REVIEW: Targeting Heterochromatin Formation to Transposable Elements in Drosophila: Potential Roles of the piRNA System
M. Sentmanat, S. H. Wang, and S. C. R. Elgin*
Department of Biology, Washington University, Campus Box 1137, One Brookings Drive, St. Louis, MO 63130-4899, USA; fax: (314) 935-4432; E-mail: selgin@biology.wustl.edu* To whom correspondence should be addressed.
Received January 21, 2013; Revision received February 22, 2013
Successful heterochromatin formation is critical for genome stability in eukaryotes, both to maintain structures needed for mitosis and meiosis and to silence potentially harmful transposable elements. Conversely, inappropriate heterochromatin assembly can lead to inappropriate silencing and other deleterious effects. Hence targeting heterochromatin assembly to appropriate regions of the genome is of utmost importance. Here we focus on heterochromatin assembly in Drosophila melanogaster, the model organism in which variegation, or cell-to-cell variable gene expression resulting from heterochromatin formation, was first described. In particular, we review the potential role of transposable elements as genetic determinants of the chromatin state and examine how small RNA pathways may participate in the process of targeted heterochromatin formation.
KEY WORDS: heterochromatin, genome stability, transposable elementsDOI: 10.1134/S0006297913060023
Heterochromatin is classically defined as densely packaged, peripherally localized chromatin within the cell nucleus. The repetitious sequence content of eukaryotic genomes was initially recognized by quantitative DNA reassociation analysis (generating C0t curves) using principles pioneered by Roy Britten and colleagues [1]. These studies, coupled with in situ hybridization techniques, revealed the abundance and arrangement of repetitive DNA, and ultimately led to the understanding that heterochromatin is enriched in satellite and transposable element sequences of varying copy number. Although understanding genome organization within the more complex, gene-rich euchromatic compartment took precedence for many years, heterochromatin has recently received more attention with the development of improved sequencing technologies and bioinformatics strategies. These tools have enabled improved assemblies and significant annotation of the repetitious sequences present in heterochromatin in many instances.
In a complex organism, those DNA sequences packaged as heterochromatin in all cell types are referred to as “constitutive heterochromatin”, while DNA packaged as “facultative heterochromatin” (important for developmentally controlled genes) occurs in this form in some cells but not others [2]. Along a chromosome, constitutive heterochromatin is usually found at pericentric repeats and telomeres, while facultative heterochromatin can be interspersed along the chromosome arms.
In the fruit fly, Drosophila melanogaster, heterochromatin becomes visible during nuclear cycles 11-14 of embryogenesis (3-4 h), reflecting a pattern of post-translational histone modifications that persists throughout development [3-6]. Most constitutive heterochromatic sites are enriched for histone H3 lysine 9 di- and tri-methylation (H3K9me2/3), the chromo-domain protein HP1a, and the histone methyltransferase (HMT) SU(VAR)3-9, whose catalytic SET domain delivers the H3K9me2/3 mark. In plants, mammals, and some other organisms constitutive heterochromatin is also associated with DNA methylation at CpG or CpNpG sequences. Two additional SET domain proteins have been identified in Drosophila, dSETDB1 (encoded by egg) and G9a; both are also H3K9 histone methyltransferases, although SU(VAR)3-9 and dSETDB1 appear to have the dominant roles [7].
Errors in establishing patterns of constitutive heterochromatin can lead to genome instability through a number of mechanisms. A few examples will illustrate the possibilities. Functional studies that deplete SU(VAR)3-9 homologues in mammals or in yeast have shown that the protein is important for kinetochore assembly and chromosome segregation [8, 9], while a loss of HP1a in Drosophila results in telomere fusions [10]. Another form of instability resulting from the loss of heterochromatin (HP1a in particular) in the female germline is the activation of transposable elements [11], which can lead to double strand breaks as well as the mutagenizing effects of TE insertions within protein-coding DNA. In contrast, gain-of-function mutations in Su(var)3-9 cause heterochromatin expansion, leading to female sterility in Drosophila [12].
The appropriate targeting of facultative heterochromatin is equally important, as this system plays a key role in cell identity. Well-studied examples of this phenomenon include X-inactivation in mammals and the heterochromatin formation observed during bird erythropoiesis, when the majority of the genome is silenced. A parallel, chromatin-based mechanism to achieve developmentally controlled silencing programs is associated with Polycomb group (PcG) proteins, which accomplish targeted gene silencing using an H3K27me3-based mechanism. However, this review will primarily focus on mechanisms associated with HP1a targeting. Our discussion of “heterochromatin” will be in reference to constitutive, HP1a-dependent heterochromatin unless otherwise specified. In Drosophila, heterochromatin domains include the pericentric heterochromatin flanking all centromeres, the Telomere Associated Sequences (TAS) adjacent to the HeT-A and TART elements that make up the telomeres, and the bulk of the small fourth chromosome (Muller F element) [13].
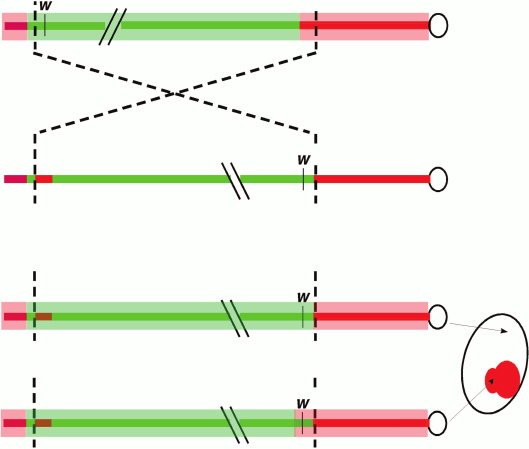
A classic and commonly used assay to dissect the cis- and trans-acting factors involved in heterochromatic silencing in Drosophila (among other systems) involves position-effect variegation (PEV), first observed in Drosophila melanogaster by Herman Muller in 1930. Following X-ray mutagenesis, Muller recovered fly lines (termed wm, white mottled) that had a variegating, red-interspersed-with-white pattern across the fly eye, rather than the normal solid red (or completely white, if mutant) appearance [14]. The phenotype is caused by an inversion that places the euchromatic white gene (which has a transport function required cell-autonomously for red eye pigmentation) proximal to repeat-rich pericentric heterochromatin. This rearrangement results in the stochastic “spreading” of heterochromatin components along the now proximally located euchromatic region that includes white (Fig. 1; see color insert). Dominant loss-of-function mutations in the genes coding for heterochromatin components suppress the PEV phenotype such that the expression of white is restored in a greater fraction of the cells; in the case of structural components such as Su(var)3-9 or HP1a, over-expression of these same genes can have the opposite effect. At the chromatin level, PEV is characterized as resulting in a relatively regular nucleosome array [15, 16], with a loss of accessibility (loss of the nucleosome-free region) at the transcription start site [17]. Biochemical analysis across the inverted breakpoint of one strain from the wm collection, wm4, shows variable enrichment of heterochromatin proteins along a 30 kb stretch or more, suggesting some sequence determinants might be more susceptible than others to ectopic heterochromatin assembly [6, 18]. Together, these observations suggest that heterochromatin assembly can spread in cis, provided a permissible sequence context and sufficient trans-acting molecules. These properties have made PEV a widely used model with which to dissect the cis- and trans-acting factors responsible for heterochromatin assembly.
