) as the pushing agent. Panels (c) and (d) show the time range 85-93 min (with nitrate) and 62-70 min (with acetate), respectively. Peaks are labeled with their putative DP values, with the exception of the trimer (DP = 3) and pentamer (DP = 5) that were determined using standards. The chromatographic parameters were: Dionex CarboPac PA200 guard and analytical columns (3 × 30 and 3 × 250 mm, respectively); flow rate, 0.5 ml/min; injection volume, 10 µl; column temperature, 30°C; eluent 100 mM NaOH with 20 mM NaNO3 for 2 min, 20 to 100 mM NaNO3 in 7 min, 100 to 160 mM NaNO3 in 30 min, 160-310 mM in 60 min (curve 5, linear) for the nitrate gradient (a, c), and 100 mM NaOH with 200 to 1000 mM sodium acetate in 69 min (curve 4, a slightly non-linear gradient delivery) for the acetate gradient (b, d).
HPAE-PAD OLIGOSACCHARIDE ANALYSIS
HPAE-PAD is an established technique for profiling glycoprotein oligosaccharides and has been used in glycoprotein discovery and characterization as well as in the assay of human therapeutic glycoproteins for lot release. The technique has been applied to N-linked and O-linked oligosaccharides as well as neutral and charged oligosaccharides. The importance of HPAE-PAD to glycoprotein oligosaccharide analysis was recognized early and was the first application of the technique to glycoprotein characterization [47, 48]. HPAE-PAD has been shown to deliver reproducible high-resolution separations of oligosaccharides, including linkage and positional isomers, and sensitive detection without the need for pre- or post-column sample derivatization. To aid in N-linked oligosaccharide identification, a list of empirical rules was published to rationalize the elution order of N-linked oligosaccharides [49].
An HPAE-PAD glycoprotein oligosaccharide analysis is typically conducted as follows. The oligosaccharides are first released from the glycoprotein. For N-linked oligosaccharides this it typically done with the amidase PNGase F. O-Linked oligosaccharides are typically released chemically by β-elimination in a reducing environment. Released oligosaccharide samples usually require a small amount of cleanup prior to HPAE-PAD, although under the simplest PNGase F digest conditions (no denaturants, such as SDS, or β-mercaptoethanol in the digest) the sample can be injected directly. Though sample pre-treatment conditions vary, guidance is available for PNGase F digests [50] and β-elimination reactions [51]. Some analysts reduce the released N-linked oligosaccharides with sodium borohydride (this eliminates reducing terminal GlcNAc to N-acetylmannosamine epimerization [2]), but most analyze native oligosaccharides. The prepared sample is then injected onto the HPAE-PAD system. This analysis typically requires only a few micrograms of protein. The oligosaccharides are separated on a Dionex CarboPac column with a mobile phase of 100 mM sodium hydroxide and a 45 to 60 min sodium acetate gradient. The acetate concentration usually starts at 10 to 20 mM and rarely exceeds 250 mM. Originally the Dionex CarboPac PA1 column was used, but improvements in subsequent years that increased oligosaccharide resolution led to first the PA100 column and finally the PA200 column being the column of choice.
