Collateral Presentation of Antigens as Physiological Prototype for Lymph Node Metastases
V. N. Manskikh1* and V. M. Perelmuter2,3
1Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: manskikh@mail.ru2Cancer Research Institute, Siberian Branch of the Russian Academy of Medical Sciences, ul. Savinykh 12/1, 634028 Tomsk, Russia
3Siberian State Medical University, Moskovskii Trakt 2, 654050 Tomsk, Russia; E-mail: pvm@ngs.ru
* To whom correspondence should be addressed.
Received September 12, 2012; Revision received November 11, 2012
The formation of lymphogenic metastases remains enigmatic. In particular, the much more pronounced predilection of carcinomas than of sarcomas to metastasizing into regional lymph nodes is an unsolved problem. We suggest that this difference could be due to the ability of epitheliocytes for a hypothetical process termed by us “collateral presentation of antigens”. Under conditions of infection of epithelium with intracellular pathogens or during inflammation, epithelial cells acquire a special receptor phenotype, undergo the epithelial–mesenchymal transition, and migrate along lymphatic vessels into lymph nodes where they present antigen to immunocytes. The collateral presentation of antigens can be of significant biological importance in the case of insufficient classical pathway of antigen presentation (by dendritic cells) or on disturbance in the death mechanisms of the infected cells. Depending on conditions of induction of the epithelial–mesenchymal transition and on possible ability of epitheliocytes to express MHC II with co-stimulating molecules, two pathways, “container-mediated” and “MHC II-dependent”, of antigen presentation in lymph nodes resulting in development of immunogenesis or anergy of immunocytes are supposed to exist. All pathways of delivery of the epithelial cells into lymph nodes and of antigen presentation by epitheliocytes terminate by death of these cells. The lymphogenic metastasizing realizes the same mechanism under conditions of tumor disease; however, this is not associated with cell death, but they actively colonize the lymph node. The proposed hypothesis allows us to explain the metastasizing of sarcomas into lymph nodes. The main prerequisite for lymphogenic metastasizing seems to be related with the mesenchymal–epithelial transition of sarcoma cells promoting their involvement in the presentation of antigens.
KEY WORDS: collateral presentation of antigens, lymphogenic metastasizing, benign epithelial inclusions, MHC II, carcinomas, sarcomasDOI: 10.1134/S0006297913030152
Abbreviations: AE1/AE3, cocktail of antibodies detecting a variety of cytokeratins; APC, antigen-presenting cell; BEI, benign epithelial inclusion; CD, clusters of differentiation; EMT, epithelial–mesenchymal transition; gp180, membrane glycoprotein 180 of intestinal epithelial cells; ICAM, intercellular adhesion molecule; IFγ, interferon γ; MHC I(II), major histocompatibility antigens I and II; TGFβ, transforming growth factor β; Th1, type 1 T-lymphocyte helpers.
There is no shortage of publications concerning mechanisms and
conditions of metastasizing. Changes that occur in tumor cells and in
places of future metastases to promote formation of secondary tumor
nodes are known in general [1-6]. In the great majority of works, the process is
considered on models of hematogenic metastasizing. Lymphogenic
metastasizing is formally similar to the hematogenic one, but it has
its own specificity. A clear manifestation of lymphogenic metastasizing
is its regular development in cases of carcinoma and the significantly
lower frequency in cases of sarcoma, notwithstanding that these two
large groups of tumors have a similar predilection for producing
hematogenic metastases [7-9].
The reason for the obvious difference between carcinomas and sarcomas
in the development of lymphogenic metastases remains unclear [8-10]. Factors that are known to
determine the appearance of lymphogenic metastases (formation of
intratumoral network of lymphatic vessels, presence of constitutive
metastatic niches and preniches, locomotion phenotype, and invasiveness
of tumor cells) are equally capable of providing the delivery and
proliferation of cells of both carcinomas and sarcomas in regional
lymph nodes [1, 5, 8-10].
It seems unlikely that the regular lymphotropism of carcinoma cells is an accidental feature inherent only in the tumor epithelium. Whatever the immediate molecular nature was of the lymphotropism of carcinoma-derived metastases, we think that this phenomenon is due to some functional feature of epithelial cells acquired during evolution and important for adaptation. Therefore, lymphogenic metastasizing is likely to possess its own physiological prototype, the knowledge of which would be favorable for understanding the formation of metastases in lymph nodes.
In general, we think that the search for and analysis of physiological prototypes of pathological processes can be very fruitful for studies on problems of pathology and especially of oncology [11-13]. There is also another essential aspect – analyzing reactions of cells and tissues under conditions of pathology can indicate the existence of yet undiscovered physiological phenomenon. In this paper, we shall try to consider lymphogenic metastasizing of tumors based on our hypothesis that it can be a manifestation (under conditions of tumor disease) of a link of the immune process, which we have termed “collateral presentation of antigens”.
COLLATERAL PRESENTATION OF ANTIGENS: DEFINITION OF THE
TERM
With this term we describe the situations when cells that formally are not components of the immune system are able to present antigens in addition1 to the classical scheme the antigen presentation2, acting as full-value analogs of macrophages (dendritic cells) or only as a “container” of the antigen for delivery into the place of localization of cells responsible for professional presentation of antigens.
1 This is the reason for the term “collateral”, i.e. “by-passing”, “additional” presentation of antigens.
2 Uptake by a macrophage (dendritic cell) of the antigen with subsequent processing, presenting of epitopes on the membrane in complex with MHC II and co-stimulating molecules, migration into lymph nodes, and presentation of antigenic determinants to T- and B-lymphocytes.
For presentation of antigens by epithelial cells at least three conditions are required: the release of the cell from the epithelial layer and acquisition of locomotion phenotype; delivery of the cell into the lymphatic vessel and lymph node; acquisition by the epithelial cell of ability for presentation of antigen Th0 to lymphocytes or phagocytosis of the epitheliocyte with associated processing and presentation of its antigens by professional APC of the lymph node.
Consequently, the collateral presentation of antigens can be realized by at least two fundamentally different hypothetical variants: “active”, when the epithelial cell itself, without mediators, acts as an APC; and “passive” (“container-mediated”), when the cell only delivers the antigen to professional APCs. Further, we shall consider these two possibilities, but initially it is reasonable to pay attention to the biological role of this hypothetical process.
