REVIEW: Domains of a- and b-Globin Genes in the Context of the Structural–Functional Organization of the Eukaryotic Genome
S. V. Razin1,2*, S. V. Ulianov2, E. S. Ioudinkova1, E. S. Gushchanskaya1,2, A. A. Gavrilov1,3, and O. V. Iarovaia1
1Institute of Gene Biology, Russian Academy of Sciences, 34/5 Vavilov str., 119334 Moscow, Russia; E-mail: sergey.v.razin@usa.net2Biological Faculty, Lomonosov Moscow State University, 119234 Moscow, Russia
3Oslo University Medical Research Center in Russia, 34/5 Vavilov str., 119334 Moscow, Russia
* To whom correspondence should be addressed.
Received December 20, 2011; Revision received January 31, 2012
The eukaryotic cell genome has a multilevel regulatory system of gene expression that includes stages of preliminary activation of genes or of extended genomic regions (switching them to potentially active states) and stages of final activation of promoters and maintaining their active status in cells of a certain lineage. Current views on the regulatory systems of transcription in eukaryotes have been formed based on results of systematic studies on a limited number of model systems, in particular, on the α- and β-globin gene domains of vertebrates. Unexpectedly, these genomic domains harboring genes responsible for the synthesis of different subunits of the same protein were found to have a fundamentally different organization inside chromatin. In this review, we analyze specific features of the organization of the α- and β-globin gene domains in vertebrates, as well as principles of activities of the regulatory systems in these domains. In the final part of the review, we attempt to answer the question how the evolution of α- and β-globin genes has led to segregation of these genes into two distinct types of chromatin domains situated on different chromosomes.
KEY WORDS: α- and β-globin genes, genomic domains, transcription regulation, globin gene evolutionDOI: 10.1134/S0006297912130019
Abbreviations: a-MRE, Major Regulatory Element of α-globin gene domain; DHS(s), DNase I Hypersensitivity Site(s); LCR, Locus Control Region; MCS, Multispecies Conserved Sequence; OR, Olfactory Receptor gene.
In the prokaryotic genome, functionally related genes are combined as
operons, which are targets for the regulatory systems responsible for
the control of transcription. In eukaryotic cells, functionally related
genes are often situated on different chromosomes. Therefore, during
many years transcription was believed to be regulated on the level of
promoters of individual genes. It is now clear that the situation is
more complicated. Studies on chromatin and epigenetic mechanisms acting
on the level of the packaging of genetic material in the cell nucleus
revealed a multilevel system of the transcription regulation in
eukaryotic cells. In addition to the control of activities of
individual promoters, it is equally important to know whether a
chromatin domain including a given gene has active or inactive status
[1]. New findings, in particular the demonstration
that histone modifications usually called the histone code play a role
in the control of DNA packaging in chromatin, have restored interest in
the domain hypothesis of eukaryotic domain organization. This
hypothesis was proposed in the late 1970s [2, 3]. In its classic variant, the hypothesis suggested
that the whole genome should consist of uniform
structural–functional units, or chromatin domains. A chromatin
domain is a rather extended region of the genome, and within its limits
the packaging of chromatin fibrils can be changed locally without
extending onto the flanking regions. A chromatin domain can include one
or several genes. Activation of a chromatin domain changes its status
with subsequent potential transcriptional activation of all genes
situated in the domain. In many cases, the activation of a chromatin
domain correlates with its transformation from a DNase-resistant to a
DNase-sensitive configuration. The domain hypothesis of eukaryotic
genome organization has been, in particular, based on results of
studies on the sensitivity to DNase I of the active genes and their
flanking sequences. It was shown in pioneer works of Weintraub’s
laboratory that actively transcribing tissue-specific genes were
preferentially processed in permeabilized nuclei treated with DNase I
[4]. Later, it was shown that the preferential
sensitivity to DNase I was specific for a rather extended genomic
region including one or several transcribing genes and the flanking DNA
sequences [5, 6]. Two
observations seemed to be especially interesting for researchers: (i)
the same genomic regions were preferentially sensitive or relatively
resistant to DNase I in cells differentiated by different pathways, and
(ii) the DNase-sensitive regions have markedly distinct limits [6]. These very observations led to the idea that the
genome should consist of uniform structural–functional units
inside which the transcriptional status of the genes could be regulated
through changes in the chromatin packaging (Fig. 1). For a long time, molecular mechanisms responsible
for the packaging and status of a chromatin fibril were studied. By now
it has been established that the compactization of a chromatin fibril
is mainly controlled through histone acetylation [7, 8]. The domain model of the
genome organization suggests that some boundary structures should exist
for isolating the domain from the environment and limiting the
chromatin reconfiguration at the boundaries of individual domains. Such
boundary elements were found and termed insulators. Insulators were
first detected on the boundary of the locus of heat shock genes
hsp70 from Drosophila melanogaster [9]. Later such elements were found in all groups of
eukaryotes studied [10, 11].
