Chemical Modification of Photosystem II Core Complex Pigments with Sodium Borohydride
M. I. Vishnev, A. A. Zabelin, V. A. Shkuropatova, M. F. Yanyushin, V. A. Shuvalov, and A. Ya. Shkuropatov*
Institute of Basic Biological Problems, Russian Academy of Sciences, ul. Institutskaya 2, 142290 Pushchino, Moscow Region, Russia; fax: (4967) 33-0532; E-mail: ashkur@mail.ru* To whom correspondence should be addressed.
Received November 27, 2012
The reaction of the irreversible chemical reduction of the 131-keto C=O group of pheophytin a (Pheo a) with sodium borohydride in reaction centers (RCs) of functionally active spinach photosystem II (PS II) core complexes was studied. Stable, chromatographically purified PS II core complex preparations with altered chromophore composition are obtained in which ~25% of Pheo a molecules are modified to 131-deoxo-131-hydroxy-Pheo a. Some of the chlorophyll a molecules in the complexes were also irreversibly reduced with borohydride to 131-deoxo-131-hydroxy-chlorophyll a. Based on the results of comparative study of spectral, biochemical, and photochemical properties of NaBH4-treated and control preparations, it was concluded that: (i) the borohydride treatment did not result in significant dissociation of the PS II core complex protein ensemble; (ii) the modified complexes retained the ability to photoaccumulate the radical anion of the pheophytin electron acceptor in the presence of exogenous electron donor; (iii) only the photochemically inactive pheophytin PheoD2 is subjected to the borohydride treatment; (iv) the Qx optical transition of the PheoD2 molecule in the RC of PS II core complexes is located at 543 nm; (v) in the Qy spectral region, PheoD2 probably absorbs at ~680 nm.
KEY WORDS: photosystem II core complex, pheophytin a, chemical modification, sodium borohydrideDOI: 10.1134/S0006297913040068
Abbreviations: ΔA, absorbance changes; Car, β-carotene; Chl, chlorophyll; Cyt b559, cytochrome b559; P680, dimer of Chl a molecules in photosystem II reaction center; Pheo, pheophytin; PheoD1 and PheoD2, Pheo bound to D1 and D2 polypeptides, respectively; PS II, photosystem II; QA, primary quinone acceptor; RC, reaction center.
The core complex of photosystem II (PS II) of oxygenic photosynthesis is
a minimal functional pigment–protein ensemble that is capable of
photoinduced charge separation in the photochemical reaction center
(RC) and performs the oxidation of water to molecular oxygen via the
oxygen-evolving complex (for a recent review see [1]). The PS II core complex consists of two antenna
proteins, CP43 and CP47, containing chlorophyll (Chl) a and
β-carotene (Car) molecules, RC proteins (D1 and D2 polypeptides,
α- and β-subunits of cytochrome (Cyt) b559), the
manganese (Mn)-cluster, and several extrinsic proteins required for
stabilization of the oxygen-evolving complex. The X-ray crystal
structure of PS II core complexes from cyanobacteria is known with
resolution to 1.9 Å [2-4]. The central RC cofactors, the dimer of Chl
a molecules (P680), two monomeric Chl a
(ChlD1 and ChlD2), two pheophytins (Pheo)
a (PheoD1 and PheoD2), and two
plastoquinones (QA and QB) are arranged in two
quasi-symmetric branches, of which only one, bound to the D1 protein,
takes part in electron transfer. The RC also contains two peripheral
Chl a and two Car molecules. Excitation of the RC pigments by
light quanta directly or by means of energy transfer from antenna lead
to formation of the radical ion pair
P680+PheoD1– [5-7]. The electron from
PheoD1– is transferred to the primary
quinone acceptor QA and further to the secondary quinone
QB; on the donor side, the positive charge is transferred
from P680+ to the Mn-cluster via the redox-active
TyrZ [1].
The detailed mechanism of primary charge separation and pathways of initial electron transfer and the nature of the primary electron donor in PS II RCs remain subjects of discussion [5-7]. To a considerable extent, this is related to a strong overlap of the Qy absorption bands of the pigments in RCs and in the antenna complexes CP43 and CP47 that makes it problematic to determine spectral characteristics of the individual pigments and significantly complicates interpretation of the photoinduced absorbance changes. One approach providing valuable information on spectral properties of the PS II chromophores, both active and inactive in electron transfer, is modification of the pigment composition of the RC by chemical methods [8-15]. Data of this kind have been used also in modeling of the electronic structure of the PS II RC in the framework of exciton theory [16-18]. However, studies on chemical modification of the PS II pigments were performed earlier mainly on isolated D1/D2/Cyt b559 RC complexes [8-15]. These complexes do not contain quinone electron acceptors; they are not capable of water oxidation, and their spectral properties are probably partially changed in the course of the isolation process (see, for example, [19, 20]). In this connection, chemical modification of pigments within more intact PS II particles is of great interest. In work [21], the chemical reaction between sodium borohydride and pigments in spinach PS II complex, which contains the core complex together with some tightly-bound Chl a/b proteins, was investigated. The spectral effects associated with the reduction of Chl a and Chl b to their hydroxy-derivatives were revealed. In that work, however, there was no information on modification of pheophytin molecules, and no attempts to purify the modified complexes and to analyze the degree of their functional activity were undertaken.