FOR WHAT CAN COLLATERAL PRESENTATION OF ANTIGENS BE
REQUIRED?
We think that the collateral presentation of antigens arising under certain conditions and delivering antigenic determinants of pathogens into regional lymph nodes can be important during infections caused by intracellular pathogens, first of all, by viruses.
Infection is a complicated interaction of the micro- and macroorganism, and during infection the main purpose of the pathogen is to ensure the survival of the species due to genetically variable proliferation [14]. On the contrary, the evolutionary purpose of the macroorganism is to create the most efficient anti-infection resistance preventing the persistence, proliferation, and spreading of the pathogen in the organism and minimization of the pathogen-induced damage. Respectively, we may suppose that collateral presentation should reach two alternative purposes: to enhance the efficiency of immunogenesis, or, on the contrary, to create immunological tolerance (anergy) preventing inadequate immune response.
First, consider the place of collateral presentation of antigens among mechanisms responsible for elimination of pathogens. It is known that during evolution a number of defense systems appeared that are capable of eliminating from the macroorganism of intracellular pathogens, whereas these pathogens acquired some features allowing them to avoid the elimination. In addition to purely intracellular mechanisms, such as RNA interference [15], there are some processes that occur on the cell level. An important pathway for defense against an infectious agent is death of the infected cell, because this death simultaneously destroys the virus and inhibits the spreading of the viral infection [16]. Four types of death of the host’s infected cells are known (apoptosis, necrosis, pyroptosis, and autophagy) [17], and the role of apoptosis is characterized in most detail [16]. In some cases, apoptosis is effective also against intracellular bacterial pathogens [18].
Aside from apoptosis, autophagy can also be associated with viral infection. In some cases (infection with human immunodeficiency virus) autophagy precedes apoptosis [19], and in other cases (infection with influenza virus) inhibition of autophagy (e.g. by virus matrix protein 2) promotes apoptosis of the virus-infected cells [20].
Autophagy can also act as a defense mechanism destroying an intracellular pathogen without cell death [21]. It should be noted that the role of autophagy in resistance against intracellular pathogens is not limited to their destruction (associated with the death or with retained viability of the infected cells). Autophagy is also involved in processing of endogenously synthesized proteins (including those of the virus) followed by joining them to MHC II and presentation on the cell surface [22]. Pyroptosis and necrosis are the roughest responses to a pathogen resulting in the release of the cell contents into the extracellular medium and development of inflammation with severe alteration of tissues [17].
However, intracellular infectious agents rather frequently have features allowing them to escape elimination as a result of cell death (Fig. 1). In particular, infection with some viruses induces expression of antiapoptotic proteins, which results in replication of the viral genome before the death of the cell. Moreover, in such situations apoptosis promotes the exit of the virus from the cell and acts as a mechanism of development of the disease [16, 23, 24]. The genomes of some viruses have genes whose products inhibit autophagy, in particular, via signaling pathways associated with protein Beclin-1, and thus they prevent degradation in autophagosomes [25].
Fig. 1. Variant of sanogenesis due to death of both the infected cell and the intracellular pathogen (a) and mechanism of escaping it (b).
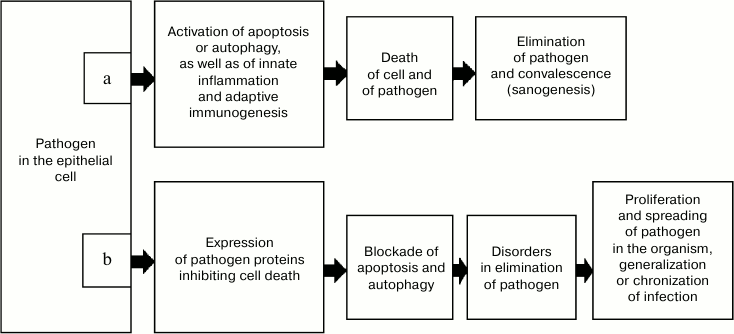
In addition to virus-initiated cell death, antivirus defense is also guaranteed by the inborn and adaptive immunity mechanisms [26]. Adaptive antivirus immunity is activated due to APCs. In APCs, viral proteins are treated with production of antigenic peptides, which in complex with MHC II stimulate CD4+ and CD8+ T- and B-lymphocytes [27]. However, similarly to the case of cell death, during evolution pathogens have developed mechanisms capable of actively preventing their recognition by the immune system, including the earliest stage of antigen presentation. Thus, such viruses as the herpes causative agent and possibly the varicella virus can cause apoptosis of Langerhans cells [28] and thus sharply decrease the efficiency of immunogenesis during these diseases. During the development of infection, disorders can arise in MHC I expression or in proteasomal degradation of proteins of the pathogen in infected cells, which makes impossible their interaction with effector CD8+ T-lymphocytes responsible for elimination of the infected cells.
It seems obvious that epithelial tissues are in closer contact with the environment than other tissues, and thus they undergo the greatest risk of contamination by intracellular pathogens. The load with pathogens is especially great for epidermis. This is indirectly indicated by its possessing the Langerhans cell network, which functions as “professional” APCs [28]. Taking these data and considerations into account, it seems reasonable that during evolution a mechanism would arise responsible for activation of immune sanogenesis even on insufficiency of apoptosis (and autophagy) in epithelial cells at the infection gate and also on failure of the classical pathway of antigen presentation. It seems possible that in this situation, epithelial cells containing an intracellular pathogen due to some inherent features can realize the collateral presentation of the antigen, i.e. to transfer it into a place with the most favorable conditions for immunogenesis. Such a place in the organism are the regional lymph nodes, and the infected epitheliocytes can act as both simple antigen-carrying “containers” and realize the full-value MHC II-dependent presentation of the antigen to Th0-lymphocytes. The immune response developing in this case leads to elimination of the pathogen from the organism, and thus it ensures efficient sanogenesis even on failure of the pathogen-dependent cell death and initial stages of the classical immunogenesis at the infection gate.