Fig. 1. Position-effect variegation in Drosophila melanogaster. Schematic depiction of the chromosomal inversion (X chromosome) generating the white-mottled four line (ln(1)wm4) recovered by Muller [14]. The inversion places the euchromatic white gene (coding for a transporter protein required for red eye pigment deposition) adjacent to pericentric heterochromatin. The light red bar represents heterochromatin, while the light green bar represents a euchromatic chromatin state. The chromosomal inversion results in gene silencing in some cells (white) due to heterochromatin spreading over the w gene in a stochastic process that allows expression to be maintained in other cells (red).
Localized distribution of heterochromatin in the genome implies an underlying sequence determinant for its targeted formation. The immediate question following this observation asks for a mechanistic explanation for the targeting process. In recent years, work from plants and the fission yeast Schizosaccharomyces pombe have established that many of the heterochromatin components in these systems are associated with RNA-directed transcriptional silencing [19]. In these systems, RNA transcribed from repetitive, heterochromatic loci is processed into small RNAs that ultimately become the targeting signal for heterochromatin assembly. Such a targeting mechanism, in which the targeting signal is generated from heterochromatin (the target) itself, allows plasticity. This is thought to be necessary to accommodate imprecision during DNA replication or following new TE invasions that change the system’s DNA composition, while ensuring functional precision (faithful heterochromatin assembly).
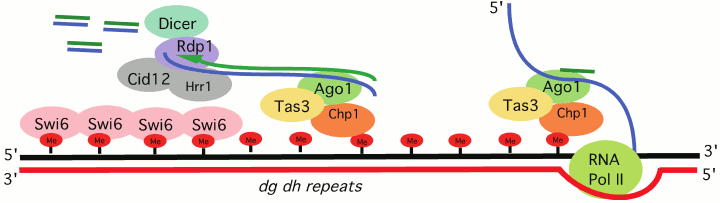
Schizosaccharomyces pombe, a model organism for which RNA-directed transcriptional silencing is well described, serves as an excellent example of how cis-sequence determinants can work with trans-acting factors to assemble heterochromatin at repeats, generally remnants of transposable elements (TEs). Targeting of the HP1 family protein Swi6 and the H3K9 HMT Clr4 depends on the processing of RNA Pol II transcripts generated from heterochromatic loci. The RNAi-induced transcriptional silencing complex (RITS) contains the chromo domain protein Chp1, as well as the RNAi component Ago1, which binds small RNAs generated from target sites (e.g. dg/dh repeats, the cis-acting signals) located in pericentric heterochromatin [20] (Fig. 2; see color insert). Mutations in the slicer activity of Ago1 result in a loss of silencing for reporters located at heterochromatic sites [21], indicating that Ago1 is an essential trans-acting factor for heterochromatin assembly in S. pombe, and that processing the long RNA cis-acting signal from dg/dh repeats into smaller fragments is required. The small RNAs generated by Ago1 provide a primer for RNA-dependent RNA polymerase, which generates additional dsRNA products to be processed by Dicer1. The amplified small RNA is used to achieve additional RITS targeting. However, whether such a mechanism also operates in metazoan systems remains an open question.
Fig. 2. RNAi-transcriptional silencing in S. pombe. Transcripts of dg/dh pericentric repeats are targeted by the RNA-induced transcriptional silencing complex (RITS). RITS consists of the chromo domain protein Chp1, Tas3 and the small RNA-associated protein Ago1. A second complex, the RNA-directed RNA polymerase complex (RDRC) consists of the RNA-directed RNA polymerase 1 (Rdp1), a putative polyA polymerase Cid12, and helicase Hrr1. RDRC is recruited to dh/dg repeats by a physical interaction with RITS to synthesize double stranded RNAs; these are targeted by Dicer to make additional siRNAs to reinforce RITS recruitment. See Slotkin and Martiensson [19] and Kloc and Martiesson [20] for review of the supporting evidence.
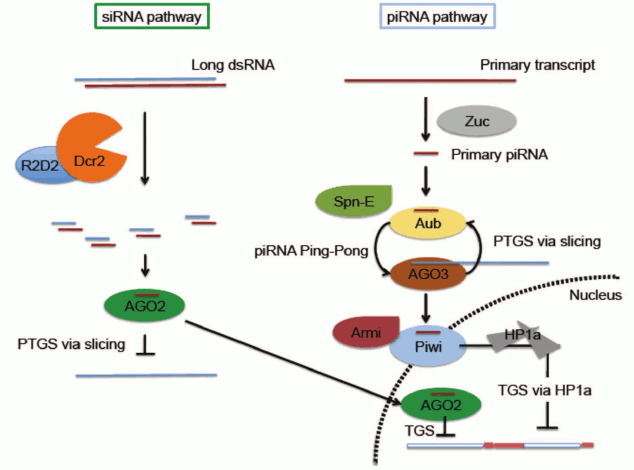
It is important to distinguish between RNA-based silencing systems (here referred to as RNA interference, or RNAi) which are associated with Post-Transcriptional Gene Silencing (PTGS), generally accomplished by mRNA degradation, and those implicated in chromatin-based silencing (Transcriptional Gene Silencing, TGS) (Fig. 3; see color insert). In Drosophila, RNAi mechanisms primarily require two families of proteins: Argonaute proteins (AGO1, AGO2, AGO3, Piwi, and Aub) and RNase III helicases (DICER-1 and DICER-2). The Argonaute family comprises two clades, the more ubiquitously expressed AGO clade (AGO1 and AGO2) and the primarily germ line PIWI clade (AGO3, Aub and Piwi). AGO1 and DICER-1 work together to generate microRNAs, derived from imperfect stem-loop transcripts that participate in translational repression or degradation of mRNA target transcripts (reviewed in [22, 23]). Short-interfering RNA (siRNA) is derived from exogenous or endogenous dsRNA processed by AGO2 and DICER-2 [24, 25]. Although siRNA is generally considered to function through a post-transcriptional silencing mechanism in the cytoplasm, both AGO2 and DICER-2 have recently been documented to associate with elongating RNA Pol II and with insulator elements in some somatic nuclei, suggesting a role in helping to define transcriptional domains (Fig. 3) [26, 27]. PIWI-interacting RNAs, piRNAs, are derived from master clusters of transposon sequences [28, 29]. Both transcriptional and post-transcriptional silencing mechanisms have been reported for transposon silencing by piRNA (Fig. 3) [11, 28-30].
Fig. 3. Small RNA-mediated silencing in D. melanogaster. Only siRNA and piRNA pathways are illustrated. Note that while the piRNA pathway is most active in the reproductive system, the siRNA pathway has a broader distribution. Both pathways have been implicated in a small RNA-mediated heterochromatin targeting process. In the siRNA pathway, small RNA generated by Dcr2 is loaded on to AGO2 RISC. The AGO2 complex can suppress expression through either of two mechanisms: slicing target mRNA in the cytoplasm through a well-characterized post-transcriptional gene silencing (PTGS) mechanism, or utilizing a yet to be characterized chromatin-based transcriptional silencing mechanism (TGS) in the nucleus. In the piRNA pathway, primary piRNA generated by a process involving Zuc is fed into the Ping-Pong cycle involving Aub and AGO3 to generate secondary piRNA. This process is proposed to function simultaneously to amplify antisense secondary piRNA and suppress transposon expression via slicing. Spn-E is required for secondary piRNA production, although the detailed mechanism is unclear. Secondary piRNAs loaded onto Piwi, likely by Armitage, allow nuclear localization of Piwi and downstream recruitment of HP1a to induce heterochromatin silencing of a subset of TEs. See text for pertinent citations; reviewed in Huisinga and Elgin [22] and Dai et al. [23].