In this study, the chemical reaction of chlorins with NaBH4 was used to modify Pheo a in highly purified, functionally active PS II core complex preparations from spinach. On this basis, new data on a contribution of the inactive PheoD2 to the absorption spectrum of the PS II reaction center as an integral component of the PS II core complex were obtained.
MATERIALS AND METHODS
Oxygen-evolving PS II core complexes containing about 35 Chl molecules per RC were isolated from spinach PS II membrane fragments [22] by the method described in works [23, 24]. Chromatographically purified core complex preparations were suspended in a 20 mM (or 100 mM) BTS400 buffer containing 20 mM (or 100 mM) Bis-Tris (pH 6.5), 20 mM MgCl2, 5 mM CaCl2, 80 mM MgSO4, 400 mM sucrose, and 0.03% (w/v) n-dodecyl-β-D-maltoside. The PS II core complexes used in this work included the primary quinone acceptor QA, but did not contain a functional secondary quinone QB [19, 23]. Cyt b559 is fully oxidized in these preparations [19, 23]. The photochemical activity of the complexes in the primary charge separation reaction was confirmed by femtosecond absorption difference spectroscopy [7].
A procedure similar to that described earlier for isolated D1/D2/Cyt b559 RC complexes [10] was used for modification of PS II core complexes with sodium borohydride. Gradually increasing amounts of solid NaBH4 (Sigma, USA) were added to the core complexes (9-16 µg Chl/ml) suspended in 100 or 20 mM BTS400 buffer, and the titration process was followed spectrophotometrically by measuring the absorption spectra at 285 K and by calculating difference spectra (NaBH4-treated-minus-original sample). The modified preparation of the complexes was dialyzed against BTS400 buffer, and then it was subjected to chromatographic purification on a Q-Sepharose FF (Sigma) column and suspended in 20 mM BTS400 buffer. A sample of PS II core complexes subjected to exactly the same procedures of incubation, dialysis, and additional purification as the modified preparation but in the absence of added NaBH4 was used as a control. All procedures of isolation, purification, and treatment of PS II core complexes were carried out at 5ºC under dim green light; preparations were stored at –80ºC until use. Chlorophyll concentrations were determined according to a published method [25].
SDS-PAGE was performed according to [26] with 6 M urea in 15% polyacrylamide resolving gel. Samples were mixed with equal volumes of buffer containing 200 mM Tris-HCl (pH 7.0), 2% SDS, 100 mM dithiothreitol, and 8 M urea. The gels were stained with colloidal Coomassie G-250 after one hour fixing in ethanol [27].
Rates of O2 evolution were measured with a Clark-type oxygen electrode (Hansatech, Great Britain) at 24ºC on samples containing 10 µg Chl/ml, under continuous saturating red light illumination (λ > 600 nm). K3[Fe(CN)6] (1 mM) and 2,6-dichloro-1,4-benzoquinone (0.25 mM) were used as artificial electron acceptors.
Photoaccumulation of reduced pheophytin was measured at room temperature in a vacuum-tight quartz cuvette in the presence of sodium dithionite (3 mM) and methyl viologen (1 µM) [10, 28]. Difference (light-minus-dark) spectra were obtained by subtracting the absorption spectrum measured in the dark from the spectrum measured under illumination of a sample during 7 s with continuous red light through a water heat filter (λ > 600 nm; ~160 mW/cm2).
Light-induced (light-minus-dark) difference spectra at low (100 K) temperature were obtained by subtraction of the absorption spectrum measured in the dark from the spectrum registered immediately after 30-s illumination of a sample with continuous red light through a water heat filter (λ > 600 nm; ~45 mW/cm2).
Absorption spectra were registered with Cary 4000 (Varian, USA), UV-1601 PC (Shimadzu, Japan), and Agilent 8453 (Agilent, USA) spectrophotometers. For measurements of absorption spectra at low temperature, PS II core complexes were mixed with 60% glycerol (v/v).
RESULTS
The experiments were designed to reveal spectral effects and to analyze pigment composition changes of isolated PS II core complexes caused by the chemical interaction of Pheo a and Chl a molecules with sodium borohydride. Considering the high lability of PS II to external influences, the protein composition of the modified preparations was also characterized, and their functional state was estimated.