However, another variant can also be realized when the antigen delivery by epitheliocytes in the complex with the MHC II but without co-stimulating molecules is accompanied by anergy of T-lymphocytes and inhibition of inadequate immune inflammation. In this case, collateral presentation acts as a regulatory mechanism minimizing immune damage to the tissues but not being immediately associated with the elimination of the pathogen. This hypothesis is based on numerous reports that under certain conditions MHC II-positive epitheliocytes can induce antigen-specific anergy of T-lymphocytes (see below).
Based on the available information, we shall attempt to consider in more detail the probable mechanisms for delivery of the antigenic information into lymph nodes by epitheliocytes.
“CONTAINER-MEDIATED” PRESENTATION OF ANTIGENS BY
EPITHELIAL CELLS
We think that in some cases epithelial cells can act as a transporting container for antigen delivery into lymph nodes. This pathway might occur by two hypothetical variants.
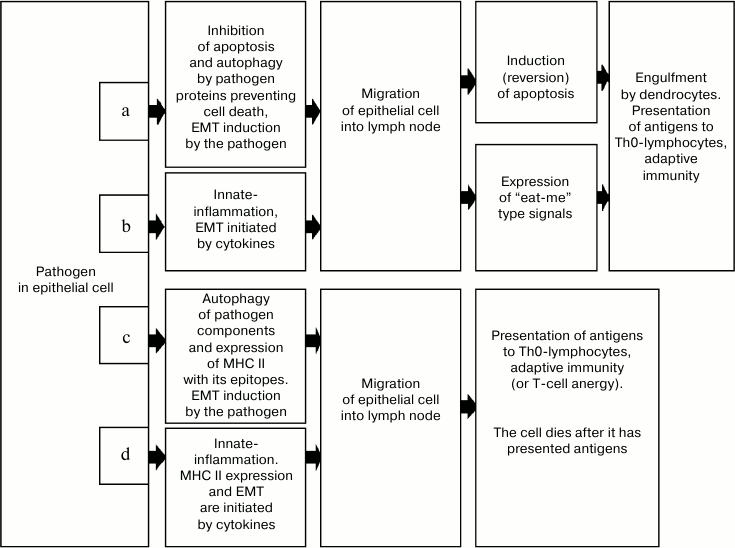
In the first case (Fig. 2a), the process is induced by appearance in the cell of viral proteins, which initiate the cell release from the layer [29] and acquisition of the locomotion phenotype [30, 31]. This reaction is supposed to be the most specific for infected cells with inhibited apoptotic or autophagic response to the pathogen. The epithelial—mesenchymal transition (EMT) seems to be the most probable mechanism of acquiring the locomotion phenotype, and this mechanism can be activated by viral proteins [30, 31]. The natural apoptotic reaction to releasing of epithelial cells from the layer, anoikis, is automatically inhibited due to EMT [32, 33], which ensures the brief retention of viability of such a cell.
Fig. 2. Variants of collateral presentation of antigens providing development of immunogenesis and sanogenesis during infections accompanied by the endocellular location of a pathogen. Inhibition of apoptosis or autophagy of the cell-carrier by proteins synthesized upon induction by pathogens can trigger mechanisms of collateral presentation of the pathogen’s antigens. a) The “container” variant associated with triggering EMT through the intracellular signaling system. The cell-carrier discharged from the bonds with other epithelial cells and possessing locomotion phenotype migrates into the regional lymph node. In the lymph node the cell dies due to reversion of the ability for apoptosis or due to expression of “eat-me” type signals. In any variant, the dying cell-carrier is engulfed by a dendritic cell performing the antigen processing and presentation to Th0-lymphocytes. The classical adaptive immunity develops. b) “The container” variant associated with development of innate-inflammation in the tissue region with infected epithelial cells. Under the influence of cytokines (TGFβ) of inflammatory effector cells, EMT occurs succeeded by processes described in variant (a). c) MHC II-dependent variant associated with autophagy of pathogen components in the cell-carrier and expression of epitopes in complex with MHC II. The EMT is induced through the intracellular signaling system. Upon immigration into the lymph node, the epithelial cell presents the antigen immediately to a Th0-lymphocyte and then dies. In this variant of collateral presentation of antigens, the epithelial cell-carrier acts as an APC, which expresses not only the antigen–MHC II complex but also the co-stimulating molecules. Adaptive immunity (or specific anergy in some cases) develops. d) MHC II variant associated with innate-inflammation. Under the influence of cytokines, the MHC II is expressed in the complex with epitopes, the EMT is induced, and then processes described in variant (c) occur.
It seems that epitheliocytes can be most easily released and delivered into lymph nodes in epithelial stem cells (cells of the intestinal crypt bottom, of the basal layer of squamous epithelium), which have some EMT features even under normal conditions [34, 35]. It is essential that, just these cells are most frequently contaminated with viruses [36]. The mechanism of subsequent directed migration of the epithelial cell into lymphatic vessels and regional lymph nodes has to be similar to the mechanism used by dendritic cells upon reception of antigenic and Toll-like signals at the infection gate [28]. In this connection, note that the directed migration of lymphocytes, macrophages, and tumor cells metastasizing into lymph nodes was recently shown to depend on the same macrophage mannose-binding receptor [37].
On coming into the lymph node, epithelial cells can undergo immunogenic apoptosis [38-41] or be totally phagocytized (due to expression of “eat-me” type signals on the membrane) by local dendritic cells [42], which are always numerous in lymph nodes [43]. Antigens inside the engulfed cell or in the apoptotic body find themselves in dendritic cells, where they undergo processing and presentation to Th0-lymphocytes in complex with MHC II. Activated immunogenesis in this case is likely to occur by the Th1-type because just this type is the most essential for elimination of infected cells.
The second variant of the “container-mediated” delivery (Fig. 2b) is different from the first by the mechanism of induction of the locomotion phenotype. In this case, the EMT is achieved under the influence of cytokines (e.g. TGFβ) in the inflammation focus. Afterwards, the infected cell directly migrates into the lymph node using a mechanism similar to that, which is used by macrophages, lymphocytes, and dendritic cells [28, 37]. The lymph node seems to lack the cytokine background responsible for induction and maintaining of the mesenchymal state, and this results in EMT reversion and the cell acquiring sensitivity to proapoptotic signals. This leads to cell death caused by anoikis, because inside the lymph node the epitheliocyte is deprived of adequate contacts either with the basal membrane or with other epithelial cells. Apoptotic bodies produced during these events contain molecules stimulating immunogenesis; therefore, their engulfment by dendritic cells is accompanied by development of immune reaction to the antigens inside the bodies. The cell can also be phagocytized in total due to ligand interaction with the “eat-me” type receptors with the subsequent development of immunogenesis according to the above-described scenario.