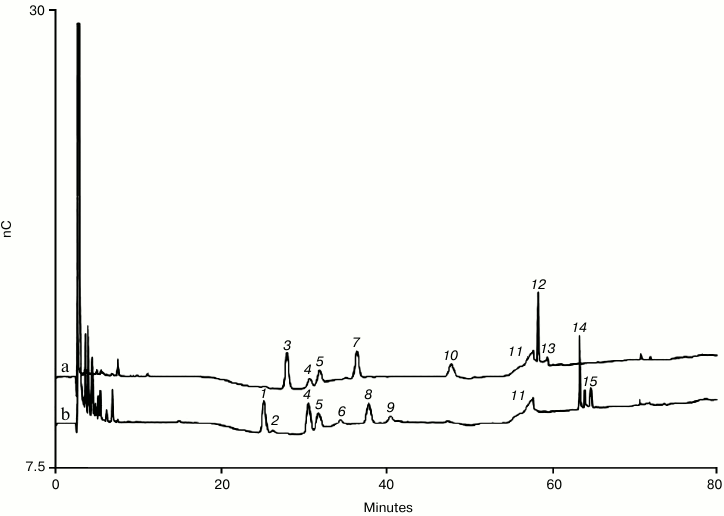
In recent years the biggest development in HPAE-PAD oligosaccharide separations is the use of sodium hydroxide concentrations less than 100 mM to achieve better resolution of neutral N-linked oligosaccharides, specifically those typically found on IgGs [52-54]. As the number of IgG-type monoclonal antibodies approved and in development for therapeutic use has rapidly increased, there has been more demand for higher resolution N-linked oligosaccharide separations, especially for the neutral oligosaccharides. In Adamo et al., comparison of oligosaccharide methods for the analysis of a fully human monoclonal antibody, they suggested that the resolution of HPAE-PAD was not sufficient [26]. Their work was done with the Dionex CarboPac PA100 column and the PA200 column run under typical conditions improves resolution for both neutral and charged oligosaccharides. The new lower hydroxide conditions offer even greater resolution for the IgG neutral oligosaccharides. This was first demonstrated by Grey et al. [52]. They showed that by reducing the sodium hydroxide concentration of the mobile phase to 55 mM and using a weak and shallow gradient of sodium acetate good resolution of ten neutral and eight charged N-linked oligosaccharides found on IgGs could be separated on a Dionex CarboPac PA200 column. This separation featured resolution superior to earlier separations that had a 100 mM sodium hydroxide mobile phase. The biggest improvement was for the neutral oligosaccharides. Most notable was the separation of the two monogalactosylated biantennary core-fucosylated isomers (G1F isomers). Working independently, Zheng et al. demonstrated the same improvement but with 50 mM sodium hydroxide and a different acetate gradient [53, 54]. The emphasis was again on separating IgG N-linked oligosaccharides. Figure 4 shows the PNGase F-released N-linked oligosaccharides from human serum polyclonal IgG and a set of oligosaccharide standards using the conditions of Zheng et al. [53, 54]. The method is longer than the method of Grey et al., but there is better resolution of the neutral oligosaccharides including the G1F isomers and GlcNAc2Mannose5 (Man5) oligosaccharide from the non-galactosylated core-fucosylated biantennary oligosaccharide (G0F).
Fig. 4. Neutral and charged N-linked oligosaccharides released from polyclonal human serum IgG by PNGase F. Note the resolution of the oligomannose species Man5 and Man6 from neighboring oligosaccharides. a) Neutral oligosaccharide standards; b) polyclonal IgG oligosaccharides. Peaks (Mab acronym): 1) G0F; 2) Man5; 3) G0; 4) G1F (1-6); 5) G1F (1-3); 6) G0bF (G0F with bisecting GlcNAc) and G1; 7) Man6; 8) G2F; 9) G1bF; 10) G2bF; 11) gradient artifact; 12) Man9; 13) Man9 (ManNAc epimer); 14) A1F (monosialylated, digalactosylated, core-fucosylated biantennary); 15) A1F (ManNAc epimer). Conditions: Dionex CarboPac PA 200 with guard at 30°C. Injection volume of 10 µl. The four-potential waveform and a conventional gold working electrode were used for PAD. Eluent flow rate of 0.35 ml/min. Gradient of 1.0-6.5 mM sodium acetate in 50 mM sodium hydroxide from 0-50 min, 6.5-25 mM sodium acetate in 50 mM sodium hydroxide from 50-51 min. Gradient of 25-130 mM sodium acetate in 50 mM sodium hydroxide from 51-80 min. Column wash of 500 mM sodium acetate in 100 mM sodium hydroxide at 0.45 ml/min from 81-91 min. Column equilibration at 1 mM sodium acetate in 50 mM sodium hydroxide from 92-100 min at 0.45 ml/min followed by 10 min of equilibration at 0.35 ml/min before injection. A 10% signal offset has been applied.
Another paper using HPAE-PAD for IgG oligosaccharide analysis used it to monitor the variation of human IgG oligosaccharides during IgG production with 105 stable cell lines [55]. They used a Dionex CarboPac PA1 separation (with a Dionex CarboPac PA100 guard) to monitor the six major neutral oligosaccharides (non-galactosylated biantennary (G0), G0F, G1F (two isomers not resolved), digalactosylated biantennary core-fucosylated (G2F), and Man5). This assay allowed them to determine that glycosylation was consistent with the same cell line using controlled conditions. They also noted conditions that led to undesirable Man5 glycosylation. Another application of HPAE-PAD to recombinant glycoprotein analysis showed the profiling the N-linked oligosaccharides from erythropoietin expressed in human ovarian carcinoma cell line SKOVA3 [56]. The authors used a Dionex CarboPac PA200 column to profile oligosaccharides both before and after neuraminidase treatment. They also used HPAE-PAD monosaccharide analysis with a PA20 column as part of their erythropoietin characterization.