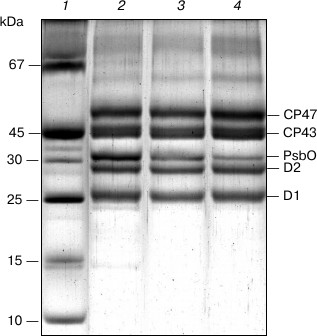
Figure 1a presents the absorption spectrum at 285 K of the original PS II core complex preparation in 100 mM BTS400 buffer used in this work for modification with sodium borohydride. The spectrum corresponds well to the absorption spectrum earlier described for similar isolated spinach PS II core complexes at room temperature [24], showing a broad band at ~675 nm that includes contributions from the Qy optical transitions of Chl a and Pheo a molecules. A weak absorption band at ~543 nm (Fig. 1a) belongs to the spectrally isolated Qx transition of the reaction center Pheo a molecules. Absorption bands of Car molecules are observed at ~465 and ~490 nm. There is only a minor contribution to the absorption spectrum from Chl b molecules, as follows from the presence of only very weakly pronounced shoulder at ~650 nm (Fig. 1a); this indicates that the core complexes were virtually free from the light-harvesting Chl a/b proteins. In accordance with this conclusion, SDS-PAGE (Fig. 2, lane 2) revealed almost complete absence in the complexes of the main light-harvesting complex LHCII (though residual amounts of the minor protein CP29 are probably present [23]). The polypeptide composition of PS II core complexes included subunits CP43, CP47, D1 and D2, as well as the Mn-stabilizing 33 kDa protein (PsbO) (Fig. 2, lane 2). The presence in the complexes of Cyt b559 was confirmed by observation of the band at 559 nm and a differential signal at 412(–)/427(+) nm in the difference absorption spectrum dithionite reduced-minus-oxidized complexes (not shown). As expected [23], the core complexes did not contain the extrinsic 17 and 23 kDa polypeptides (Fig. 2, lane 2). The rate of oxygen evolution by the complexes was 1100-1300 µmol O2/h per mg Chl.
Fig. 1. a) Absorption spectrum of the original preparation of PS II core complexes from spinach measured at 285 K. b) Difference spectra (285 K) of PS II core complexes obtained by subtracting the absorption spectrum of the original preparation from the absorption spectra recorded during the treatment of complexes with increasing additions of solid NaBH4 (see text for details). The arrows show the directions of absorbance changes at the corresponding wavelengths.
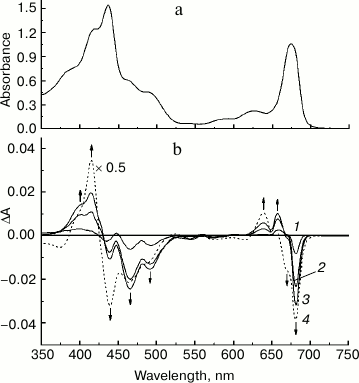
Fig. 2. SDS-PAGE of PS II core complexes: 1) marker proteins; 2) original preparation; 3) control preparation; 4) NaBH4-treated preparation. The NaBH4-treated preparation was obtained by the incubation of complexes with sodium borohydride in 100 mM BTS400 buffer. The gels were loaded with complexes containing approximately equal amounts of chlorophyll.
The difference spectra “NaBH4-treated-minus-original sample” presented in Fig. 1b (curves 1-3) show that at an early stage of the titration of PS II core complexes with sodium borohydride absorption bands develop at 657 and ~400 nm, which can be attributed respectively to the Qy and Soret optical transitions of the 131-deoxo-131-hydroxy-Pheo a (131-OH-Pheo a) formed in the reaction of irreversible chemical reduction of the 131-keto C=O group of Pheo a to a hydroxyl group [10, 11, 29, 30]. The 131-OH-Pheo a Qx absorption band expected in the region of 500 nm [10, 11, 29, 30] is apparently masked in the difference spectra by bleaching of the absorption bands of Car molecules at 465 and 491 nm, due probably to their modification or leaving from the core complexes. A somewhat slower reaction of BH4– with the 131-keto C=O group of Chl a in core complexes led to the appearance of absorption bands at 639 and 414 nm (Fig. 1b) belonging to 131-deoxo-131-hydroxy-chlorophyll a (131-OH-Chl a) [21, 29]. Remarkably, the single bleaching band at 681.5 nm apparently includes contributions from the chemical modification of both Pheo a and Chl a. The modification of the pigment was not accompanied by the appearance of ideal isosbestic points; however, in the early stages of treatment a nearly isosbestic point was observed at 667 nm (Fig. 1b).
After a considerable slowing of the 131-OH-Pheo a absorption increase at 657 nm (Fig. 1b, curve 3) that corresponded to NaBH4 concentration of 1.3 mg/ml and pH 7.1 (at 5°C), the main portion of the modified preparation was subjected to dialysis and subsequent purification on a column with Q-Sepharose (see “Materials and Methods”). This produced a NaBH4-treated preparation containing a significant amount of 131-OH-Pheo a with a relatively small contribution from 131-OH-Chl a (see below). For comparison, a small portion of the modified preparation was further treated with sodium borohydride. Figure 1b (curve 4) shows that along with the increase in the absorption bands of 131-OH-Chl a, more intensive treatment led to the appearance in the difference spectrum of an additional negative shoulder at ~670 nm and a strong increase in the bleaching at 439 nm associated with the modification of Chl a.