Fig. 1. Scheme illustrating the main idea of the domain hypothesis of genome organization.
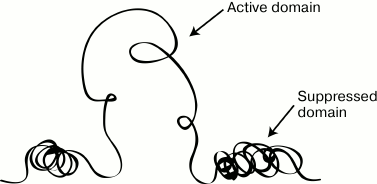
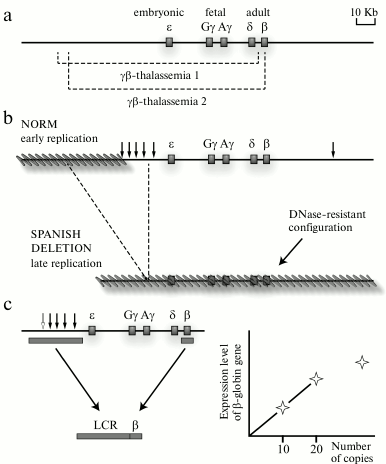
The detection of a locus control region of the human β-globin gene domain and then also of locus control regions of some other genomic domains was crucial for the model of domain organization of the genome. Experimental data on the differential sensitivity of extended chromatin domains to nucleases and the detection of a special class of regulatory elements (insulators) that, in particular, are responsible for delimitation of genomic domains could be sufficiently explained from the standpoint of the hypothesis of domain organization of the genome; nevertheless, the regulation of the transcriptional status of the domain as a whole remained unclear. It was reasonable to suppose that some regulatory elements should exist to control the transcriptional status of the genomic/chromatin domain. Such regulatory element was found in the domain of human β-globin genes. This element was identified by two lines of observations. Analysis of natural deletions influencing the expression of human β-globin genes (Fig. 2a) revealed that the so-called “Spanish deletion” resulting in the removal from the genome of a rather extended DNA fragment including sequences located in the 5′-flanking region of the β-globin gene cluster but lacking just the β-globin genes and their promoters leads to termination of transcription of all β-globin genes situated on the deletion-containing chromosome. Moreover, the domain of β-globin genes loses its preferential sensitivity to DNase I in erythroid cells, and the domain replication time changes (Fig. 2b). Under normal conditions, the domain of β-globin genes in erythroid cells is replicated in the beginning of the S-phase (that is specific for the majority of transcribing genes), whereas in the chromosome carrying the Spanish deletion the domain is replicated in the end of the S-phase (that is specific for heterochromatin and tissue-specific genes inactive in this cell lineage) [12]. The Spanish deletion is associated with the removal from the genome of a cluster of DNase I-Hypersensitive Sites (DHSs) situated at a distance of 6-22 kb before the cluster of β-globin genes [12] (Fig. 2b). It was supposed that this cluster of DHSs should contain a regulatory element that controls the transcriptional status of the β-globin gene domain. To test this hypothesis, transgenic mice were produced whose genome was integrated with a mini-domain containing one of the human β-globin genes and the above-mentioned cluster of DHSs removed from the genome due to the Spanish deletion (Fig. 2c). Analysis of these transgenic mice revealed that the presence in the construct of the DHS cluster was responsible for a high and independent of integration position (proportional to the number of integrated copies) level of expression of the human β-globin gene in the erythroid cells of the mice. The authors concluded that the studied DHS cluster included a regulatory element capable of creating an autonomous erythroid-specific mini-domain in the genome of the transgenic mice [13]. The detection of the regulatory element operating at the domain level and termed Locus Control Region (LCR) in the domains of β-globin genes of other vertebrates [14] was favorable for the wide acknowledgement of main ideas of the domain hypothesis of genome organization [2, 3]. The domain of human β-globin genes and similarly structured domains of β-globin genes of different vertebrates concurrently became the most popular models used for analyzing different regulatory mechanisms of gene expression. Concentration of the efforts of many research groups