HPAE-PAD oligosaccharide analysis for IgG N-linked oligosaccharides was compared to matrix-assisted laser desorption time-of-flight mass spectrometry (MALDI-TOF-MS) in the two of the previously discussed publications [26, 52]. Both publications concluded that the two techniques produced similar results, but in both cases HPAE-PAD was preferred. Adamo et al. [26] stated that MALDI has difficulties with low abundance oligosaccharides and sialylated oligosaccharides, while Grey et al. [52] stated that HPAE-PAD was more robust and the precision was significantly better than MALDI.
COUPLING HPAE-PAD TO MS
Growth in the popularity of MS for carbohydrate analysis has increased interest in coupling HPAE-PAD, or at least HPAE, to MS. HPAE offers resolution and selectivity not available in other LC techniques. The mobile phases used for HPAE-PAD are not MS-friendly, but even in the early days of HPAE-PAD this did not stop analysts from coupling the two techniques using eluent suppression technology from IC [57, 58], or the eventual commercialization of a product for changing the sodium hydroxide/sodium acetate mobile phase to a MS-friendly weak acetic acid solution, the Carbohydrate Membrane Desalter (CMD) [59]. In recent years there have been additional publications that demonstrate HPAE-PAD coupled to MS [60-64]. These publications have focused primarily on using smaller diameter Dionex CarboPac columns and IC suppressors designed for smaller diameter columns. The basic experimental parameters were outlined by Bruggink et al. [60]. They used a Dionex CarboPac PA200 column set with a 2 mm anion suppressor and then added lithium chloride prior to the MS. The PA200 column has a 3 mm diameter and therefore has a lower optimum flow rate compared to a 4 mm column. This makes it possible for an electrolytic suppressor to produce the needed hydronium ions to convert the sodium hydroxide/sodium acetate mobile phase to weak acetic acid, without the need to add acid across the regenerant chamber of the suppressor. The lithium chloride addition produces lithium-oligosaccharide adducts, which produce a greater signal in the MS compared to native oligosaccharides. They also compared sensitivity to PAD by splitting the flow after the column, and for this experiment PAD was 3-10 times more sensitive than a single quadrupole MS with positive mode electrospray (ES) sample introduction. A similar approach was taken but with a prototype capillary Dionex CarboPac PA200 column (0.381 × 250 mm), a prototype capillary suppressor, and a modification of the electrochemical cell to reduce the dead volume [61]. Instead of adding lithium chloride, sodium chloride was added prior to an ion-trap MS to generate sodium-oligosaccharide adducts. The prototype suppressor was able to tolerate higher sodium concentration than commercial suppressors. The principle authors applied the same approach to determining urinary oligosaccharides in order to detect lysosomal storage disorders [62]. With this approach they identified 54 oligosaccharides present in urine.
The final two examples do not involve glycoproteins, but do show other approaches for coupling HPAE to MS. In a more classical approach investigating cell surface bacterial polysaccharides, a 2 × 250 mm Dionex CarboPac PA1 column was paired with a 4 mm suppressor [63]. The authors chose the PA1 column rather than the PA200 column because smallest diameter of the PA200 column is 3 mm. They did not supply the suppressor effluent with a metal ion before the MS and noted variable amounts of hydronium and sodium adducts for each oligosaccharide. Boschker et al. took a different approach when they paired HPAE with isotope ratio MS (IRMS) to investigate the 13C/12C ratios of carbohydrates typically found in plants [64]. They used a CarboPac PA20 column (3 × 150 mm) to determine monosaccharides with 1 mM sodium hydroxide mobile phase and sugar acids with a 1 mM sodium hydroxide and 2 mM sodium nitrate mobile phase. In this work the column effluent undergoes a wet chemical oxidation under acidic conditions and the released carbon dioxide is transferred to the IRMS to measure the 13C/12C ratio.