MHC II-DEPENDENT PRESENTATION OF ANTIGENS BY
EPITHELIOCYTES
Epithelial cells are usually rather immunologically active elements that can immediately or distantly interfere in immune processes and modulate them. Thymus stroma epithelium is known to influence the maturation of T-lymphocytes, regulation of functions of theliolymphocytes by enterocytes, and release by epithelial cells of proinflammatory (and also other) cytokines upon interaction of pathogens with Toll-like receptors, which play an important role in activation and inhibition of immune reactions [43]. The role of epitheliocytes as antigen-presenting cells is also well known, especially their MHC I-mediated interaction with CD8+ T-lymphocyte-killers [43]. There are also some data on possible presentation of antigens to T-lymphocytes by epitheliocytes with involvement of “noncanonical” antigen-presenting molecules [44].
The ability of epithelial cells to interact with CD4+ T-lymphocytes-helpers that is important for inducing a full-value adaptive immunogenesis is less clear. Under conditions of death or functional incompetence of “professional” APCs when the antigen delivery into lymph nodes is inhibited, induction of antigen-presenting abilities seems to be reasonable just in epithelial cells, which in this case act as independent full-value APCs. However, the ability for the antigen processing and its expression in complex with MHC II and co-stimulating molecules are necessary for this [43].
Antigens of intracellular infectious agents can be processed in epitheliocytes either through proteasomal degradation with a subsequent expression in complex with MHC I, or through autophagy, which can not only destroy intracellular pathogens, but also present their antigens on the membrane together with MHC II. The latter pathway is especially important because just the antigen–MHC II complex can interact with the interface of CD4+ T-lymphocyte-helper [43]. Presentation of epitopes processed upon autophagy and expressed in the complex with MHC II is considered to be an addition when MHC I-mediated presentation is impossible because aggregation of antigens prevents their degradation in proteasomes [22]. Therefore, triggering of autophagy in infected cells can lead to stimulation of CD4+ T-lymphocytes specific for the pathogen’s antigens. Various epithelial tissues were recently shown to express MHC II on membranes, especially under pathological conditions3. MHC II expression on keratinocytes is shown in demodecosis [46], leishmaniasis [47], papilloma virus infection [48], and some experimental autoimmune skin inflammatory lesions in laboratory animals [49, 50]. Epithelium in both the large and small intestine can constitutively express MHC II, and receptors are located only on the basolateral surface of enterocytes [51]. MHC II expression by enterocytes sharply increases on stimulation with proinflammatory cytokines (IFγ) [51, 52]. It was supposed that intestinal epithelial cells capable of processing antigens and presenting them in complex with MHC II, and also of expressing various co-stimulating molecules, could interact with inter-epithelial T-lymphocytes and with T-lymphocytes located under the basal membrane of the intestine, and could regulate the T-cellular response in the intestinal mucosa [51]. It is reasonable to suppose that, on the presence of MHC II in the epithelium, the triggering of autophagy in infected cells can lead to stimulation of CD4+ T-lymphocytes specific for antigens of the infectious agent.
Thus, we can conventionally put forward two variants of MHC II-dependent collateral presentation of antigens. In the first variant (Fig. 2c), expression of antigen–MHC II complex is induced by activation of autophagy, and in the second variant (Fig. 2d) the expression is induced by cytokines released by the cells during the development of inflammation in the place of the pathogen’s introduction. The further stages of the process – migration into the lymph node and presentation of antigens to immunocytes, as well as the biological interpretation of these stages – are the same for both variants.
3 MHC II in vitro induction has been described also in fibroblasts; however, this complex is shown to promote the activation of fibroblasts themselves and not their regulation of functions of T-lymphocytes [45].
The targeted delivery of epithelial cells into the lymph node is the central postulate of the hypothesis. The probability of meeting of an epitope with definite specificity presented by an epithelial cell at the infection gate, e.g. in the intestinal mucosa, with T-lymphocyte with the T-cellular receptor with the same specificity, is extremely low even under conditions of active migration of immunocytes due to immune inflammation. Peyer’s patches and solitary follicles, which also are possible places of local migration of epithelial cells, seem to be the only exceptions. Therefore, the detected capability of various epithelial cells for processing the antigen and presenting it in complex with MHC II and co-stimulating cells receives great functional meaning if there is a mechanism for delivery of such cells into the regional lymph node. The role of such a mechanism can be played by EMT, which can occur in epithelial cells either under the influence of proteins encoded by the viral genome (Fig. 2c), or under the action of cytokines in the inflammation focus (Fig. 2d).
Upon receiving the locomotion phenotype, the cell can actively reach lymphatic vessels and lymph nodes and present there the antigen in complex with MHC II using the same mechanisms as professional antigen presenters – dendritic cells [28, 43]. There are reasons to think that such a phenomenon can occur especially frequently under conditions of inflammation accompanied by dysregenerations exemplified by pseudoepitheliomatous hyperplasia associated with infiltrative growth of the epithelium, which is very similar to an invasive growth of carcinoma. A cell that finds itself inside the lymph node and presents the antigen has to die either through apoptosis (e.g. through anoikis because of absence of cytokine context maintaining the mesenchymal state) or through phagocytosis due to the ligand interaction with the “eat-me” type receptors.
The effect of antigen presentation by the epithelial cell is believed to depend on the molecular context. Antigen delivery by cells possessing MHC II and capable of presenting antigen but deprived of co-stimulating molecules (CD40, CD80, CD86) is known to stimulate antigen-specific anergy of CD4+ T-lymphocytes [51, 53]. We shall show below that in some cases the antigen presentation is arranged in just the same manner. Although such an “alternative” role of the collateral presentation of antigens seems less realizable, it can be significant as a mechanism of suppressing inadequate immune response. Therefore, it should be noted that the IFγ, which is an inducer of MHC II expression, is concurrently a factor inhibiting the classical presentation of antigens by Langerhans cells [54]. It seems not accidental that thymus, which is mainly responsible for immunological tolerance, is the only organ of the immune system possessing epithelial stroma capable of expressing MHC II [43].