for characterizing the same group of model systems revealed many basic principles of transcription regulation in eukaryotes. We shall discuss this in more detail in the following sections of this review. But here we would like to emphasize that focusing studies on the same group of model system for some years prevented the realization that the eukaryotic genome imagined as a mosaic of uniform chromatin domains, either active or inactive, was too simplified, and even false in the general case. The incompetence of the classic model of genome domain organization [2, 3] became obvious when the structure of α-globin gene domains in mammals and birds could not be explained within the framework of the model. These domains are located in chromosomal segments enriched with constantly expressed genes (housekeeping genes) and are highly sensitive to DNase I in both erythroid and non-erythroid cells (i.e. irrespectively of the transcriptional status of the α-globin genes) [15]. The α-globin gene domain is not isolated by insulators from the adjacent regions of the genome. Moreover, the major regulatory element of the α-globin gene domain is situated within the intron of the housekeeping gene adjacent to the domain [16]. All these features of organization of the tissue-specific domain of α-globin genes were difficult to explain from the standpoint of the classic model of domain organization of the genome. The overlapping of genomic domains and even of transcription units occurred not uncommonly in the genome of higher eukaryotes [17]. Obviously, such genes could not be situated within isolated structural–functional domains similar to the domain of the β-globin genes. Therefore, the domain model of genome organization had to be revised. This resulted in the concept of open and closed genomic domains (this should not be confused with “open” and “closed” conformation of chromatin) [14, 18]. The open genomic domain (a domain with vague boundaries) is determined, first of all, on the functional level, and it can be overlapped on a chromosome with other similar domains. Such domains often include genes that are not related functionally and phylogenetically. It is clear that regulatory systems of open genomic domains (those with vague boundaries) have to be essentially different from regulatory systems of domains with fixed boundaries (closed domains).
Fig. 2. Identification of the locus control region (LCR) of human β-globin genes. a) Natural deletions leading to appearance of different thalassemias due to physical elimination of all or of the majority of the β-globin genes. Deleted fragments are shown by dotted lines under the scheme of the domain. b) Scheme illustrating the Spanish deletion position and consequences of this deletion. Vertical arrows show positions of DNase I hypersensitivity sites. Inactive regions of the genome are indicated by the slanted gray lines. As a result of the Spanish deletion eliminating the cluster of DNase I hypersensitivity sites and the genome segment adjacent to this cluster at the 5′-end, the β-globin gene domain is completely inactivated. c) Scheme demonstrating the strategy of creating an erythroid-specific mini-domain and results of analyzing the level of expression in the genome of transgenic mice of the human β-globin gene from this mini-domain. At relatively low number of mini-domain copies, the total amount of the human β-globin gene product is proportional to the number of mini-domain copies carrying this gene that are integrated into the transgenic mouse genome.
In the following chapters of this review, we analyze in detail specific features of regulatory mechanisms of transcription in open and closed domains exemplified by domains of the α- and β-globin genes.