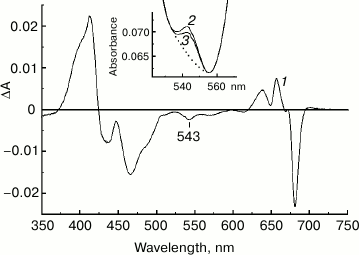
Figure 3 (curve 1) represents a difference between the absorption spectra of the chromatographically purified NaBH4-treated and control preparations of PS II core complexes. The absorption spectra were measured at 285 K and normalized to each other at 667 nm to provide comparable concentrations of the complexes. It is seen that the difference spectrum includes characteristic absorbance changes reflecting the formation of the 131-hydroxy-derivatives of Pheo a and Chl a, and it also demonstrates a pronounced negative band in the region of the Qx absorption of Pheo a at 543 nm. Comparison of the integrated absorption of pheophytin molecules in this spectral region (Fig. 3, inset) shows that the dipole strength of the 543 nm band decreased after borohydride treatment of the complexes by about 25%, which corresponds to the conversion of ~25% of the total of Pheo a molecules into 131-OH-Pheo a. Estimations based on the ratio of amplitudes of the absorbance changes at 639 and 657 nm (Fig. 3, curve 1) using the ratio of the differential extinction coefficients for the 131-OH-derivatives of Chl a and Pheo a synthesized in reaction with NaBH4 in methanol solutions (data not shown) showed the presence of ~0.5 molecule of 131-OH-Chl a per molecule of 131-OH-Pheo a in the modified preparation. The reasons for the appearance in the difference spectrum (Fig. 3, curve 1) of a small negative peak at 667-669 nm and a minor positive maximum at 671-672 nm, as well as of an absorption increase at ~700 nm, remain unknown. It might be due to some imperfection in the normalization of the spectra to the absorption at 667 nm.
Fig. 3. 1) Difference absorption spectrum “NaBH4-treated-minus-control preparation” obtained for PS II core complexes after dialysis and chromatographic purification. The inset shows the Qx region of pheophytins in the absorption spectra of control (2) and NaBH4-treated (3) preparations used to obtain the difference spectrum 1. The absorption spectra were measured at 285 K and normalized at 667 nm (the absorption at the maximum of the Qy band was ~1.2). The dotted line in the inset shows the assumed “baseline” used to determine the absorbance of Pheo a molecules.
SDS-PAGE (Fig. 2) revealed that the preparations of PS II core complexes used in the work retained antenna proteins CP43 and CP47 and reaction center subunits D1 and D2. As seen from the figure, the control preparation (lane 3) contains a smaller amount of the PsbO protein than the original core complexes (lane 2). This points to partial loss of this polypeptide under the experimental conditions that were required for the modification (long exposure of a sample to 5°C, dialysis, chromatographic purification), and it is consistent with a decrease in the rate of oxygen evolution in the control compared to the original complexes (by 20-40%). A small further decrease in the amount of PsbO apparently took place for the NaBH4-treated preparation (Fig. 2, lane 4), which could reflect the effect of borohydride on this protein. In this case, there was a considerable inhibition of O2-evolving activity of the complexes (the rate of oxygen evolution by the NaBH4-treated preparation was ~30% of that of the control sample). Additions of NaBH4 to the core complexes suspended in 100 mM BTS400 buffer led to a relatively small increase in the pH of the incubation medium (to pH 7.1 at 5°C). Taking this fact into account, the most likely explanation for the reduced oxygen evolution rate is that BH4– could partially reduce the functional Mn-cluster in PS II core complexes and then release the Mn(II) ions from the cluster [31].
Figure 4 compares light-induced difference (light-minus-dark) spectra for photoaccumulation of the reduced pheophytin electron acceptor in the control and NaBH4-treated PS II core preparations in the presence of sodium dithionite and methyl viologen. The spectra were measured at room temperature and normalized to equal absorption of the samples at 667 nm. A similar result was obtained by normalizing the spectra to the absorption of Cyt b559 at 559 nm, which is fully reduced under these conditions with sodium dithionite. The presented difference spectra correspond well to the spectra of pheophytin photoreduction previously described for PS II enriched chloroplast fragments [28]. Formation of reduced Pheo is identified in the difference spectrum (Fig. 4) by selective bleaching of the Qx (0–0) absorption band of Pheo at 545 nm and by an appearance of absorption bands of the radical anion Pheo– at 449, 654, and ~ 800 nm, and also by an absorption decrease at 684 nm and a positive peak at 675 nm [28]. A bleaching of the Qx (0–1) absorption band of Pheo at ~515 nm is apparently masked by the negative signal at ~507 nm (Fig. 4), which is probably related to Car.
Fig. 4. Light-induced difference (light-minus-dark) spectra of photoaccumulation of reduced pheophytin in control (1) and NaBH4-treated (2) preparations of PS II core complexes in the presence of sodium dithionite and methyl viologen. The spectra were measured at room temperature and normalized to equal absorption of the preparations at 667 nm (the absorbance at the maximum of the Qy band was ~0.6). The actinic light intensity (λ > 600 nm) was ~160 mW/cm2.