In spite of their hazardous potential, transposons are among the genome’s most important tools, providing the host with new material for cis-acting regulatory features, protein-coding capacity, and perhaps other uses [31]. The paradox of a necessity to maintain genome integrity, while also achieving diversity within a population, has been empirically linked to RNAi-mediated transposon regulation [32]. Thus, RNAi systems in Drosophila, particularly the piRNA pathway, can be thought of as a regulatory switchboard, with a primary task of TE repression, but with many other potential functions. Whether TE repression driven by the piRNA system occurs at the chromatin level is the topic of this review.
We recognize that a multiplicity of targeting mechanisms has been observed for sites with similar chromatin marks in systems that possess well-documented RNAi-mediated transcriptional silencing, such as S. pombe and Neurospora crassa. Perhaps this is not surprising, given the importance of maintaining silencing of TEs. In S. pombe, all of the major heterochromatic domains are targeted for silencing by proteins that recognize specific DNA sequences in addition to the RNA-based mechanisms for targeting heterochromatin formation [19]. In some cases, these DNA recognition systems appear to have evolved from the ability of transposases to recognize their own TEs. For example, CENP-B, apparently derived from a transposase, recruits histone deacetylases to silence Tf2 retrotransposons [33]. TE elements have been used for different recognition events as well, with transposase-derived chromatin modifiers documented in Drosophila as well as in mammals. Specifically, BEAF-32, derived from the hAT transposase, is a chromatin insulator protein that binds the scs chromatin boundary element [34].
Mechanisms similar to those documented in the fungi may have evolved in Drosophila to specifically target heterochromatin factors to TEs. Such targeting might occur through protein recognition events, RNAi-based events, or both. One potential mechanism for silencing utilizes the AT-hook, DNA binding protein D1, which has been found to localize to centromeric heterochromatin; mutation of the locus suppresses PEV [35]. Genome-wide mapping analysis has revealed that D1 overlaps with several combinatorial categories of chromatin marks that can be generally ascribed to silent chromatin, in particular, HP1a-dependent heterochromatin and PcG-associated silencing [36]. Indeed, D1 over-expression induces pairing among its targets in polytene chromosomes, suggesting a role in higher order chromatin organization [37].
In this review our aim is to synthesize the evidence for RNAi-induced heterochromatin targeting in Drosophila. In particular, we will focus on repetitious elements acting as cis-acting signals for trans-acting chromosomal proteins. We begin by discussing established examples of cis-acting silencing signals, which serve as precedents for sequence-specific targeting of chromatin modifying enzymes. Although there are several empirically established examples of TE-derived signals involved in transcriptional activation [31], we will focus on the potential of TE remnants to act as silencing signals to be used by RNAi pathway component effectors to direct heterochromatin formation.
cis-ACTING ELEMENTS
Transposable elements and their remnants comprise 22% of the Drosophila melanogaster genome [38] and roughly half of the human genome [39]; they reside primarily in repressive, heterochromatic regions. The non-random distribution and evolutionary conservation of heterochromatic TE clusters suggests that this residence is functionally required. TEs inherently possess regulatory signals or may acquire them de novo, and this, combined with their capacity for insertional mutagenesis, more often than not results in a substantial blow to the system during mobilization events. Thus, repression of these elements takes precedence under most circumstances. Indeed, the flux of TEs in the genome requires a rapidly adaptive targeted silencing system for survival. In flies, deep sequencing of small RNA libraries has shown that TEs are expressed, and become targets for small RNA-mediated silencing [25, 29]. Although small RNA pathways are better known for their function in a post-transcriptional capacity, evidence for chromatin-based silencing in Drosophila has been reported [22]. Both piRNA and chromatin structural proteins (and/or their mRNAs) are present in the early embryo (0-6 h) [40] during the early stages when heterochromatin formation occurs [6]. Thus, piRNA sequence elements could help define some heterochromatic domains, specifically those with the subset of repeats represented in the piRNA repertoire.
Chromosome organization per se suggests that TEs could be targets for silencing, as most Drosophila PEV reporters showing the variegating phenotype typical of heterochromatic domains map to repeat-rich regions of the genome, found to be heterochromatic by other criteria as well. Studies aimed at mapping heterochromatic domains on the repeat-rich fourth chromosome of Drosophila melanogaster using an hsp70-white reporter have shown that 20-60 kb deletions or duplications of flanking DNA can be sufficient to shift a red eye phenotype to variegating phenotype (and vice versa), indicating local variation in chromatin packaging at that scale [41]. Genomic analysis of these variegating lines found a correlation between the presence of the DNA transposable element remnant 1360 and silencing of an inserted reporter.
Follow-up experiments using FLP-mediated excision to control the presence or absence of a 1360 remnant upstream of an hsp70-white reporter revealed that 1360 is indeed capable of supporting heterochromatin formation (as shown by increased silencing) in a repeat-rich area of the genome (~30% repeats) [42]. Further, 1360 has been found to be sufficient to induce ectopic, HP1a-dependent heterochromatin assembly in a domain of annotated euchromatin [13] that is close to a heterochromatic mass near the base of chromosome arm 2L [43]. Variegation in both contexts, repeat-rich and euchromatic, is suppressed in su(var)205 and piwi mutants, suggesting that RNAi components may facilitate the HP1a targeting event. RNAi-based heterochromatin targeting in both S. pombe and plants is thought to act through RNA–RNA recognition events. A similar mechanism is suggested by the finding that read-through transcripts of the P element insert containing 1360 are present in 0-10 h embryos in the above case, providing a plausible RNA targeting signal. Further, deletion of piRNA hotspots within the 1360 element (having homology to piRNA sequences abundantly found in Drosophila) compromises 1360-induced PEV. These results directly implicate the piRNA pathway in 1360-induced silencing at this 2L site [43].
Given that the piRNA pathway generates the most complex small RNA population in the fly – needed to target hundreds of TEs – it is likely that additional TEs will behave similarly at a 1360-sensitive site. This has been confirmed using the retro-element Invader4, which recapitulated 1360-sensitive PEV. Deletion of sites complementary to abundant piRNAs again compromised the effect [43]. The combined results support a model in which a small RNA targeting event utilizing read-through (or other) transcripts for RNA–RNA recognition participates in the HP1a-dependent assembly of heterochromatin at this site.
Reporter insertion sites that exhibit 1360-sensitive heterochromatin formation appear to be limited to sites proximal to pericentric repeats, or in some cases within mapped pericentric regions. As noted above, the presence of a single copy of 1360 within the euchromatic arms (which have a low repeat density, <10%) is insufficient to trigger a variegating phenotype. However, 1360 is sufficient to promote heterochromatin over the hsp70-w reporter when inserted within or proximal to HP1a-dense regions. These observations suggest that piRNA pathway target sites are likely HP1a-target sites (as 1360-sensitive silencing is an HP1a-dependent phenomenon), but limited to a subset of domains. A need for a reporter insertion site that results in read-through transcription of the 1360 element could also limit the set of reporter loci demonstrating this form of targeted silencing. Whether read-through transcription of the 1360 insert is necessary to promote local HP1a accumulation will require further investigation.