Expression of co-stimulating molecules (CD40, CD58, CD80, CD86, gp180, ICAM-1) in epithelial cells has been described not once and under different conditions; however, regularities of its arising are not established. In some cases, these molecules were expressed constitutively (CD58 on enterocytes, CD86 on keratinocytes) [48, 51, 55], whereas in other cases expression of all the molecules or only of some of them was activated during inflammation [51]. And, finally, sometimes this expression was not observed at all, or only corresponding mRNAs were detected in the cytoplasm of epitheliocytes [53]. Similarly, different experimental situations revealed the ability of enterocytes and keratinocytes with MHC II to present antigens accompanied by activation of CD8+ T-lymphocytes [50, 51], CD4+ T-lymphocytes [49, 56], anergy of T-cells, and even without any effect [50, 52]. Obviously, conditions determining consequences of antigen presentation by epithelial cells are insufficiently known.
Thus, although conditions determining effects of epithelial presentation of antigens are not known completely, the probability of such process and the presence in epitheliocytes of all necessary components have been convincingly shown in different models. The two variants described – depending either on autophagy activation or on stimulation by cytokines in the inflammation focus – would be able to promote production of migrating cells – derivatives of the epithelial layer capable of delivering antigen into the lymph node, of full-value presenting it, and of realizing sanogenesis (or anergy of T-cells) if this component of adaptive immunity is incompetent.
EPITHELIAL INCLUSIONS IN LYMPH NODES: “BENIGN”
LYMPHOGENIC METASTASES
It was reported in old works [57, 58] that lymph nodes contain epithelial islets, which were termed “benign epithelial inclusions” (BEIs). By now some data have accumulated, mainly concerning differential diagnosis of such formations and of lymphogenic micrometastases of tumors [59-67]. The incidence of BEIs is not known exactly. Up to now, there is no distinct concept about the origin of BEIs. Some authors consider them to be embryonic dystopias [57, 63, 65, 66, 68], whereas others think that they are epithelial cells accidentally coming into lymphatic tracts under conditions of massive alteration of tissue or during inflammation [58, 66, 67]. From the viewpoint of collateral presentation of antigens, BEIs seem to indirectly prove the delivery into lymph nodes of epithelial cells that have succeeded in avoiding the death caused by apoptosis (anoikis), but rather have proliferated and formed a small colony. As differentiated from separate epithelial cells, which can be easily confused with macrophages and even with plasmocytes, such islets are definite morphological evidence of the presence of epithelial elements in lymph nodes4. The rapid death of epitheliocytes and their engulfment by dendritic cells seem to explain the rare detection of BEIs by pathologists in lymph nodes, similarly to apoptotic bodies under physiological conditions. The morphological detection of the collateral presentation of antigens could fail because separate epithelial cells could lose during EMT the typical morphology and acquire apparent similarity with small mesenchymal cells.
4 We observed some cases of squamous epithelial clusters of cells including small keratinized complexes in axillary lymph nodes examined because of invasive cancer of mammary gland duct. And in the primary tumor there were no signs of squamous metaplasia or squamous component of the carcinoma (unpublished data).
Metastasizing into lymph nodes from an undetected primary site is another phenomenon of malignant tumor progression that is generally known but nevertheless still unexplained. The frequency of this variant of tumor disease described by Foulds [69] that demonstrates relative independence of metastasizing on development of the primary tumor is rather high, up to 5-8% [70, 71]. As a rule, metastases without a detected primary tumor node are carcinomas5. It seems that in some such cases the primary site cannot be detected because it is formed by malignant epithelial cells of “inclusions” inside the lymph nodes.
5 We observed the case with a period of 5 years between detection of the first and second cervical lymph nodes affected by squamous carcinoma, whereas the primary tumor site was not found (unpublished data).
LYMPHOGENIC METASTASIZING OF CARCINOMAS AS MANIFESTATION OF
COLLATERAL PRESENTATION OF ANTIGENS
Now the comparison of stages of supposed collateral presentation of antigens (the cell is released from the layer, acquires locomotion phenotype, and migrates into lymphatic vessels and regional lymph nodes) reveals their similarity with events leading to formation of lymphogenic metastases.
In the framework of the concept of collateral presentation of antigens, it is reasonable to suppose that in some cases lymphogenic metastasizing has to be triggered by infection of cellular elements of carcinomas with intracellular pathogens, especially with viruses. Due to synthesis of factors inhibiting apoptosis or autophagy, these viruses prevent the death of the cell and initiate the delivery of the tumor elements into the lymph node (“container-mediated presentation of antigens”). Physiological mechanisms of tumor cell migration into lymph node can be activated by autophagy associated with degradation of pathogens resulting in presentation of antigens in complex with MHC II or in activation of MHC II and EMT by cytokines under conditions of inflammation (“MHC II-dependent presentation”).
There is a fundamental difference between normal and tumor epithelium: the carcinoma cells occurring into the lymph node have an unlimited ability for proliferation and a stable (and not short-term) blockade of the anoikis mechanism. Just this specific feature always leads to formation in the lymph node of a large colony of tumor cells, but not to inevitable death as in the case of non-malignant cells.
Thus, according to our hypothesis, metastasizing into lymph nodes in some cases is a result of infection with intracellular pathogens (especially viruses) that is essential for searching for targets and monitoring this process. Thus, decontamination of mucous membranes of organs during preneoplastic and early neoplastic processes, especially under conditions of chronic inflammation, can be an approach for preventing metastasizing of carcinoma on its possible arising from these organs.
MESENCHYMAL—EPITHELIAL TRANSITION AS A PREREQUISITE FOR
SPORADIC LYMPHOGENIC METASTASIZING OF SARCOMAS
From the standpoint of the hypothesis, it is important to analyze rather rare situations when sarcomas can give lymphogenic metastases. The frequency of sarcoma metastases into lymph nodes is 3-10%. The lymphogenic metastasizing is more often observed in highly malignant sarcomas, and among them especially often in rhabdomyosarcoma (11-36% of cases), epithelioid sarcoma (17-80%), clear cell sarcoma (25-50%), synovial sarcoma (2-17%), and angiosarcoma (11-40% of cases) [72-78].