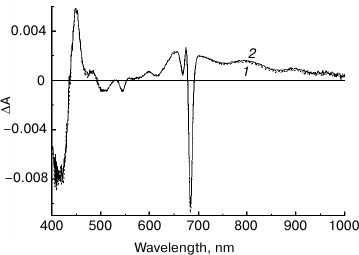
Figure 4 shows that the spectra of photoaccumulation of Pheo– for the NaBH4-treated preparation and the control sample are virtually identical to each other both in shape and amplitude. This suggests that the photochemical activity was comparable for the two preparations and that a Pheo a molecule in the PheoD1-binding site that takes part in the primary charge separation was apparently not involved in the chemical reaction with borohydride. Otherwise, a reduced yield of the PheoD1– photoaccumulation reaction would be expected for the NaBH4-treated preparation because according to the previous estimations the free energy level of the P680+131-OH-Pheo a– state lies considerably above the level of P680* due to the low reduction potential of 131-OH-Pheo a [11]. This means that only the photochemically inactive PheoD2 was subjected to borohydride modification and, accordingly, that ~50% of complexes in the NaBH4-treated preparation contained 131-OH-Pheo a in the PheoD2-binding site instead of Pheo a. Prevailing replacement of PheoD2 by 131-OH-Pheo a was observed earlier also for isolated D1/D2/Cyt b559 PS II RC complexes treated with NaBH4 [10]. Apparently, in both types of complexes the PheoD1 molecule is less accessible to NaBH4 than the PheoD2 molecule.
It is known that illumination of PS II at cryogenic temperatures under conditions when Cyt b559 is in the oxidized state leads to the formation of long-lived charge-separated states that include the reduced primary quinone QA– and the oxidized secondary electron donor (Car+, Chl+) [32-35]. The difference absorption spectra related to the formation of these states include an electrochromic short-wavelength shift of the Qx absorption band of PheoD1 at ~545 nm (the so-called C550 shift) that is an indicator of QA– formation [36, 37]. We used this shift to estimate a relative amount of the photoreducible QA in the NaBH4-treated PS II core complex preparation, comparing the amplitude of the C550 signal in the difference spectra of the treated and the control preparations (Fig. 5) measured at 100 K and normalized to equal sample absorption at the maximum at 670 nm (inset to Fig. 5). The pigment molecules that were not subjected to modification appear to mainly absorb in the region of 670 nm (Fig. 1b, curves 1-3; Fig. 3, curve 1), and thus the normalization used corresponds to a comparable concentration of core complexes in the samples.
Fig. 5. Light-induced difference (light-minus-dark) absorption spectra for control (1) and NaBH4-treated (2) preparations of PS II core complexes, measured at 100 K in the range of 450-1100 nm. Samples were illuminated with actinic light (λ > 600 nm; ~45 mW/cm2) for 30 s. The inset shows the Qy region (640-700 nm) of the absorption spectra at 100 K of control (3) and NaBH4-treated (4) preparations used to obtain the difference spectra 1 and 2, respectively. The difference spectra were smoothed and normalized to the equal absorption of the preparations at the maximum of the band at 670 nm, as shown in the inset.
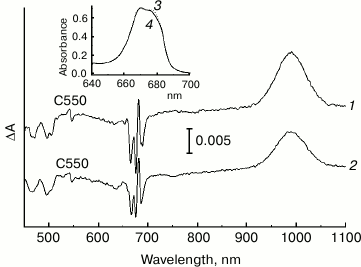
From Fig. 5, the amplitude of the C550 signal in the spectrum of the treated preparation is about 80% of that registered for the control sample. As the share of the complexes with chemically reduced PheoD2 in the treated preparation was ~50% (see above), at least a portion of the modified complexes are likely to include the functionally active QA. In agreement with literature data, the difference spectra presented in Fig. 5 contain bleaching bands of Car in the region of 450-520 nm and an intensive absorption band of the radical cation Car+ at ~990 nm [38]. Taking into account the partial removal (or modification) of the carotenoid molecules upon treatment of the complexes with borohydride (Figs. 1 and 3), one can suggest that, along with a possible dissociation of QA from RC, a loss of Car as a secondary electron donor to P680+ in the state P680+QA– could be one of the reasons for the reduced C550 signal in the NaBH4-treated preparation. The absorbance changes in the region of 620-720 nm (Fig. 5) reflect oxidation of Chl a (bleaching at 668 nm) and electrochromic band shifts of the RC pigments caused by the negative charge on QA– and the positive charge on Car+ or Chl+ [32, 33]. A small bleaching at ~637 nm observed in the difference spectrum of the treated preparation (Fig. 5) seems to indicate the ability of 131-OH-Chl a also to play a role of electron donor to P680+.