The repertoire of possible cis targets for the piRNA system is considerable, but only some TEs have been associated with chromatin-based changes in piwi mutants. Knockdown of germline Piwi has been shown to compromise HP1a deposition at promoters of HeT-A, Blood, Bari1, and Invader1, among a small set of TEs tested in Drosophila melanogaster ovaries [11, 30]. The high copy number of most TEs and the lack of a complete genome assembly in heterochromatic regions have hampered efforts to identify specific targets. Genomic context at a larger scale (at least over 10 kb, and perhaps much more) may prove to be an important factor in identifying additional cis-acting determinants of heterochromatin formation. Recent genome-wide efforts utilizing piwi knockdowns have demonstrated considerable reductions in H3K9me3 over TEs in ovarian somatic and germ cells, reinforcing the notion of piRNA-mediated TGS [44, 45]. Ultimately, it will be of interest to identify if and how this pathway participates in re-establishing heterochromatin at repetitive elements during early embryogenesis.
trans-ACTING MACHINERIES FOR SMALL RNA TARGETING
A small RNA-mediated targeting model [22] provides a mechanism of remarkable simplicity and adaptability, utilizing sequence information encoded in small RNAs to achieve highly specific target site recognition. The coding capacity of a 20-30 nucleotide long RNA allows a wide range of potential target sequences to be identified.
Recently, both endo-siRNA and piRNA (small RNA populations defined by size and by the RNAi machinery that generates them) have been implicated in heterochromatin targeting [11, 46]. In both cases, however, many critical questions remain to be answered; in particular, whether changes observed at the chromatin level in endo-siRNA and piRNA pathway mutants are a result of direct or indirect effects remains an open question. The potential redundancy and/or cross talk between the two pathways further confound our ability to interpret results from genetic perturbation experiments. We will discuss first evidence supporting a role for the endo-siRNAs, and second that supporting a role for piRNAs in heterochromatin formation.
In flies, endo-siRNAs have been identified and characterized by sequencing the small RNAs associated with AGO2 and/or those small RNAs bearing 2′O-methylation at their 3′ terminus, working from somatic cells [24, 25]. These small RNAs are enriched in transposon and intergenic sequences, and their production is strongly impacted by mutations disrupting the siRNA pathway. Interestingly, the involvement of these small RNAs in heterochromatin targeting had been suggested even before their identification. It had already been shown, mostly by cytological assays, that mutations in ago2 result in defects in centromeric heterochromatin formation [47]. Given the well-established role of AGO2 in a small RNA-based silencing mechanism, and a potential parallel mechanism in S. pombe (describing small RNA targeting of heterochromatin formation; see Fig. 2), these observations pointed to the enticing possibility of siRNA targeting for heterochromatin formation in flies. The model is particularly attractive when taken together with the observed enrichment of transposon sequences in endo-siRNAs. A test of this model, looking at perturbation of heterochromatin formation under conditions where endo-siRNA production is disrupted, provides encouraging support. It has been shown that both the sequestering of endo-siRNA by viral proteins, and introduction of mutations impacting endo-siRNA production, result in dominant suppression of a stubble PEV reporter, SbV (a translocation of Sb to the 2R pericentric region) [46]. Further, trans-heterozygous mutations in components needed for endo-siRNA production, such as ago2 and dcr2, result in strong suppression of wm4 PEV. These genetic results are supported by cytological studies, showing that endo-siRNA component mutations have an impact on localization of HP1a and H3K9me2/3, observed by immunofluorescent staining of polytene chromosomes. While a clear impact on the distribution of these marks is observed for a majority of samples, pericentric heterochromatin remains visibly stained in all cases. These results argue that while the endo-siRNA pathway does play a role in determining the localization pattern of heterochromatin, the specific targeting of heterochromatin formation at the pericentric region is either independent of the endo-siRNA pathway, or (more likely) the role of endo-siRNA in this process is redundant with other mechanisms. Note that while dominant mutations in these same genes have little or no impact on PEV at some reporter sites [42], inserts with reporter transgenes in other regions of the genome show significant suppression. It appears that involvement of endo-siRNA in targeting heterochromatin formation is likely to be context dependent.
One conundrum of the endo-siRNA targeting model for heterochromatin formation is the fact that the siRNA pathway is better known for its function in post-transcriptional silencing in the cytoplasm. There is therefore little work that might help to draw a direct mechanistic link to a nuclear targeting process for heterochromatin formation. However, two recent studies have independently demonstrated that AGO2 protein can be chromatin-bound [26, 27], albeit in larval or adult tissues. While a direct mechanistic link is still missing (i.e. it remains unclear what is recruited by AGO2 to initiate heterochromatin formation), the results to date argue that the endo-siRNA pathway is involved in heterochromatin assembly and/or maintenance, in at least some regions of the heterochromatin-packaged genome.
As noted above, there are five genes encoding Argonaute proteins in the fly genome: piwi, aub (aubergine), ago1 (argonaute 1), ago2, and ago3. Of these, the one protein product conspicuously localized in the nucleus in both germline and the ovarian soma is Piwi, of the PIWI sub-family of Argonaute proteins [29, 48]. Piwi was first described to be involved in the maintenance of germline stem cells, functioning in the stem cell niche of the ovarian soma [49], but it is clearly a protein with many roles. Because of its nuclear localization, Piwi is regarded as the primary candidate to be involved in heterochromatin targeting in Drosophila. The Piwi protein associates with piRNAs, 26-30 nt small RNAs that are enriched for TE sequences. The Drosophila melanogaster genome contains many “piRNA loci”, transcribed regions that can be several kilobases long, containing a diverse mixture of TE sequences. The piRNA loci are postulated to be discrete regulatory loci, proposed to generate a transposon defense system via processing of such transcripts to generate piRNAs. piRNA loci and endo-siRNA clusters predominantly map to the edges of pericentric and telomeric regions—which are highly enriched in transposable element remnants and other repetitious sequences. Thus Piwi has the necessary attributes to play a role in heterochromatin formation: a nuclear location, and association with a pool of small RNAs (piRNAs) rich in TE sequences commonly found in heterochromatic regions. Indeed, mutations in PIWI family proteins piwi and aub impact two types of variegating reporters (tandem arrays of the mini-white gene, and hsp70-white transgene reporters) at multiple genomic loci [50]. In a study of Spn-E, a putative helicase involved in the piRNA pathway (Fig. 3), Gvozdev and colleagues have demonstrated a loss of heterochromatic structure at transposon sites due to this perturbation [51].
Further evidence supporting a piRNA-targeting model comes from biochemical experiments showing a direct interaction between Piwi and HP1a [52]. This direct interaction utilizes the hydrophobic binding surface generated by the HP1a dimer, and is dependent on the PXVXL motif at the Piwi N-terminus. A point mutation in this domain disrupts the interaction between Piwi and HP1a in a yeast two-hybrid setting and in vitro [53]. This observation connects the targeting model directly to the well-established HP1a-centric model for the spread of heterochromatin [54], and provides a theoretical framework for understanding the heterochromatin formation process in flies.