DOMAIN OF BETA-GLOBIN GENES
General characteristics. Domains of human, mouse, and chicken β-globin genes are the best studied (Fig. 3). In humans and mice, the domains of the β-globin genes are situated, respectively, on the 11th and 7th chromosome and are components of an essentially more extended cluster of olfactory receptor genes. The length of these domains is not determined accurately because erythroid-specific DHSs that seem to indicate positions of regulatory elements of the β-globin genes domains are located rather distantly before the β-globin gene cluster inside the chromosome segment containing the genes of the olfactory receptors [14]. Enhancers of β-globin genes are present also in the 3′-terminal flanking region of the cluster of human β-globin genes. The approach of these enhancers to genes encoding the fetal β-globin chains as a result of some natural deletions leads to development of some thalassemias (diseases associated with absence or imbalance of expression of different subunits of hemoglobin) [19]. In erythroid cells of mice, the region of preferential sensitivity to DNase I is significantly larger than the insulator-delimited cluster of β-globin genes, and it includes a number of genes of olfactory receptors located in both the 5′- and 3′-terminal flanking regions [20]. But it should be emphasized that the olfactory receptor genes situated within the boundaries of this DNase-sensitive domain are not expressed in erythroid cells, and this is a good illustration of the principle of the multilevel regulation of gene activities. In human erythroid cells, the domain of the preferential sensitivity to DNase I, which includes the β-globin genes cluster, has not been analyzed in detail. Nevertheless, there are indirect indications that in humans this domain is also rather extended and is significantly larger than the genomic segment delimited by the locus control region and the 3′-terminal insulator [21].
Vertebrates have several globin genes (of both a- and b-type), which are differentially expressed at different stages of the ontogenesis. For the clusters of β-globin genes of humans and other mammals, the locations of these genes on the chromosome in the order of their activation during the ontogenesis is an important specific feature (Fig. 3). The embryonic gene ε is the nearest to the LCR [22].
Fig. 3. Genomic surrounding of clusters of β-globin genes of human, mouse, and chicken. The DHSs located within LCR limits are indicated by numerals increasing with distance from the cluster. The 3′-DHS is a conserved DHS situated on the 3′-end of the β-globin gene cluster and co-localized with the insulator. Erythroid-specific DHSs located at great distances before the cluster of β-globin genes of the human and mouse genomes are shown by arrows with indicated distances (in kb) from the β-globin gene cluster. In the scheme, the β-globin genes are represented with their traditional names that are widely used in the modern literature, although in databases other names of these genes are used (see text). OR, olfactory receptor genes; β/ε enh, enhancer located between the β and ε genes, which is present only in the domain of chicken β-globin genes; FOLR, folate receptor gene.
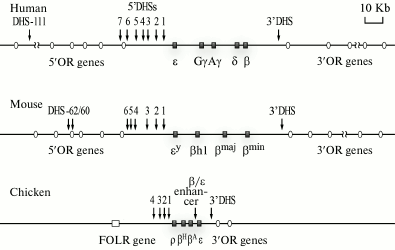
The domain of chicken β-globin genes seems to be the most typical representative of closed domains. This extended domain of 33 kb is located on the first chromosome between the folate receptor gene separated from the globin genes by a heterochromatin region and the cluster of olfactory receptor genes (Fig. 3). The functional domain, which involves the locus control region and is delimited with the 5′- and 3′-insulators, is rather accurately co-localized with the erythroid-specific domain of the preferential sensitivity to DNase I and the erythroid-specific domain of histone hyperacetylation [23]. Between the LCR of the β-globin gene domain and the folate receptor gene, there is a permanently condensed region (facultative heterochromatin) [24]. The domain includes a cluster consisting of four β-globin genes: ρ (HBG1), βH (HBE1), βA (HBG2), and ε (HBE), as well as some regulatory elements coinciding with the hypersensitivity sites to DNase I and required for regulating the transcription, replication time, and chromatin domain status [25].