Modified preparations with spectral and photochemical properties similar to those described above were also obtained upon treatment with sodium borohydride of PS II core complexes suspended in 20 mM BTS400 buffer. In this case, a small differential signal at 667-669(–)/671-672(+) nm similar to the signal presented in the difference spectrum “NaBH4-treated-minus-control preparation” in Fig. 3 (curve 1) was more pronounced (not shown). It should be noted that the addition of NaBH4 to 20 mM BTS400 causes a stronger increase in the pH of the medium (up to pH 8.7 at 5ºC) than in the case of 100 mM BTS400 buffer (see above), which has greater buffer capacity. As expected from the literature [39, 40], the shift of the medium pH above 8 during incubation of core complexes in 20 mM BTS400 was accompanied by an almost complete loss of the PsbO protein in the modified preparations (data not shown). Oxygen-evolving activity of such preparations was completely inhibited due possibly to the influence of NaBH4 as a reducing agent on the Mn-cluster [31] and also as a result of the increase in the pH [39, 40] caused by the addition of this reagent.
DISCUSSION
To study the spectral properties of PS II RC pheophytin in intact PS II particles, isolated O2-evolving PS II core complexes from spinach were treated with sodium borohydride, which can irreversibly reduce the 131-keto C=O group of Pheo a to form 131-OH-Pheo a [10, 11, 29, 30]. Using a small difference in reactivity to borohydride between Pheo a and Chl a, we obtained modified preparation in which ~25% of the total of Pheo a molecules were replaced by 131-OH-Pheo a with a relatively low content of 131-OH-Chl a. The modified complexes contained the antenna polypeptides CP43 and CP47 and the reaction center subunits (D1, D2, Cyt b559), and in the case of 100 mM BTS400 buffer they also retained a significant portion of the PsbO protein of the oxygen-evolving complex (Fig. 2). This shows that the borohydride treatment does not lead to significant dissociation of the PS II core complex protein ensemble.
A comparison of the difference spectra for photoaccumulation of Pheo– in the NaBH4-treated preparation and the control sample (Fig. 4) showed that only the inactive PheoD2 was modified with borohydride in ~50% of RCs. Based on this result, the negative signal at 543 nm observed in the difference spectrum “NaBH4-treated-minus-control preparation” (Fig. 3, curve 1) can be identified as a selective bleaching of the Qx absorption band of the inactive PheoD2 as a result of its conversion to 131-OH-Pheo a under the influence of NaBH4. We conclude therefore that the Qx transition of the PheoD2 molecule is located at 543 nm.
As follows from Fig. 1b (curves 1-3), at an early stage of the treatment of PS II core complexes with sodium borohydride, when Pheo a is preferentially subjected to reduction with the appearance of the Qy absorption band of newly formed 131-OH-Pheo a at 657 nm, the bleaching band at 681.5 nm develops in parallel in the difference spectra. This observation indicates that, in the Qy spectral region, the native Pheo a molecule located in the PheoD2-binding site seems to absorb in the region near 680 nm. Clearly, however, some of the chemically reducible by NaBH4 Chl a molecules also absorb in the same spectral range, because the development of the Qy absorption band of 131-OH-Chl a at 639 nm in the beginning of the reaction was not accompanied by the appearance of a new bleaching band (Fig. 1b).
Comparing these results to the literature, it should be noted that previous attempts to determine the spectral characteristics of Pheo in PS II RCs mainly focused on isolated D1/D2/Cyt b559 RC complexes. There is as yet no consensus on the spectral position of the optical transitions of the PheoD2 molecule in this type of complexes. It is proposed that the Qx absorption band of PheoD2 is located at 542-543 nm [10, 12, 13, 41, 42] or in the region of 537-541.5 nm [20, 43-47]; the absorption at 670-672 nm [20, 41, 43, 44, 48, 49] or in the range of 676-680 nm [10, 12, 13, 45, 50] is ascribed to the Qy transition. A much smaller number of works are devoted to the study of the spectral properties of pheophytins in PS II core complexes [18, 51]. This is especially true of the PheoD2 molecule, which is inactive in the charge separation process, and its absorption spectrum is apparently not subjected to significant changes in response to the electron transfer and the formation of the radical ion pairs in the photoactive cofactor branch. Recently, the Qx absorption band of PheoD2 in similar PS II core complexes from spinach was identified at 541.2 nm [18]. Some discrepancy between these data and our assignment of the PheoD2 Qx transition at 543 nm could be due to the difference in the temperature of the samples (~285 K in this work and 1.7 K in [18]). Note that an entirely different approach to determine the PheoD2 Qx absorption profile based on the decomposition of the composite Qx absorption band of PheoD1 and PheoD2 into Gaussian components was used in work [18]. Earlier, a Qy absorption band with maximum at 669.3 nm at 77 K was ascribed to the PheoD2 molecule in PS II core complexes of Synechocystis PCC 6803 on the basis of analysis of the absorbance changes induced by photoreduction of the primary quinone QA [51]. Our measurements do not confirm this assignment for spinach PS II core complexes because at an early stage of the treatment of the complexes with NaBH4 there was no absorption decrease in the region of 670 nm of the difference spectra (Fig. 1b, curves 1-3), which would correspond to the appearance of the Qy band of 131-OH-Pheo a at 657 nm. A pronounced shoulder at ~670 nm on the slope of the bleaching band at 681.5 nm was observed only in a later stage of treatment (Fig. 1b, curve 4), probably reflecting the presence of Chl a pools in PS II that are characterized by a different reactivity to borohydride [21]. The absorption of the inactive Pheo in the region of ~680 nm proposed in the present work is consistent with recent results of modeling of the optical spectra of PS II core complexes in the framework of exciton theory, which show that PheoD2 dominantly contributes to the exciton absorption band at ~678 nm [16] or ~685 nm [18].