In 2007, two groups independently proposed that a “ping-pong” amplification loop is responsible for piRNA biogenesis from primary transcripts [28, 29]. Deep sequencing of piRNAs initially positioned Piwi alongside Aub in the Ping-Pong amplification cycle for generating secondary piRNAs, as both bind primarily anti-sense piRNAs, and could thus partner with Ago3, which binds primarily sense strand piRNAs [29]. This model was later modified in response to results from sequencing piRNA in piwi mutant ovaries [55]; these results showed that secondary piRNA production is unaffected in the absence of Piwi. A functional test in the female germline of the role of Piwi in heterochromatin formation at TEs has placed Piwi downstream of piRNA production in the deposition of HP1a at the putative promoter regions for most of the transposons tested [11]. A model in which Piwi functions by carrying piRNAs into the nucleus to direct heterochromatin formation is also supported by an independent study using an N-terminal truncation mutant of Piwi, which fails to localize in the nucleus. This mutation results in the failure of transposon silencing and of the enrichment of heterochromatic markers at a subset of transposon sites, demonstrating the critical importance of Piwi nuclear localization for this assembly pathway [30]. Saito and colleagues had earlier demonstrated that piRNA binding is required for Piwi nuclear localization [56]. Taken together, the results from these studies make a compelling case that Piwi bound to piRNA enters the nucleus where it plays a role in transcriptional silencing of transposons by a mechanism utilizing heterochromatin assembly (Fig. 3).
Evidence supporting the transcriptional silencing model for Piwi-dependent transposon suppression also arises from an independent report showing an increase in HeT-A transcription using nuclear run-on assays performed in ovaries depleted for Piwi [57]. However, earlier reports from Zamore and colleagues found a lack of impact on the transcription rate for transposons (e.g. mst40) in armitage mutants, suggesting a post-transcriptional silencing mechanism [58]. Consistent with this observation, silencing of the transposon Jockey is not impacted by HP1a depletion, indicating that it is not regulated by a chromatin-based mechanism, even though its silencing is dependent on Piwi [11]. Thus, a post-transcriptional component is clearly part of the piRNA silencing mechanism, and may be particularly relevant to a subset of TEs. However, given the predominant nuclear localization pattern of Piwi, and the concordance between TE over-expression and depletion of HP1a at a significant group of TEs, we argue that a transcriptional silencing mechanism mediated through a piRNA-directed heterochromatin targeting process is likely to be a major mechanism for transposon silencing by piRNA. Indeed, recent genome-wide mapping of H3K9me3 in piwi knockdown cells supports the idea that this pathway is required to sustain this mark at many TEs [44, 45].
The physical interaction between Piwi and HP1a, which connects the RNAi targeting model with the spreading model of heterochromatin formation, is a substantive link. However, an attempt to verify the importance of this direct interaction in transposon silencing in vivo indicated unexpected complexities. On replacing the wild type Piwi in the germline with a single residue mutant form (V30A) that fails to interact with HP1a in a yeast two-hybrid experiment, one finds no obvious impact on transposon silencing [11]. It is possible that additional proteins bridge the Piwi and HP1a interaction in vivo, perhaps in a way that mimics the role of Tas3 in the S. pombe RITS complex, and that this creates a more robust system. Alternatively, chromosomal proteins other than HP1a might be initially targeted to the TEs by Piwi. A tudor-domain-containing histone methyltransferase, EGG, appears to be a promising candidate for this role; this key protein is prominently associated with piRNA loci, and is necessary to maintain the heterochromatic status of these loci and many others [59]. Further biochemical work will likely be needed to yield insights into these potential protein–protein interactions.
In future studies, experiments using constructs bypassing the need for small RNA targeting of Piwi could be informative in deciphering how Piwi recruits relevant downstream factors, if indeed it does. Tethering of HP1a to sites adjacent to euchromatic reporters has been reported to induce ectopic heterochromatin over the reporter and is sufficient to induce new chromosomal interactions with other endogenous heterochromatic sites [60, 61]. Similar studies should be carried out for Piwi. A strong claim could potentially be made from this type of sufficiency (if observed), but the results from these experiments may be difficult to interpret due to the context-dependent nature of heterochromatin silencing. (For example, a variegating phenotype may only occur when Piwi is tethered adjacent to a repeat cluster or other heterochromatic mass.) Given the discussion above, a context-dependent impact from tethering Piwi is the likely outcome.
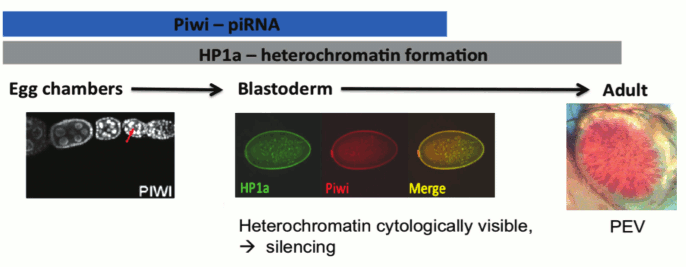
One critical question concerning the piRNA targeting model for heterochromatin formation stems from the fact that piRNA is thought to be restricted to the reproductive system and the early zygote [29]. However, heterochromatin is critical for maintaining genome stability and adequate chromosome segregation during mitosis throughout the lifetime of the individual; thus, the lack of a heterochromatin targeting/assembly mechanism in most tissue types does not seem plausible. While the endo-siRNA pathway could potentially be an alternative targeting mechanism in the soma, many of the studies cited above show an impact of piRNA component mutations in the larval and adult tissues normally scored in PEV assays. How might Piwi have an impact at these stages, a developmental period and tissue where it is not normally expressed? One intriguing possibility turns on the epigenetic inheritance of chromatin structure through mitosis. Heterochromatin formation is first observed during embryonic stage four (nuclear cycle 11-14) and is thought to maintain complete silencing until the relaxation phase during the late third instar larval stage [4]. The observations above suggest that the impact of Piwi depletion in the early zygote can be maintained epigenetically through mitosis to lead to the observed phenotype (suppression of PEV) in later developmental stages of the zygote (Fig. 4; see color insert). We note that while the TEs are an important component of heterochromatin, satellite DNA sequences are also a significant part of the whole, and are likely targeted by other mechanisms (see discussion of protein D1 above). Studies of mitotic inheritance upon ectopic heterochromatin formation induced by conditional (temporal) tethering of Piwi could provide a strong argument for the epigenetic inheritance model described here.
Fig. 4. Model for piRNA-directed silencing in Drosophila: Piwi and HP1a are both present as nuclear proteins during blastoderm, when heterochromatin becomes visible at 1.5 h followed by silencing at 2.5 h development. Piwi and HP1a are both required for silencing of a subset of TEs during oogenesis. Piwi is loaded into the oocyte and co-localizes with HP1a during blastoderm. This is a critical time point for heterochromatin assembly and silencing; the impact of perturbations at this stage can be seen at PEV loci in adults [4]. Illustrations: left, nuclear localization of Piwi in female germ line [11]; middle, co-localization of Piwi and HP1a in blastoderm nuclei; right, PEV from a transgene reporter, scored in the adult.