HPAE-PAD TO FOLLOW GLYCOSIDASE AND GLYCOSYLTRANSFERASE
REACTIONS
HPAE-PAD has proven to be a powerful technique for following the progress of glycosidase and glycosyltransferase reactions. Because HPAE-PAD does not rely on a fluorescent or absorbent label for detection, the fate of the substrate(s) and the product(s) can be followed, allowing the analyst to judge the success of the reaction. These methods typically determine a monosaccharide or sialic acid and an oligosaccharide in a single sample analysis. Two recent glycosyltransferase examples of this application include screening for trans-sialidase activity [65] and characterization of Caenorhabditis elegans α-1,3-fucosyltransferases [66].
Trypanosomatids cannot synthesize their own sialic acid, so they express trans-sialidases that remove sialic acids from host glycoconjugates and transfer them to their own surface glycoconjugates that are then used to invade the host. Sartor et al. developed an HPAE-PAD assay for trypanosomal trans-sialidase activity that requires no radioactive labeling [65]. Their method used a Dionex CarboPac PA100 column with 100 mM NaOH/50 mM sodium acetate mobile phase to measure the disappearance of the substrate (sialyllactose) and the appearance of lactose and the sialylated acceptor products. The other substrate, the benzylated acceptor disaccharide, was not measured or identified in the chromatogram, though the chromatography suggests it eluted at or near the void. To determine whether five C. elegans genes with homology to known α-1,3-fucosyltransferases were in fact fucosyltransferases, the genes were expressed and then incubated with fucose and an acceptor substrate and the reaction followed by HPAE [66]. In this example the authors followed a Dionex CarboPac PA1 column separation by scintillation counting rather than PAD, but the method separated the acceptor substrate and the product so that both the appearance of one and the disappearance of the other could be monitored.
OTHER HPAE-PAD GLYCOPROTEIN ASSAYS
Two other common HPAE-PAD glycoprotein assays are the determination of the M-6-P content of a glycoprotein and the determination of monosaccharide alditols. The latter are typically present after a reductive step for oligosaccharide release and subsequent acid hydrolysis to produce monosaccharides. In this manner the monosaccharide that is responsible for linking the oligosaccharide to the protein can be determined. This is most commonly done for O-linked oligosaccharides due to the lack of an enzyme similar to PNGase F (for N-linked oligosaccharides) that is specific for the attachment. For O-linked oligosaccharides the preparation is reductive β-elimination followed by acid hydrolysis. This sample is then injected onto the HPAE-PAD system with a Dionex CarboPac MA1 column to determine which monosaccharide alditol is present. The MA1 column was designed for monosaccharide sugar alcohol separations. Two recent examples have used this assay to determine the linking monosaccharide of an O-linked oligosaccharide [67, 68]. In one study the HPAE-PAD assay was used to determine the level of O-mannosylation in two types of neural stem cell [67]. The other paper studied a trypanosomal α-N-acetylglucosaminyltransferase used to initiate the O-linked oligosaccharide on the parasite’s surface, which later becomes sialylated by the trans-sialidase [68]. In this work the authors used the HPAE-PAD assay to confirm that the enzyme transferred a GlcNAc residue to threonine.
M-6-P is a terminal monosaccharide on some N-linked oligosaccharides. It is important for targeting and recognition on a number of lysosomal glycoproteins and is present on recombinant therapeutic glycoproteins that are made to treat certain congenital disorders of glycosylation. As M-6-P is charged at neutral pH, its determination, like that of sialic acid, requires sodium acetate in the mobile phase. Two publications by scientists at Genzyme Corporation show that HPAE-PAD is effective for measuring the M-6-P content of a recombinant glycoprotein [69, 70]. In one publication the authors assayed the content of M-6-P as part of a study to determine if recombinant human glucocerebrosidase from two manufacturers could be distinguished [69]. After an acid hydrolysis typical of monosaccharide analysis, M-6-P was separated on a Dionex CarboPac PA10 column with a 10 min 170-400 mM acetate gradient in 100 mM sodium hydroxide. The same approach as used to determine M-6-P in a recombinant version of human glucocerebrosidase in which the number of mannose residues had been altered [70].