In conclusion, the identification of the Qx transition of PheoD2 in PS II core complexes at 543 nm and probable localization of its Qy absorption band in the region of ~680 nm correspond to a spectral assignment made earlier for this pigment on the basis of experiments on chemical modification of Pheo molecules in isolated D1/D2/Cyt b559 RC complexes [10-13].
This work was supported by the Russian Foundation for Basic Research (grant No. 10-04-308), the Ministry of Education and Science of the Russian Federation (state contract No. 02.740.11.029), the Program of Basic Research of the Russian Academy of Sciences “Molecular and Cell Biology” (project No. 6), and a President of the Russian Federation (grant NSh-32.2010.4).
REFERENCES
1.Renger, G. (2008) in Primary Processes of
Photosynthesis: Principles and Apparatus (Renger, G., ed.) Pt. II,
Royal Society Chemistry, Cambridge, pp. 237-290.
2.Zouni, A., Witt, H. T., Kern, J., Fromme, P.,
Krauss, N., Saenger, W., and Orth, P. (2001) Nature, 409,
739-743.
3.Ferreira, K. N., Iverson, T. M., Maghlaoui, K.,
Barber, J., and Iwata, S. (2004) Science, 303,
1831-1838.
4.Umena, Y., Kawakami, K., Shen, J. R., and Kamiya,
N. (2011) Nature, 473, 55-61.
5.Holzwarth, A. R., Muller, M. G., Reus, M.,
Nowaczyk, M., Sander, J., and Rogner, M. (2006) Proc. Natl. Acad.
Sci. USA, 103, 6895-6900.
6.Romero, E., van Stokkum, I. H. M., Novoderezhkin,
V. I., Dekker, J. P., and van Grondelle, R. (2010) Biochemistry,
49, 4300-4307.
7.Shelaev, I. V., Gostev, F. E., Vishnev, M. I.,
Shkuropatov, A. Ya., Ptushenko, V. V., Mamedov, M. D., Sarkisov, O. M.,
Nadtochenko, V. A., Semenov, A. Yu., and Shuvalov, V. A. (2011) J.
Photochem. Photobiol. B, 104, 44-50.
8.Gall, B., Zehetner, A., Scherz, A., and Scheer, H.
(1998) FEBS Lett., 434, 88-92.
9.Zehetner, A., Scheer, H., Siffel, P., and Vacha, F.
(2002) Biochim. Biophys. Acta, 1556, 21-28.
10.Shkuropatov, A. Ya., Khatypov, R. A.,
Volshchukova, T. S., Shkuropatova, V. A., Owens, T. G., and Shuvalov,
V. A. (1997) FEBS Lett., 420, 171-174.
11.Shkuropatov, A. Ya., Khatypov, R. A.,
Shkuropatova, V. A., Zvereva, M. G., Owens, T. G., and Shuvalov, V. A.
(1999) FEBS Lett., 450, 163-167.
12.Germano, M., Shkuropatov, A. Ya., Permentier, H.,
Khatypov, R. A., Shuvalov, V. A., Hoff, A. J., and van Gorkom, H. J.
(2000) Photosynth. Res., 64, 189-198.
13.Germano, M., Shkuropatov, A. Ya., Permentier, H.,
de Wijn, R., Hoff, A. J., Shuvalov, V. A., and van Gorkom, H. J. (2001)
Biochemistry, 40, 11472-11482.
14.Tomo, T., Hirano, E., Nagata, J., and Nakazato,
K. (2005) Photosynth. Res., 84, 77-83.
15.Liu, S., Dong, F. Q., Yang, C. H., Tang, C. Q.,
and Kuang, T. Y. (2006) J. Integr. Plant Biol., 48,
1330-1337.
16.Raszewski, G., Diner, B. A., Schlodder, E., and
Renger, T. (2008) Biophys. J., 95, 105-119.
17.Novoderezhkin, V. I., Dekker, J. P., and van
Grondelle, R. (2007) Biophys. J., 93, 1293-1311.
18.Cox, N., Hughes, J. L., Steffen R., Smith, P. J.,
Rutherford, A. W., Pace, R. J., and Krausz, E. (2009) J. Phys. Chem.
B, 113, 12364-12374.
19.Smith, P. J., Peterson, S., Masters, V. M.,
Wydrzynski, T., Styring, S., Krausz, E., and Pace, R. J. (2002)
Biochemistry, 41, 1981-1989.