Boundary elements of the β-globin gene domain. The boundary elements of the genome (insulators) were already mentioned in the first section of this review (see also [11, 26]). Classic insulators display both enhancer-blocking activity (preventing the influence of enhancer on a promoter situated behind the insulator [9]) and barrier activity (preventing processive propagation of covalent modifications of histones along the chromatin fiber [27]). Defective insulators also exist having only one of the above-mentioned activities. They should be more correctly termed either enhancer-blocking or barrier elements. Unfortunately, not all authors follow this rule, and therefore the term “insulator” is rather commonly used in the literature to indicate both enhancer-blocking elements lacking the barrier activity and barrier elements lacking the enhancer-blocking activity.
The insulator situated on the 5′-end of the LCR in the domain of chicken β-globin genes and co-localized with DHS4 (Fig. 3) is the best-studied insulator of vertebrates. This insulator is competent because it possesses both the enhancer-blocking and barrier activities [28, 29]. The minimal DNA fragment possessing the insulator activity (the core element) co-localized with DHS4 has the size of 250 bp and represents a CpG-island, which is somewhat like promoters of housekeeping genes [30]. Within boundaries of the minimal insulator, five binding sites of different proteins have been mapped [30]. One of these sites, the so-called footprint II (FII), is necessary and sufficient for manifesting the enhancer-blocking activity. This site is associated with the multifunctional transcriptional factor CTCF [31]. Deletion of the FII results in a loss of enhancer-blocking activity of the insulator. Recent works have revealed that the activity of the enhancer-blocking element in addition to CTCF is contributed by the protein cohesin. The CTCF-binding site is overlapped with the cohesin-binding site. Knockdowns by both CTCF and cohesin are associated with removal of the enhancer-blocking activity of DHS4 [32]. A physical interaction of CTCF and cohesin is necessary for enhancer-blocking activity [33].
The barrier activity of the insulator is retained on deletion of the CTCF-binding site [31]. This means that the barrier and enhancer-blocking activities are provided for by different structural elements of the insulator, and the barrier activity of the insulator depends on interaction with proteins other than CTCF [29], in particular with protein USF1, which binds with the insulator and attracts to it complexes of H3K4- and H4R3-specific histone methyltransferases, histone acetyltransferases, and chromatin remodeling complexes [34, 35]. Thus, the idea of the insulator as a passive element preventing the propagation of various signals (“traffic jam” theory) is far from reality. In fact, the insulator is a nucleation point where different enzymatic complexes are assembled, which is responsible for chromatin remodeling and histone modification.
The 3′-boundary of the domain of chicken β-globin genes is significantly less studied. At the distance of ~5 kb behind the ε gene, the constitutive site of hypersensitivity to DNase I (3′-DHS) was found. This site is somewhat similar in structure and functions to the 5′-terminal insulator of the DHS4 domain. Inside 3′-DHS, some CTCF-binding sites have been mapped. Correspondingly, 3′-DHS has enhancer-blocking activity [36]. However, unlike the DHS4, 3′-DHS is unable to protect the transgene against the positional effect, i.e. it does not have the barrier function that is inherent in classic insulators.
CTCF-dependent enhancer-blocking elements have been also identified in the domains of β-globin genes of human and mouse, where they are situated more or less similarly to the domain of chicken β-globin genes –at the 5′-end of the locus control region and in the 3′-terminal flanking region of the cluster of β-globin genes [37-39]. Other authors have shown that the 5′-insulator of the domain of human β-globin genes can protect transgenes against positional effects, i.e. similarly to the 5′-DHS4, the chicken β-globin gene domain insulator possesses both the enhancer-blocking and barrier activities [40].
The functional role of the enhancer-blocking elements located at the 5′-end of the LCR in the domains of β-globin genes of human and mouse is not quite clear. As discussed above (subsection “General Characteristics” of this section), the regulatory elements of these domains are also present outside the region delimited by these enhancer-blocking elements.