After 25 years HPAE-PAD continues to be a popular choice for determining the monosaccharide, sialic acid, M-6-P, and oligosaccharide contents of a glycoprotein. The method is sensitive without the need for sample derivatization. Recently we have witnessed improvements that contribute to method ruggedness, including longer lasting disposable electrodes, greater knowledge in their use, and greater knowledge concerning the preparation, storage, and use of HPAE-PAD mobile phases. Improved methods for separating neutral N-linked oligosaccharides have been reported. There also have been improvements in separation speed, mirroring a current trend in LC, and miniaturization with the introduction of a capillary HPAE-PAD system. In the future we expect to see HPAE-PAD have greater application in determining comparability of glycoprotein biopharmaceuticals and assaying the quality of biosimilars and biobetters.
REFERENCES
1.Rocklin, R. D., and Pohl, C. A. (1983) J. Liq.
Chromatogr., 6, 1577-1590.
2.Hardy, M. R., Townsend, R. R., and Lee, Y. C.
(1988) Anal. Biochem., 170, 54-62.
3.Chen, L-M., Yet, M-G., and Shao, M.-C. (1988)
FASEB J., 2, 2819-2824.
4.Spellman, M. W. (1990) Anal. Chem.,
62, 1714-1722.
5.Lee, Y. C. (1990) Anal. Biochem.,
189, 151-162.
6.Lee, Y. C. (1996) J. Chromatogr. A,
720, 137-149.
7.Cataldi, R. I., Campa, C., and De Benedette, G. E.
(2000) Fresenius J. Anal. Chem., 368, 739-758.
8.Rohrer, J. S. (2000) Anal. Biochem.,
283, 3-9.
9.Hardy, M. R., and Rohrer, J. S. (2007) in
Comprehensive Glycoscience (Kamerling, J. P., ed.) Vol. 2,
Elsevier, Netherlands, pp. 303-327.
10.Higgins, E. (2010) Glycoconj. J.,
27, 211-225.
11.Behan, J. L., and Smith, K. D. (2011) Biomed.
Chromatogr., 25, 39-46.
12.Rocklin, R. D., Clarke, A. P., and Weitzhandler,
M. (1998) Anal. Chem., 70, 1496-1501.
13.Rohrer, J. S. (1998) Thermo Fisher Scientific
Dionex Technical Note 21 (http://www.dionex.com/en-us/webdocs/5050-TN21_LPN034889-03.pdf).
14.Hurum, D., Christenson, T., Perati, P.,
Basumallick, L., and Rohrer, J. (2011) Thermo Fisher Scientific
Dionex Technical Note 110 (http://www.dionex.com/en-us/webdocs/111176-TN110-IC-Carb-HPAEPADdisposAuPTFE-12Oct2011-LPN2952-R2.pdf).
15.Rohrer, J. S. (2007) Thermo Fisher Scientific
Dionex Technical Note 71 (http://www.dionex.com/en-us/webdocs/58087-TN71-Eluent-Prep-HPAE-PAD-16Sept2009-LPN1932-02.pdf).
16.Kotnik, D., Novic, M., LaCourse, W. R., and
Pihlar, B. (2011) J. Electroanal. Chem., 683, 30-35.
17.Kotnik, D., Novic, M., Novic, M., Pihlar, B., and
Neubauer, D. (2012) Poster at the Int. Ion Chromatography Symp.,
Berlin, Germany.
18.Hurum, D. C., and Rohrer, J. S. (2011) Anal.
Biochem., 419, 67-69.
19.Cheng, J. (2012) Presentation at the 2012
Pittsburgh Conference, Orlando, FL USA.
20.Rohrer, J. S. (2012) in Applications of Ion
Chromatography in the Analysis of Pharmaceutical and Biological
Products (Bhattacharyya, L., and Rohrer, J. S., eds.) John Wiley
and Sons Inc., Hoboken, New Jersey, pp. 339-350.
21.Basumallick, L., and Rohrer, J. (2012) Thermo
Fisher Scientific Dionex Technical Note 40 (http://www.dionex.com/en-us/webdocs/5052-TN40-IC-Glycoprotein-Monosaccharide-23May2012-LPN1632-.pdf).
22.Zhang, Z., Khan, N. M., Nunez, K. M., Chess, E.
K., and Szabo, C. M. (2012) Anal. Chem., 84,
4104-4110.
23.Herrmann, A., Konig, S., Lechtenberg, M.,
Sehlbach, M., Vakhrushev, S. Y., Peter-Katalinic, J., and Hensel, A.
(2012) Glycobiology, 22, 1424-1439.
24.Bergfeld, A. K., Pearce, O. M. T., Diaz, S. L.,
Pham, T., and Varki, A. (2012) J. Biol. Chem., 287,
28865-28881.