CONCLUDING REMARKS AND FUTURE PERSPECTIVE
In this review we have focused our discussion on the targeting aspect of heterochromatin formation, the decision to package a particular domain in heterochromatic form. We have reviewed the tremendous progress made during the past decade using the fruit fly as the model organism. Clearly, small RNAs are instrumental in the targeting process required to silence transposons (TEs). The strongest support for small RNA-mediated TE PTGS and TGS has come from investigations using D. melanogaster ovarian tissue [11, 29, 30]. Indeed, protecting the genomic integrity of germ line cells is the primary line of defense aimed at preserving transcriptional programs in subsequent generations. However, the degree to which TGS components in the oocyte affect chromatin packaging in the embryo and/or the adult remains an open question. We propose that small RNA, particularly that derived from the piRNA pathway, and heterochromatin components loaded into the oocyte facilitate the establishment of a subset of heterochromatin marks in the developing embryo. Both Piwi and HP1a are present during nuclear cycle 11-14 [48] when heterochromatin first becomes cytologically visible [3]. It is during this critical stage (early gastrulation) that the gene silencing consequences of heterochromatin formation, as observed by PEV, become apparent and mitotically heritable during differentiation [4] (Fig. 4). piRNAs, using either an RNA–RNA or RNA–DNA recognition process, could be directing the deposition of HP1a at sites of matching TEs, silencing these specific elements.
However, a reoccurring theme throughout the review is that most of the reported experimental observations are dependent on genome context (proximity to heterochromatic masses, etc.), thus making the derivation of a general rule difficult. For example, the impacts of mutations in the genes for RNAi pathway components show a differential response when tested on PEV reporter inserts present at different genomic loci. This no doubt reflects the mosaic nature of heterochromatin, and could also relate to the special features of the piRNA loci, which are certainly packaged as heterochromatin in somatic cells [13]. The effectiveness of 1360 to enhance or drive HP1a-dependent silencing also varies depending on the site tested (see discussion above). It is apparent that complex interactions among multiple mechanisms must be occurring, preventing the derivation of simple rules from the collected observations. Given the importance of limiting TE movement in the genome, multiple silencing mechanisms would seem advantageous. Moreover, from an evolutionary point of view, the involvement of transposons in heterochromatin formation itself suggests a convoluted mechanism, as observed. There is no doubt an “arms race” between the host species and the invading transposable elements through the evolutionary time scale, similar to that reported for viral defense systems. Whatever strategy succeeds in helping the host cope with an invasive new transposon will result in a further (potentially redundant) mechanism being built into the system.
The idea of heterochromatin targeting originated earlier, at a time when two types of chromatin were considered to make up the bulk of the genome. In this scenario, while the majority of the Drosophila genome is composed of euchromatin, the formation of the localized heterochromatic regions must be specifically targeted. The dichotomous classification of chromatin structure, while a good starting point and still useful in many cases, is insufficient to describe observations made from recent experiments. Different domains or subtypes of heterochromatin have therefore been reported to describe the differences between pericentric and telomeric heterochromatin [62, 63]. The fourth chromosome and Y chromosome also have distinctive properties (see for example [64]). More recently, results from genome-wide chromatin immunoprecipitation mapping of chromosomal proteins and histone modifications has suggested other informative ways of classifying chromatin structure across the genome. For example, the nine-state model can be used to adequately identify enhancer regions, transcription start sites, and Polycomb-regulated regions in addition to classic heterochromatin, and to map the distribution of such domains within the fourth chromosome or other large regions considered heterochromatic by classical criteria [13]. These new additions to our knowledge have in many ways made the distinction between euchromatin and heterochromatin more nuanced. As the resolution of chromatin states continues to improve, the definitions of these states will likely require modifications.
While there is no doubt that certain targeting events are needed to ensure proper heterochromatin silencing, as supported by ample evidence reviewed here, the evidence also suggests that no single unifying mechanism for heterochromatin targeting is likely to apply. We propose, instead, that multiple mechanisms function in a complex network to ensure proper chromatin structure formation in the genome. This interactive network forms the basis of the context-dependent effects that we so often see in genetic dissections of chromatin biology. To gain predictive power from the outcomes from simple perturbation experiments, we will need to embrace the inherent complexity of the system and utilize the wealth of genomic information derived from high throughput technologies. These sorts of approaches should enable us to develop a deeper understanding of how genomes balance their relationship with transposable elements, both silencing and using these invaders to adapt and evolve.
We thank Brent Brower-Toland (Monsanto Corp.) for permission to use the middle picture in Fig. 4.
Preparation of this manuscript has been supported by General Medical Sciences, National Institutes of Health (USA) under award number R01 GM068388 to SCRE. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
1.Britten, R. J., and Kohne, D. E. (1968)
Science, 161, 529-540.
2.Beisel, C., and Paro, R. (2011) Nat. Rev.
Genet., 12, 123-135.
3.Foe, V. E., and Alberts, B. M. (1983) J. Cell.
Sci., 61, 31-70.
4.Lu, B. Y., Ma, J., and Eissenberg, J. C. (1998)
Development, 125, 2223-2234.
5.Vlassova, I. E., Graphodatsky, A. S., Belyaeva, E.
S., and Zhimulev, I. F. (1991) Mol. Gen. Genet., 229,
316-318.
6.Rudolph, T., Yonezawa, M., Lein, S., Heidrich, K.,
Kubicek, S., Schafer, C., Phalke, S., Walther, M., Schmidt, A.,
Jenuwein, T., and Reuter, G. (2007) Mol. Cell, 26,
103-115.
7.Brower-Toland, B., Riddle, N. C., Jiang, H.,
Huisinga, K. L., and Elgin, S. C. R. (2009) Genetics,
181, 1303-1319.
8.Aagaard, L., Laible, G., Selenko, P., Schmid, M.,
Dorn, R., Schotta, G., Kuhfittig, S., Wolf, A., Lebersorger, A., Singh,
P. B., Reuter, G., and Jenuwein, T. (1999) EMBO J., 18,
1923-1938.
9.Ekwall, K., Nimmo, E. R., Javerzat, J. P.,
Borgstrom, B., Egel, R., Cranston, G., and Allshire, R. (1996) J.
Cell. Sci., 109 (Pt. 11), 2637-2648.
10.Fanti, L., Giovinazzo, G., Berloco, M., and
Pimpinelli, S. (1998) Mol. Cell, 2, 527-538.
11.Wang, S. H., and Elgin, S. C. (2011) Proc.
Natl. Acad. Sci. USA, 108, 21164-21169.
12.Kuhfittig, S., Szabad, J., Schotta, G., Hoffmann,
J., Mathe, E., and Reuter, G. (2001) Genetics, 157,
1227-1244.
13.Kharchenko, P. V., Alekseyenko, A. A., Schwartz,
Y. B., Minoda, A., Riddle, N. C., Ernst, J., Sabo, P. J., Larschan, E.,
Gorchakov, A. A., Gu, T., Linder-Basso, D., Plachetka, A., Shanower,
G., Tolstorukov, M. Y., Luquette, L. J., Xi, R., Jung, Y. L., Park, R.
W., Bishop, E. P., Canfield, T. K., Sandstrom, R., Thurman, R. E.,
MacAlpine, D. M., Stamatoyannopoulos, J. A., Kellis, M., Elgin, S. C.,
Kuroda, M. I., Pirrotta, V., Karpen, G. H., and Park, P. J. (2011)
Nature, 471, 480-485.