Regulation of β-globin gene transcription. The LCR is the major regulatory element that controls the expression of β-globin genes. At first, the LCR attracted researchers’ attention because this regulatory element was supposed to control the genomic domain packaging in chromatin, but data on the activity of this LCR are very contradictory. On one hand, the LCR undoubtedly is able to form an active erythroid-specific chromatin domain in the ectopic position [41, 42]. On the other hand, a directed total deletion of the LCR from the normal genomic position results only in a sharp decrease in the expression of globin genes, whereas the preferential sensitivity of the β-globin gene domain to DNase I in erythroid cells and an increased level of histone acetylation are retained [21, 43]. Causes of pronounced differences between the LCR activities detected in the normal and ectopic positions are unknown. Most probably, the 5′-flanking region of the β-globin gene domain includes some regulatory elements, each of which can maintain an active status of this domain in erythroid cells. In this case, the elimination of the LCR itself will not lead to the loss of the DNase-sensitive domain, whereas the elimination of a more extended DNA fragment involving the LCR and unidentified additional regulatory elements will have more dramatic consequences, which are actually observed in the case of the Spanish deletion [12].
The LCR of the human β-globin gene domain consists of a number of modules co-localized with individual hypersensitivity sites to DNase I (DHS2-5; Fig. 3). DHS1 is not eliminated by the Spanish deletion (Fig. 2b) [12]; therefore, it is usually not considered to be a part of the LCR. DHS2-5 can be considered to be LCR subdomains. One of these subdomains (DHS5) is an insulator, as discussed in the previous section. DNA fragments involving DHS2-4 turned out to be erythroid-specific enhancers, as demonstrated experimentally by transfection of constructs with the reporter gene [44]. The most powerful enhancer was mapped in the DHS2 region [44]. In DHS2-4, binding sites for different transcription factors were mapped, in particular, GATA-1, NF-E2, AP1, and CP2 [45, 46]. In the genome of transgenic mice, short DNA fragments (~1 kb) originating from DHS2 and DHS4 can directly produce erythroid-specific DHSs in ectopic positions [47]. More careful analysis of a DNA fragment from DHS4 resulted in identification of a minimal fragment (101 bp) capable of directing the production of erythroid-specific DHSs in ectopic positions. Within this DNA fragment, the binding sites of SP1, GATA1, AP1, and NF-E2 were identified, and the GATA1-binding sites were represented by a tandem of inversed repeats located at a distance of ~50 bp after the AP1/NF-E2 binding site [47]. Later such organization of the AP1/NF-E2 and GATA1 binding sites was also revealed in DHS1-3 [48]. Mutations preventing the binding of NF-E2 and AP1 led to inability to direct the production of DHSs in ectopic positions [48].
In domains of β-globin genes of different vertebrates, the LCR is organized similarly to its organization in the domain of human β-globin genes [49-51]. In all these domains, LCR is situated before the gene cluster (in the order of transcription of the globin genes) and consists of some functional blocks in positions coinciding with those of DHSs. There is a significant homology between the DNA sequences in individual functional blocks within the boundaries of the same LCR and between the LCRs of domains of β-globin genes of different organisms. First of all, it concerns the binding sites of crucial erythroid-specific transcription factors – their composition and mutual positions [48].
After the LCR had been shown to be a block of tissue-specific enhancers, the question of the mechanism of action of LCR was reduced to the question of the action mechanism of enhancers. In the case of the domain of β-globin genes, it is also necessary to explain the activation selectivity of different genes in the course of the organism’s development (the so-called expression switching of globin genes). There are currently two groups of most popular models of enhancer action: (i) direct enhancerpromoter interaction, and (ii) formation of an activator complex on the enhancer with its subsequent transfer onto the promoter. The first group of models is based on the hypothesis that the enhancer physically interacts with the promoter and thus provides the stabilization of a produced pre-initiating transcription complex [52]. Since enhancers are commonly situated rather distantly from promoters, these regulatory elements can directly interact only upon the looping of the chromatin fibril fragment that separates them. Therefore, the models postulating the direct enhancerpromoter interaction in the English literature are termed “looping models”.