25.Everest-Dass, A. V., Jin, D., Thaysen-Andersen,
M., Nevalainen, H., Kolarich, D., and Packer, N. H. (2012)
Glycobiology, 22, 1465-1479.
26.Adamo, M., Qui, D., Dick, L. W., Jr., Zeng, M.,
Lee, A-H., and Cheng, K-C. (2009) J. Pharm. Biomed. Anal.,
49, 181-192.
27.Machado, E., Kandzia, S., Carilho, R., Altevogt,
P., Conradt, H. S., and Costa, J. (2011) Glycobiology,
21, 376-386.
28.Yu, T., Guo, C., Wang, J., Hao, P., Sui, S.,
Chen, X., Zhang, R., Wang, P., Yu, G., Zhang, L., Dai, Y., and Li, N.
(2011) Glycobiology, 21, 206-224.
29.Clarke, A. J., Sarabia, V., Keenleyside, W.,
MacLachlan, P. R., and Whitfield, C. (1991) Anal. Biochem.,
199, 68-74.
30.Harazono, A., Kobayashi, T., Kawasaki, N., Itoh,
S., Tada, M., Hashii, N., Ishii, A., Arato, T., Yanagihara, S., Yagi,
Y., Koga, A., Tsuda, Y., Kimura, M., Sakita, M., Kitamura, S.,
Yamaguchi, H., Mimura, H., Murata, Y., Hamazume, Y., Sato, T., Natsuka,
S., Kakehi, K., Kinoshita, M., Watanabe, S., and Yamaguchi, T. (2011)
Biologicals, 39, 171-180.
31.Padler-Karavani, V., Yu, H., Cao, H., Chokhawala,
H., Karp, F., Varki, N., Chen, X., and Varki, A. (2008)
Glycobiology, 18, 818-830.
32.Raju, T. S., Briggs, J. B., Borge, S. M., and
Jones, A. J. (2000) Glycobiology, 10, 477-486.
33.Baker, K. N., Rendall, M. H., Hills, A. E.,
Hoare, M., Freedman, R. B., and James, D. C. (2001) Biotechnol.
Bioeng., 73, 188-202.
34.Manzi, A. E., Diaz, S., and Varki, A. (1990)
Anal. Biochem., 188, 20-32.
35.Hara, S., Yamaguchi, M., Takemori, Y., Nakamura,
M., and Ohkura, Y. (1986) J. Chromatogr. B, 377,
111-119.
36.Hara, S., Yamaguchi, M., Takemori, Y., Furuhata,
K., Ogura, H., and Nakamura, M. (1989) Anal. Biochem.,
179, 162-166.
37.Hurum, D. C., and Rohrer, J. S. (2012) Gen.
Eng. Biotech. News, 32, 18-19.
38.Suriano, R., Ghosh, S. K., Chaudhuri, D.,
Mittelman, A., Banerjee, A., and Tiwari, R. K. (2009)
Glycobiology, 19, 1427-1435.
39.Suzuki, N., Su, T-H., Wu, S-W., Yamaloto, K.,
Khoo, K-W., and Lee, Y. C. (2009) Glycobiology, 19,
693-706.
40.Zhang, Y., Inoue, Y., Inoue, S., and Lee, Y. C.
(1997) Anal. Biochem., 250, 245-251.
41.Inoue, Y., and Inoue, S. (2001)
Glycobiology, 11, 759-767.
42.Lin, S.-L., Inoue, Y., and Inoue, S. (1999)
Glycobiology, 9, 807-814.
43.Vionnet, J., and Vann, W. F. (2007)
Glycobiology, 17, 735-743.
44.Nakata, D., and Troy, F. A., 2nd (2005) J.
Biol. Chem., 280, 38305-38316.
45.Wong, K. S., and Jane, J. (1995) J. Liq.
Chromatogr., 18, 63-80.
46.Basumallick, L., and Rohrer, J. (2012) Thermo
Fisher Scientific Dionex Application Note 1013 (http://www.dionex.com/en-us/webdocs/113699-AN1013-IC-Polysialic-Acid-Polymers-08Aug2012-AN70124_E-R2.pdf).
47.Chen, L-M., Yet, M-G., and Shao, M.-C.
(1988) FASEB J., 2, 2819-2824.