14.Muller, H. J. (1930) J. Genet., 22,
299-334.
15.Wallrath, L. L., and Elgin, S. C. R. (1995)
Genes Dev., 9, 1263-1277.
16.Sun, F. L., Cuaycong, M. H., and Elgin, S. C. R.
(2001) Mol. Cell Biol., 21, 2867-2879.
17.Cryderman, D. E., Tang, H., Bell, C., Gilmour, D.
S., and Wallrath, L. L. (1999) Nucleic Acids Res., 27,
3364-3370.
18.Vogel, M. J., Pagie, L., Talhout, W., Nieuwland,
M., Kerkhoven, R. M., and van Steensel, B. (2009) Epigenetics &
Chromatin, 2, 1.
19.Slotkin, R. K., and Martienssen, R. A. (2007)
Nat. Rev. Genet., 8, 272-285.
20.Kloc, A., and Martienssen, R. A. (2008) Trends
Genet., 24, 511-517.
21.Irvine, D. V., Zaratiegui, M., Tolia, N. H.,
Goto, D. B., Chitwood, D. H., Vaughn, M. W., Joshua-Tor, L., and
Martienssen, R. A. (2006) Science, 313, 1134-1137.
22.Huisinga, K. L., and Elgin, S. C. R. (2009)
Biochim. Biophys. Acta, 1789, 3-16.
23.Dai, Q., Smibert, P., and Lai, E. C. (2012)
Curr. Top. Dev. Biol., 99, 201-235.
24.Kawamura, Y., Saito, K., Kin, T., Ono, Y., Asai,
K., Sunohara, T., Okada, T. N., Siomi, M. C., and Siomi, H. (2008)
Nature, 453, 793-797.
25.Ghildiyal, M., Seitz, H., Horwich, M. D., Li, C.,
Du, T., Lee, S., Xu, J., Kittler, E. L. W., Zapp, M. L., Weng, Z., and
Zamore, P. D. (2008) Science, 320, 1077-1081.
26.Moshkovich, N., Nisha, P., Boyle, P. J.,
Thompson, B. A., Dale, R. K., and Lei, E. P. (2011) Genes Dev.,
25, 1686-1701.
27.Cernilogar, F. M., Onorati, M. C., Kothe, G. O.,
Burroughs, A. M., Parsi, K. M., Breiling, A., Lo Sardo, F., Saxena, A.,
Miyoshi, K., Siomi, H., Siomi, M. C., Carninci, P., Gilmour, D. S.,
Corona, D. F., and Orlando, V. (2011) Nature, 480,
391-395.
28.Gunawardane, L. S., Saito, K., Nishida, K. M.,
Miyoshi, K., Kawamura, Y., Nagami, T., Siomi, H., and Siomi, M. C.
(2007) Science, 315, 1587-1590.
29.Brennecke, J., Aravin, A. A., Stark, A., Dus, M.,
Kellis, M., Sachidanandam, R., and Hannon, G. J. (2007) Cell,
128, 1089-1103.
30.Klenov, M. S., Sokolova, O. A., Yakushev, E. Y.,
Stolyarenko, A. D., Mikhaleva, E. A., Lavrov, S. A., and Gvozdev, V. A.
(2011) Proc. Natl. Acad. Sci. USA, 108, 18760-18765.
31.Feschotte, C. (2008) Nat. Rev. Genet.,
9, 397-405.
32.Gangaraju, V. K., Yin, H., Weiner, M. M., Wang,
J., Huang, X. A., and Lin, H. (2010) Nat. Genet., 43,
153-158.
33.Cam, H. P., Noma, K.-I., Ebina, H., Levin, H. L.,
and Grewal, S. I. S. (2008) Nature, 451, 431-436.
34.Aravind, L. (2000) Trends Biochem. Sci.,
25, 421-423.
35.Aulner, N., Monod, C., Mandicourt, G., Jullien,
D., Cuvier, O., Sall, A., Janssen, S., Laemmli, U. K., and Kas, E.
(2002) Mol. Cell Biol., 22, 1218-1232.
36.Filion, G. J., van Bemmel, J. G., Braunschweig,
U., Talhout, W., Kind, J., Ward, L. D., Brugman, W., de Castro, I. J.,
Kerkhoven, R. M., Bussemaker, H. J., and van Steensel, B. (2010)
Cell, 143, 212-224.
37.Smith, M. B., and Weiler, K. S. (2010)
Chromosoma, 119, 287-309.
38.Kapitonov, V. V., and Jurka, J. (2003) Proc.
Natl. Acad. Sci. USA, 100, 6569-6574.
39.Lander, E. S., Linton, L. M., Birren, B.,
Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M.,
FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland,
J., Kann, L., Lehoczky, J., LeVine, R., McEwan, P., McKernan, K.,
Meldrim, J., Mesirov, J. P., Miranda, C., Morris, W., Naylor, J.,
Raymond, C., Rosetti, M., Santos, R., Sheridan, A., Sougnez, C.,
Stange-Thomann, N., Stojanovic, N., Subramanian, A., Wyman, D., Rogers,
J., Sulston, J., Ainscough, R., Beck, S., Bentley, D., Burton, J.,
Clee, C., Carter, N., Coulson, A., Deadman, R., Deloukas, P., Dunham,
A., Dunham, I., Durbin, R., French, L., Grafham, D., Gregory, S.,
Hubbard, T., Humphray, S., Hunt, A., Jones, M., Lloyd, C., McMurray,
A., Matthews, L., Mercer, S., Milne, S., Mullikin, J. C., Mungall, A.,
Plumb, R., Ross, M., Shownkeen, R., Sims, S., Waterston, R. H., Wilson,
R. K., Hillier, L. W., McPherson, J. D., Marra, M. A., Mardis, E. R.,
Fulton, L. A., Chinwalla, A. T., Pepin, K. H., Gish, W. R., Chissoe, S.