20.Acharya, K., Neupane, B., Zazubovich, V., Sayre,
R. T., Picorel, R., Seibert, M., and Jankowiak, R. (2012) J. Phys.
Chem. B, 116, 3890-3899.
21.Scheer, H., Porra, R. J., and Anderson, J. M.
(1989) Photochem. Photobiol., 50, 403-412.
22.Berthold, D. A., Babcock, G. T., and Yocum, C. F.
(1981) FEBS Lett., 134, 231-234.
23.Van Leeuwen, P. J., Nieveen, M. C., van de Meent,
E. J., Dekker, J. P., and van Gorkom, H. J. (1991) Photosynth.
Res., 28, 149-153.
24.Van Leeuwen, P. J. (1993) The Redox Cycle of
the Oxygen Evolving Complex of Photosystem II, Ph. D. thesis,
Leiden University, The Netherlands.
25.Arnon, D. I. (1949) Plant Physiol.,
24, 1-15.
26.Laemmli, U. K. (1970) Nature, 227,
680-685.
27.Kang, D., Gho, Y. S., Suh, M., and Kang, C.
(2002) Bull. Korean Chem. Soc., 23, 1511-1512.
28.Klimov, V. V., Klevanik, A. V., Shuvalov, V. A.,
and Krasnovsky, A. A. (1977) FEBS Lett., 82, 183-186.
29.Holt, A. S. (1959) Plant Physiol.,
34, 310-314.
30.Wolf, H., and Scheer, H. (1971) Liebigs Ann.
Chem., 745, 87-98.
31.Ghanotakis, D. F., Topper, J. N., and Yocum, C.
F. (1984) Biochim. Biophys. Acta, 767, 524-531.
32.Krausz, E., Hughes, J. L., Smith, P., Pace, R.,
and Arskold, S. P. (2005) Photochem. Photobiol. Sci., 4,
744-753.
33.Schlodder, E., Renger, T., Raszewski, G.,
Coleman, W. J., Nixon, P. J., Cohen, R. O., and Diner, B. A. (2008)
Biochemistry, 47, 3143-3154.
34.Faller, P., Fufezan, C., and Rutherford, A. W.
(2005) in Photosystem II: The Light-Driven Water:Plastoquinone
Oxidoreductase (Wydrzynski, T. J., and Satoh, K., eds.) Vol. 22,
Springer, Dordrecht, The Netherlands, pp. 347-365.
35.Shinopoulos, K. E., and Brudvig, G. W. (2012)
Biochim. Biophys. Acta, 1817, 66-75.
36.Erixon, K., and Butler, W. L. (1971) Biochim.
Biophys. Acta, 234, 381-389.
37.Van Gorkom, H. J. (1974) Biochim. Biophys.
Acta, 347, 439-442.
38.Schenck, C. C., Diner, B., Mathis, P., and Satoh,
K. (1982) Biochim. Biophys. Acta, 680,
216-227.
39.Kuwabara, T., and Murata, N. (1983) Plant
Cell Physiol., 24, 741-747.
40.Cole, J., Boska, M., Blough, N. V., and Sauer, K.
(1986) Biochim. Biophys. Acta, 848, 41-47.
41.Shuvalov, V. A., Heber, U., and Schreiber, U.
(1989) FEBS Lett., 258, 27-31.
42.Breton, J. (1990) in Perspectives in
Photosynthesis (Jortner, J., and Pullman, B., eds.) Kluwer Academic
Publishers, The Netherlands, pp. 23-38.
43.Mimuro, M., Tomo, T., Nishimura, Y., Yamazaki,
I., and Satoh, K. (1995) Biochim. Biophys. Acta, 1232,
81-88.
44.Jankowiak, R., Ratsep, M., Picorel, R., Seibert,
M., and Small, G. J. (1999) J. Phys. Chem. B, 103,
9759-9769.
45.Yruela, I., Torrado, E., Roncel, M., and Picorel,
R. (2001) Photosynth. Res., 67, 199-206.
46.Xiong, L., Seibert, M., Gusev, A. V.,
Wasielewski, M. R., Hemann, C., Hille, C. R., and Sayre, R. T. (2004)
J. Phys. Chem. B, 108, 16904-16911.
47.Tomo, T., Akimoto, S., Tsuchiya, T., Fukuya, M.,
Tanaka, K., and Mimuro, M. (2008) Photosynth. Res., 98,
293-302.
48.Braun, P., Greenberg, B. M., and Scherz, A.
(1990) Biochemistry, 29, 10376-10387.
49.Konermann, L., Yruela, I., and Holzwarth, A. R.
(1997) Biochemistry, 36, 7498-7502.
50.Van Kan, P. J. M., Otte, S. C. M.,
Kleinherenbrink, F. A. M., Nieveen, M. C., Aartsma, T. J., and van
Gorkom, H. J. (1990) Biochim. Biophys. Acta, 1020,
146-152.
51.Stewart, D. H., Nixon, P. J., Diner, B. A., and
Brudvig, G. W. (2000) Biochemistry, 39, 14583-14594.