48.Townsend, R. R., Hardy, M. R., Hindsgaul, O., and
Lee, Y. C. (1988) Anal. Biochem., 174, 459-470.
49.Rohrer, J. S. (1995) Glycobiology,
5, 359-360.
50.Perati, P., and Rohrer, J. (2010) Thermo
Fisher Scientific Dionex Application Update 176 (http://www.dionex.com/en-us/webdocs/88108-AU176-IC-PNGaseF-HPAEPAD-07Sep2010-LPN2576-R2.pdf).
51.Hayase, T., Sheykhanazari, M., Bhavanandan, V.
P., Savage, A. V., and Lee, Y. C. (1993) Anal. Biochem.,
211, 72-80.
52.Grey, C., Edebrink, P., Krook, M., and Jacobssin,
S. P. (2009) J. Chromatogr. B, 877, 1827-1832.
53.Zheng, T., Rohrer, J., and Rao, S. (2010)
Genet. Eng. News, 30, 42-43.
54.Zheng, T., Rao, S., Rohrer, J., and Pohl, C.
(2012) in Antibody-Mediated Drug Delivery Systems (Pathak, Y.,
and Benita, S., eds.) John Wiley and Sons Inc., Hoboken, New Jersey,
pp. 145-167.
55.Van Berkel, P. H. C., Gerritsen, J., Perdok, G.,
Valbjorn, J., Vink, T., van de Winkel, J. G. J., and Parren, P. W. H.
I. (2009) Biotechnol. Prog., 25, 244-251.
56.Machado, E., Kandzia, S., Carilho, R., Altevogt,
P., Conradt, H. S., and Costa, J. (2011) Glycobiology,
21, 376-386.
57.Simpson, R. C., Fenselau, C. C., Hardy, M. R.,
Townsend, R. R., and Lee, Y. C. (1990) Anal. Chem., 62,
248-252.
58.Conboy, J. J., and Henion, J. (1992) Biol.
Mass Spectr., 21, 397-407.
59.Thayer, J., Rohrer, J. S., Avdalovic, N., and
Gearing, R. P. (1998) Anal. Biochem., 256, 207-216.
60.Bruggink, C., Maurer, R., Herrman, H., Cavalli,
S., and Hoefler, F. (2005) J. Chromatogr. A, 1085,
104-109.
61.Bruggink, C., Wuhrer, M., Koeleman, C. A. M.,
Barreto, V., Liu, Y., Pohl, C., Ingendoh, A., Hokke, C. H., and
Deelder, A. (2005) J. Chromatogr. B, 829, 136-143.
62.Bruggink, C., Poorthuis, B. J. H. M., Deelder, A.
M., and Wuhrer, M. (2012) Anal. Bioanal. Chem., 403,
1671-1683.
63.Chataigne, G., Couderc, F., and Poinsot, V.
(2008) J. Chromatogr. A, 1185, 241-250.
64.Boschker, H. T. S., Moerdijk-Peertvliet, T. C.
W., van Breugel, P., Houtekamer, M., and Middelburg, J. J. (2008)
Rapid Commun. Mass Spectrom., 22, 3902-3908.
65.Sartor, P. A., Agusti, R., Leguizamon, M. S.,
Campetella, O., and de Lederkremer, R. M. (2010) Glycobiology,
20, 982-990.
66.Nguyen, K., van Die, I., Grundahl, K. M., Kawar,
Z. S., and Cummings, R. D. (2007) Glycobiology,
17, 586-599.
67.Zhang, P., and Hu, H. (2012) Glycobiology,
22, 235-247.
68.Heise, N., Singh, D., van der Wei, H., Sassi, S.
O., Johnson, J. M., Feasly, C. L., Koeller, C. M., Previato, J. O.,
Mendoca-Previato, L., and West, C. M. (2009) Glycobiology,
19, 918-933.
69.Lee, K., Jin, X., Zhang, K., Copertino, L.,
Andrews, L., Baker-Malcolm, J., Geagan, L., Qiu, H., Seiger, K.,
Barngrover, D., McPherson, J. M., and Edmunds, T. (2003)
Glycobiology, 13, 305-313.
70.Van Patten, S. M., Hughes, H., Huff, M. R.,
Piepenhagen, P. A., Waire, J., Qiu, H., Ganesa, C., Reczek, D., Ward,
P. V., Kutzko, J. P., and Edmunds, T. (2007) Glycobiology,
17, 467-478.