Note that metastasizing sarcomas have in common the ability to express cytokeratins or epithelial membrane antigens, i.e. markers peculiar for epithelial tissue. Their appearance in sarcomas may be considered as a manifestation of mesenchymal—epithelial transition. The possibility of such transition in sarcomas has been shown for synovial sarcoma (without discussion about the probability of its relation with lymphogenic metastasizing) [79].
The average frequency of cytokeratin expression is 5% in different subtypes of rhabdomyosarcoma [80], but in alveolar rhabdomyosarcoma expression of different cytokeratins is up to 50% [81]. In epithelioid sarcoma a wide spectrum of epithelium differentiation markers has been detected: epithelial membrane antigen, cytokeratins 8 and 18, and in some cases cytokeratin 7 and high molecular weight keratins [82]. Expression of cytokeratins is observed in 29% of clear cell sarcomas. In the majority of synovial sarcomas cytokeratins are expressed isolated or combined to one another that can be detected with AE1/AE3 antibodies, as well as cytokeratins 7, 19 and epithelial membrane antigen [83]. Markers of epithelial differentiation are also revealed in some angiosarcomas: cytokeratins in 3% and epithelial membrane antigen in 10% [84].
These data allow us to pose a very specific question: whether the totality of the above-presented observations means that the inherence in tumor cells of some important biological features common with those of epithelial cells can be a key factor of lymphogenic metastasizing. We think that under certain conditions such feature can be the ability of the epithelial cell to realize the collateral presentation of antigens in lymph nodes. Thus, according to our hypothesis, sarcomas can give lymphogenic metastases only upon a certain degree of mesenchymal—epithelial transition of their cells.
In this work we have attempted to find a physiological foundation for lymphogenic metastasizing of tumors based on the concept that various pathological processes are products of quite “normal” physiological phenomena that can arise under unusual conditions or in unusual combinations. We think that the idea of collateral antigen presentation can be useful not only because its confirmation is promising for development of new approaches and targets for treating lymphogenic metastasizing, but also because it postulates the existence of a yet unknown immunological process that can be an important regulatory link determining the development either of immune response or anergy of lymphocytes.
We believe that the hypothesis that epithelial cells are capable of collateral presentation of antigens can solve a very old and well-known enigma of oncology about the much more pronounced predilection of carcinomas for lymphogenic metastasizing as compared to mesenchymal tumors. This hypothesis promotes understanding of why epithelial tumor cells have a tendency for migration into lymph nodes, similarly to immunologically active cells of leukemias and hematopoietic solid tumors.
PREDICTIONS FOR VERIFICATION OF THE HYPOTHESIS
Inhibition of EMT of epithelial cells under conditions of inflammation has to induce a decrease in immune response parameters during infections caused by intracellular pathogens capable of blocking apoptosis and autophagy. The inverse effect can also occur due to abolishment of anergy of T-lymphocytes.
Introduction into a lymph node of epithelial cells carrying MHC II and heterologous antigen has to significantly influence immune response efficiency because, depending on the expression of co-stimulating molecules on epitheliocytes, it can induce either immunogenesis or anergy.
Contamination of carcinoma cells with intracellular pathogens inhibiting apoptosis or autophagy increases the risk of lymphogenic metastasizing and must be considered as an obligate promoter of metastatic disease and a potential target for prophylaxis of this disease.
Signs of mesenchymal–epithelial transition in sarcomas increase the risk of development of lymphogenic metastases; thus, inhibition of this transition has to lower the probability of lymphogenic metastasizing.
In the presence of pseudoepitheliomatous hyperplasia, separate epithelial cells and whole epithelial inclusions are expected to be regularly found in regional lymph nodes.
The probability of development of BEIs in lymph nodes has to depend on of the contamination of the epithelium with intracellular pathogens and/or on the presence of inflammation.
Under conditions of dysregeneration, the inflammatory infiltrate of the regional lymph node stroma and tissue can contain cells with genetic, transcriptional, and expression markers of epithelial cells produced as a result of EMT and be morphologically indistinguishable from mesenchymal cells.
REFERENCES
1.Psaila, B., and Lyden, D. (2009) Nat. Rev.
Cancer, 9, 285-293.
2.Kaplan, R. N., Riba, R. D., Zacharoulis, S.,
Bramley, A. H., Vincent, L., Costa, C., MacDonald, D. D., Jin, D. K.,
Shido, K., Kerns, S. A., Zhu, Z., Hicklin, D., Wu, Y., Port, J. L.,
Altorki, N., Port, E. R., Ruggero, D., Shmelkov, S. V., Jensen, K.
K., Rafii, S., and Lyden, D. (2005) Nature, 438,
820-827.
3.Hiratsuka, S., Watanabe, A., Aburatani, H., and
Maru, Y. (2006) Nat. Cell Biol., 8, 1369-1375.
4.Hiratsuka, S., Watanabe, A., Sakurai, Y.,
Akashi-Takamura, S., Ishibashi, S., Miyake, K., Shibuya, M., Akira, S.,
Aburatani, H., and Maru, Y. (2008) Nat. Cell Biol.,
10, 1349-1355.
5.Peinado, H., Lavotshkin, S., and Lyden, D. (2011)
Sem. Cancer Biol., 21, 139-146.
6.Kaplan, R. N., Psaila, B., and Lyden, D. (2006)
Cancer Metastasis Rev., 25, 521-529.
7.Fong, Y., Coit, D. G., Woodruff, J. M., and
Brennan, M. F. (1993) Ann. Surg., 217, 72-77.
8.Leong, S. P., Cady, B., Jablons,
D. M., Garcia-Aguilar, J., Reintgen, D., Jakub,
J., Pendas, S., Duhaime, L., Cassell,
R., Gardner, M., Giuliano, R.,
Archie, V., Calvin, D., Mensha, L., Shivers,
S., Cox, C., Werner, J. A., Kitagawa, Y., and
Kitajima, M. (2006) Cancer Metastasis Rev., 25,
221-232.
9.Nathanson, S. D. (2003) Cancer, 98,
413-423.
10.Kawada, K., and Taketo, M. M. (2011) Cancer
Res., 71, 2014-2018.