L., Wendl, M. C., Delehaunty, K. D., Miner, T. L., Delehaunty, A.,
Kramer, J. B., Cook, L. L., Fulton, R. S., Johnson, D. L., Minx, P. J.,
Clifton, S. W., Hawkins, T., Branscomb, E., Predki, P., Richardson, P.,
Wenning, S., Slezak, T., Doggett, N., Cheng, J. F., Olsen, A., Lucas,
S., Elkin, C., Uberbacher, E., Frazier, M., Gibbs, R. A., Muzny, D. M.,
Scherer, S. E., Bouck, J. B., Sodergren, E. J., Worley, K. C., Rives,
C. M., Gorrell, J. H., Metzker, M. L., Naylor, S. L., Kucherlapati, R.
S., Nelson, D. L., Weinstock, G. M., Sakaki, Y., Fujiyama, A., Hattori,
M., Yada, T., Toyoda, A., Itoh, T., Kawagoe, C., Watanabe, H., Totoki,
Y., Taylor, T., Weissenbach, J., Heilig, R., Saurin, W., Artiguenave,
F., Brottier, P., Bruls, T., Pelletier, E., Robert, C., Wincker, P.,
Smith, D. R., Doucette-Stamm, L., Rubenfield, M., Weinstock, K., Lee,
H. M., Dubois, J., Rosenthal, A., Platzer, M., Nyakatura, G., Taudien,
S., Rump, A., Yang, H., Yu, J., Wang, J., Huang, G., Gu, J., Hood, L.,
Rowen, L., Madan, A., Qin, S., Davis, R. W., Federspiel, N. A., Abola,
A. P., Proctor, M. J., Myers, R. M., Schmutz, J., Dickson, M.,
Grimwood, J., Cox, D. R., Olson, M. V., Kaul, R., Shimizu, N.,
Kawasaki, K., Minoshima, S., Evans, G. A., Athanasiou, M., Schultz, R.,
Roe, B. A., Chen, F., Pan, H., Ramser, J., Lehrach, H., Reinhardt, R.,
McCombie, W. R., de la Bastide, M., Dedhia, N., Blocker, H.,
Hornischer, K., Nordsiek, G., Agarwala, R., Aravind, L., Bailey, J. A.,
Bateman, A., Batzoglou, S., Birney, E., Bork, P., Brown, D. G., Burge,
C. B., Cerutti, L., Chen, H. C., Church, D., Clamp, M., Copley, R. R.,
Doerks, T., Eddy, S. R., Eichler, E. E., Furey, T. S., Galagan, J.,
Gilbert, J. G., Harmon, C., Hayashizaki, Y., Haussler, D., Hermjakob,
H., Hokamp, K., Jang, W., Johnson, L. S., Jones, T. A., Kasif, S.,
Kaspryzk, A., Kennedy, S., Kent, W. J., Kitts, P., Koonin, E. V., Korf,
I., Kulp, D., Lancet, D., Lowe, T. M., McLysaght, A., Mikkelsen, T.,
Moran, J. V., Mulder, N., Pollara, V. J., Ponting, C. P., Schuler, G.,
Schultz, J., Slater, G., Smit, A. F., Stupka, E., Szustakowski, J.,
Thierry-Mieg, D., Thierry-Mieg, J., Wagner, L., Wallis, J., Wheeler,
R., Williams, A., Wolf, Y. I., Wolfe, K. H., Yang, S. P., Yeh, R. F.,
Collins, F., Guyer, M. S., Peterson, J., Felsenfeld, A., Wetterstrand,
K. A., Patrinos, A., Morgan, M. J., de Jong, P., Catanese, J. J.,
Osoegawa, K., Shizuya, H., Choi, S., and Chen, Y. J. (2001)
Nature, 409, 860-921.
40.Aravin, A. A., Lagos-Quintana, M., Yalcin, A.,
Zavolan, M., Marks, D., Snyder, B., Gaasterland, T., Meyer, J., and
Tuschl, T. (2003) Dev. Cell, 5, 337-350.
41.Sun, F.-L., Haynes, K. A., Simpson, C. L., Lee,
S. D., Collins, L., Wuller, J., Eissenberg, J. C., and Elgin, S. C. R.
(2004) Mol. Cell Biol., 24, 8210-8220.
42.Haynes, K. A., Caudy, A. A., Collins, L., and
Elgin, S. C. (2006) Curr. Biol., 16, 2222-2227.
43.Sentmanat, M. F., and Elgin, S. C. (2012)
Proc. Natl. Acad. Sci. USA, 109, 14104-14109.
44.Sienski, G., Donertas, D., and Brennecke, J.
(2012) Cell, 151, 964-980.
45.Le Thomas, A., Rogers, A. K., Webster, A.,
Marinov, G. K., Liao, S. E., Perkins, E. M., Hur, J. K., Aravin, A. A.,
and Toth, K. F. (2013) Genes Dev., in press.
46.Fagegaltier, D., Bouge, A.-L., Berry, B., Poisot,
E., Sismeiro, O., Coppee, J.-Y., Theodore, L., Voinnet, O., and
Antoniewski, C. (2009) Proc. Natl. Acad. Sci. USA, 106,
21258-21263.
47.Deshpande, G., Calhoun, G., and Schedl, P. (2005)
Genes Dev., 19, 1680-1685.
48.Rouget, C., Papin, C., Boureux, A., Meunier,
A.-C., Franco, B., Robine, N., Lai, E. C., Pelisson, A., and Simonelig,
M. (2010) Nature, 467, 1128-1132.
49.Cox, D. N., Chao, A., Baker, J., Chang, L., Qiao,
D., and Lin, H. (1998) Genes Dev., 12, 3715-3727.
50.Pal-Bhadra, M., Leibovitch, B. A., Gandhi, S. G.,
Rao, M., Bhadra, U., Birchler, J. A., and Elgin, S. C. R. (2004)
Science, 303, 669-672.
51.Klenov, M. S., Lavrov, S. A., Stolyarenko, A. D.,
Ryazansky, S. S., Aravin, A. A., Tuschl, T., and Gvozdev, V. A. (2007)
Nucleic Acids Res., 35, 5430-5438.
52.Brower-Toland, B., Findley, S. D., Jiang, L.,
Liu, L., Yin, H., Dus, M., Zhou, P., Elgin, S. C. R., and Lin, H.
(2007) Genes Dev., 21, 2300-2311.
53.Mendez, D. L., Kim, D., Chruszcz, M., Stephens,
G. E., Minor, W., Khorasanizadeh, S., and Elgin, S. C. (2011)
Chembiochem, 12, 1084-1096.
54.Girton, J. R., and Johansen, K. M. (2008) Adv.
Genet., 61, 1-43.
55.Li, C., Vagin, V. V., Lee, S., Xu, J., Ma, S.,
Xi, H., Seitz, H., Horwich, M. D., Syrzycka, M., Honda, B. M., Kittler,
E. L. W., Zapp, M. L., Klattenhoff, C., Schulz, N., Theurkauf, W. E.,
Weng, Z., and Zamore, P. D. (2009) Cell, 137,
509-521.
56.Saito, K., Ishizu, H., Komai, M., Kotani, H.,
Kawamura, Y., Nishida, K. M., Siomi, H., and Siomi, M. C. (2010)
Genes Dev., 24, 2493-2498.
57.Shpiz, S., Olovnikov, I., Sergeeva, A., Lavrov,
S., Abramov, Y., Savitsky, M., and Kalmykova, A. (2011) Nucleic
Acids Res., 39, 8703-8711.
58.Sigova, A., Vagin, V., and Zamore, P. D. (2006)
Cold Spring Harb. Symp. Quant. Biol., 71, 335-341.
59.Rangan, P., Malone, C. D., Navarro, C., Newbold,
S. P., Hayes, P. S., Sachidanandam, R., Hannon, G. J., and Lehmann, R.
(2011) Curr. Biol., 21, 1373-1379.
60.Seum, C., Delattre, M., Spierer, A., and Spierer,
P. (2001) EMBO J., 20, 812-818.
61.Li, Y., Danzer, J. R., Alvarez, P., Belmont, A.
S., and Wallrath, L. L. (2003) Development, 130,
1817-1824.
62.Doheny, J. G., Mottus, R., and Grigliatti, T. A.
(2008) PLoS One, 3, e3864.
63.Cryderman, D. E., Morris, E. J., Biessmann, H.,
Elgin, S. C., and Wallrath, L. L. (1999) EMBO J., 18,
3724-3735.
64.Haynes, K. A., Gracheva, E., and Elgin, S. C. R.
(2007) Genetics, 175, 1539-1542.