11.Perelmuter, V. M. (1983) in Current Problems
of Modern Oncology (Vasil’ev, N. V., ed.) [in Russian], Tomsk
University Publishers, Tomsk, pp. 47-51.
12.Manskikh, V. N. (2009) Dokl. Ross. Akad.
Nauk, 425, 126-128.
13.Perelmuter, V. M., and Manskikh, V. N. (2012)
Biochemistry (Moscow), 77, 111-117.
14.Odintsov, Y. N., and Perelmuter, V. M. (2002)
Sib. Med. Zh., 1-2, 44-46.
15.Lopez-Fraga, M., Martinez, T., and
Jimenez, A. (2009) BioDrugs, 23, 305-332.
16.Kaminskyy, V., and Zhivotovsky, B. (2010) J.
Intern. Med., 267, 473-482.
17.Labbe, K., and Saleh, M. (2008) Cell Death
Differ., 15, 1339-1349.
18.Dockrell, D. H. (2001) J. Infect.,
42, 227-234.
19.Espert,
L., Denizot, M., Grimaldi, M., Robert-Hebmann, V., Gay,
B., Varbanov, M., Codogno, P., and
Biard-Piechaczyk, M. (2006) J. Clin. Invest., 116,
2161-2172.
20.Gannage, M., Dormann,
D., Albrecht, R., Dengjel, J., Torossi, T., Ramer,
P. C., Lee, M., Strowig, T., Arrey,
F., Conenello, G., Pypaert, M., Andersen, J.,
Garcia-Sastre, A., and Munz, C. (2009) Cell Host Microbe,
6, 367-380.
21.Levine, B., and Deretic, V. (2007) Nat. Rev.
Immunol., 7, 767-777.
22.Munz, C. (2006) Cell Microbiol., 8,
891-898.
23.Hay, S., and Kannourakis, G. (2002) J. Gen.
Virol., 83, 1547-1564.
24.Richard, A., and Tulasne, D. (2012) Cell Death
Dis., 3, 277.
25.Deretic, V., and Levine, B. (2009) Cell Host
Microbe, 5, 527-549.
26.Gregoire, C., Chasson, L., Luci,
C., Tomasello, E., Geissmann, F., Vivier, E., and
Walzer, T. (2007) Immunol. Rev., 200, 169-182.
27.Heath, W. R., Belz, G. T., Behrens,
G. M., Smith, C. M., Forehan, S. P., Parish,
I. A., Davey, G. M., Wilson, N. S., Carbone, F.
R., and Villadangos, J. A. (2004) Immunol. Rev.,
199, 9-26.
28.Cunningham, A. L., Abendroth,
A., Jones, C., Nasr, N., and Turville, S. (2010)
Immunol. Cell Biol., 88, 416-423.
29.Ray, R. B., Basu, A., Steele, R., Beyene, A.,
McHowat, J., Meyer, K., Ghosh, A. K., and Ray, R. (2004)
Virology, 321, 181-188.
30.Horikawa, T., Yang, J., Kondo,
S., Yoshizaki, T., Joab, I., Furukawa, M., and
Pagano, J. S. (2007) Cancer Res., 67, 1970-1978.
31.Horikawa,
T., Yoshizaki, T., Kondo, S.,
Furukawa, M., Kaizaki, Y., and Pagano, J. S. (2011)
Br. J. Cancer, 104, 1160-1167.
32.Taddei, M. L., Giannoni, E., Fiaschi, T., and
Chiarugi, P. (2012) J. Pathol., 226, 380-393.
33.Guo, W., and Giancotti, F. G. (2004) Nat. Rev.
Mol. Cell Biol., 10, 816-826.
34.Shipitsin, M., Campbell, L.
L., Argani, P., Weremowicz, S., Bloushtain-Qimron,
N., Yao, J., Nikolskaya, T., Serebryiskaya,
T., Beroukhim, R., Hu, M., Halushka, M.
K., Sukumar, S., Parker, L. M., Anderson, K.
S., Harris, L. N., Garber, J. E., Richardson, A.
L., Schnitt, S. J., Nikolsky, Y., Gelman, R.
S., and Polyak, K. (2007) Cancer Cell, 3,
259-273.
35.Fillmore, C., and Kuperwasser, C. (2007)
Breast Cancer Res., 9, 303.
36.Shindoh, M., Chiba, I., Yasuda, M., Saito, T.,
Funaoka, K., Kohgo, T., Amemiya, A., Sawada, Y., and Fujinaga, K.
(1995) Cancer, 76, 1513-1521.
37.Marttila-Ichihara, F., Turja, R.,
Miiluniemi, M., Karikoski, M., Maksimow, M., Niemela,
J., Martinez-Pomares, L., Salmi, M., and Jalkanen, S. (2008)
Blood, 112, 64-72.
38.Savill, J., and Fadok, V. (2000) Nature,
407, 784-788.
39.Yrlid, U., and Wick, M. J. (2000) J. Exp.
Med., 191, 613-624.
40.Schaible, U. E., Winau, F., Sieling, P. A.,
Fischer, K., Collins, H. L., Hagens, K., Modlin, R.
L., Brinkmann, V., and Kaufmann, S. H. (2003) Nat.
Med., 9, 1039-1046.
41.Albert, M. L. (2004) Nat. Rev. Immunol.,
4, 223-231.
42.Zitvogel, L., Apetoh, L., Ghiringhelli, F., and
Kroemer, G. (2008) Nat. Rev. Immunol., 8, 59-73.
43.Male, R., Brostoff, J., Rott, D. B., and Roitt,
I. M. (2006) Immunology, 7th Edn., Mosby,
London-Philadelphia-St. Louis-Tokyo.
44.Groh, V., Steinle, A., Bauer,
S., and Spies, T. (1998) Science, 5357,
1737-1740.
45.Ohyama, H., Nishimura, F., Meguro, M.,
Takashiba, S., Murayama, Y., and Matsushita, S. (2002) Cytokine,
17, 175-181.
46.Huisinga, M., Failing, K., and
Reinacher, M. (2007) Vet. Immunol.
Immunopathol., 118, 210-220.
47.Fondevila, D., Vilafranca, M., and
Ferrer, L. (1997) Vet. Immunol. Immunopathol., 56,
319-327.
48.Ortiz-Sanchez, E., Chavez-Olmos,
P., Pina-Sanchez, P., Salcedo, M., and Garrido, E.
(2007) Int. J. Gynecol. Cancer, 17, 571-580.
49.Fan, L., Busser, B. W., Lifsted, T.
Q., Oukka, M., Lo, D., and Laufer, T. M.
(2003) Proc. Natl. Acad. Sci. USA, 100,
3386-3391.
50.Kim, B. S., Miyagawa, F., Cho, Y.
H., Bennett, C. L., Clausen, B. E., and Katz, S. I.
(2009) J. Invest. Dermatol., 129, 2805-2817.
51.Hershberg, R. M., and Mayer, L. F.
(2000) Immunol. Today, 21, 123-128.
52.Nickoloff, B. J., Mitra, R. S., Green,
J., Zheng, X. G., Shimizu, Y., Thompson, C., and
Turka, L. A. (1993) J. Immunol., 150, 2148-2159.
53.Byrne, B., Madrigal-Estebas,
L., McEvoy, A., Carton, J., Doherty, D. G., Whelan,
A., Feighery, C., O’Donoghue, D. P., and
O’Farrelly, C. (2002) Hum. Immunol., 63,
977-986.
54.Ozawa, H., Aiba, S., Nakagawa, S., and Tagami, H.
(1996) Eur. J. Immunol., 26, 648-652.
55.Framson, P. E., Cho, D. H., Lee, L.
Y., and Hershberg, R. M. (1999) Gastroenterology,
116, 1054-1062.
56.Kaiserlian, D., Vidal, K., and
Revillard, J. P. (1989) Eur. J. Immunol., 19,
1513-1516.
57.Petrov, N. N. (1910) General Teaching about
Tumors (Pathology and Clinics) in Hygiene and Sanitaria [in
Russian], St. Petersburg.
58.Abrikosov, A. I. (1947) Special Pathological
Anatomy, Vol. 1 [in Russian], Medgiz, Moscow-Leningrad.
59.Paull, G., and Mosunjac, M. (2003)
Diagn. Cytopathol., 29, 163-166.
60.Cohn, D. E., Folpe, A. L., Gown,
A. M., and Goff, B. A. (1998) Gynecol.
Oncol., 68, 210-213.
61.Brooks, J. S., LiVolsi, V. A., and
Pietra, G. G. (1990) Am. J. Clin. Pathol., 93,
741-748.
62.Jadusingh, I. H. (1992) J. Clin.
Pathol., 45, 1023-1026.
63.Fisher, C. J., Hill, S., and
Millis, R. R. (1994) J. Clin. Pathol., 47,
245-247.
64.Douglas-Jones, A. G. (1994) J. Clin.
Pathol., 47, 868-869.
65.Kadowaki, M., Nagashima,
T., Sakata, H., Sakakibara, M., Sangai,
T., Nakamura, R., Fujimoto,
H., Arai, M., Onai, Y., Nagai, Y., Miyazawa,
Y., and Miyazaki, M. (2007)
Breast Cancer, 14, 425-428.
66.Jaffer, S., Lin, R., Bleiweiss, I.
J., and Nagi, C. (2008) Arch. Pathol. Lab. Med.,
132, 1940-1942.
67.Paner, G. P., Turk, T. M., Clark,
J. I., Lindgren, V., and Picken, M. M. (2005)
Arch. Pathol. Lab. Med., 129, 317-321.
68.Resetkova, E., Hoda, S. A., Clarke, J.
L., and Rosen, P. P. (2003) Arch. Pathol. Lab. Med.,
127, e25-e27.
69.Foulds, L. (1969) Neoplastic Development,
Academic Press, London.
70.Fizazi, K., Greco, F. A., Pavlidis,
N., and Pentheroudakis, G.; ESMO Guidelines Working
Group (2011) Ann. Oncol., 22, Suppl. 6, vi64-68.
71.Tan, W. W. (2012) Metastatic Cancer with
Unknown Primary Site (Harris, J. E., ed.) http://emedicine.medscape.com/article/280505-overview
72.Guzzetta, A. A., Montgomery, E.
A., Lyu, H., Hooker, C. M., Meyer, C.
F., Loeb, D. M., Frassica, D., Weber, K. L.,
and Ahuja, N. (2012) J. Surg. Res., 177,
116-122.
73.Skinner, K. A., and Eilber, F. R. (1996) Surg.
Oncol. Clin. North Am., 5, 121-127.
74.Fong, Y., Coit, D. G., Woodruff,
J. M., and Brennan, M. F. (1993) Ann. Surg.,
217, 72-77.
75.Mazeron, J. J., and Suit, H. D. (1987)
Cancer, 60, 1800-1808.
76.Weingrad, D. N., and Rosenberg, S. A. (1978)
Surgery, 84, 231-240.
77.Ross, H. M., Lewis, J.
J., Woodruff, J. M., and Brennan, M. F. (1997) Ann.
Surg. Oncol., 4, 491-495.
78.Blazer, D. G., 3rd, Sabel, M. S., and
Sondak, V. K. (2003) Surg. Oncol., 12, 201-206.
79.Subramaniam, M. M., Navarro, S., and
Llombart-Bosch, A. (2011) Arch. Pathol. Lab. Med.,
135, 1001-1009.
80.Coindre, J. M., de Mascarel,
A., Trojani, M., de Mascarel, I., and Pages, A.
(1988) J. Pathol., 155, 127-132.
81.Bahrami, A., Gown, A. M., Baird, G. S.,
Hicks, M. J., and Folpe, A. L. (2008) Mod. Pathol., 21,
795-806.
82.Humble, S. D., Prieto, V. G., and Horenstein, M.
G. (2003) J. Cutan. Pathol., 30, 242-246.
83.Wick, M. R., and Hornick, J. L. (2009) in
Diagnostic Immunohistochemistry: Theranostic and Genomic
Applications, 3rd Edn. (Dabbs, D. J., ed.) Elsevier Inc,
Philadelphia, pp. 83-136.
84.Al-Abbadi, M. A., Almasri, N. M., Al-Quran, S.,
and Wilkinson, E. J. (2007) Arch. Pathol. Lab. Med., 131,
288-292.