Just statistically, the probability of a direct contact of the LCR with any promoter must be inversely proportional to the distance between them. Therefore, the sequential positions of β-globin genes according to the order of their activation during the ontogenesis that is typical for clusters of β-globin genes of mammals [53] can be functionally important. Due to its proximity to the LCR, the embryonic ε gene must be transcribed preferentially as compared to other β-globin genes, which could be efficiently transcribed only upon forced switching out the ε gene through some epigenetic mechanism. In fact, experiments on transgenic mice whose genome contained restructured copies of the domain of the human β-globin genes have shown that the gene position inside the cluster really determines the time of its expression during ontogenesis [39, 54].
In the framework of the classical model of direct interaction of the LCR with the promoter, it is difficult to explain the concurrent expression of at least two globin genes (genes σ and b in humans) in “adult” erythroblasts. It was supposed that the LCR should produce in turn short-living alternative complexes with the promoters of the activated genes. This hypothesis was confirmed by analysis of the transcription of β-globin genes in individual cells using immunofluorescence staining of intron sequences in the primary transcripts [55, 56].
An experimental approach has been recently elaborated that allows researchers to directly analyze the spatial configuration of different chromosomal loci. This procedure, named Chromosome Confirmation Capture (3C), allows to determine the relative interaction frequencies between different pairs of genomic elements [57]. Using the 3C approach, the LCR was shown to directly interact with promoters of the activated genes in the domain of mouse β-globin genes. It was also revealed that these genes were in complexes not only with the LCR, but also between themselves. This became the basis for a model of an active chromatin hub [58, 59]. The active chromatin hub is a complicated complex of DNA regulatory sequences and transcriptional factors bound to them. Remote regulatory elements and promoters of controlled genes are in immediate physical contact as they are connected through interactions of the transcription factors, some of which recognize the gene promoters, whereas others are bound with the regulatory elements. The active chromatin hubs are dynamic complexes whose stability depends on the spectrum of transcription factors involved in their formation [58, 60]. The chromatin hub is not assembled all at once. Consider the active chromatin hub of the mouse β-globin gene domain as an example [61]. This domain involves four genes with activities depending on the developmental stage. The genes βmaj and βmin are expressed in the adult organism, whereas the genes εy and βh1 are active during embryogenesis. At the stage of erythroid cell differentiation, which precedes the activation of the globin gene transcription, an immature chromatin hub involving the LCR and some remote regulatory sequences (5′DHS – 85, 5′DHS – 60/–62) are accumulated in the locus of the β-globin genes. At the beginning of the terminal differentiation of the cells, the promoters of active β-globin genes are also attracted to the hub. Strictly speaking, the enhancerpromoter complex can be considered as a “minimal” hub; however, in all known systems the mature chromatin hub formed with involvement of the locus control region always includes some regulatory elements modulating the activity of the hub [61]. Although the chromatin hub model is now generally accepted [61-63], it cannot explain some known facts. For example, it is difficult to explain from the viewpoint of this model the significance of the strictly determined positions of the globin genes in the order of their activation during ontogenesis. The mechanism of expression switching of the globin genes also remains unexplained. The authors of these works can only establish that the hub of embryonic erythroid cells contains embryonic β-globin genes, whereas the hub of adult erythroid cells contains “adult” globin genes [58, 59].
The LCR is characterized by a high concentration of binding sites of transcriptional factors [51]. When bound to the LCR, these transcriptional factors can attract to the LCR various multienzyme complexes capable of acetylating histones and remodeling chromatin [64, 65]. Thus, the LCR can be a center of formation of an active chromatin domain [66, 67]. Generally, an active domain has to spread to both sides. However, the described LCRs of the β-globin gene domains contain an insulator at the 5′-end. This leads to polarity in the spreading of chromatin activating modifications. The polarity of propagation of an activating signal can be also provided otherwise. The LCR is shown to be a site for initiation of so-called intergenic transcription [68]. The transcriptional complex is known to attract histone acetylases and chromatin remodeling complexes to the transcribed region [69, 70]. Moving towards the cluster of globin genes, the transcriptional complex will promote reorganization of the whole transcribed domain [71]. The processive propagation of the activating signal from the LCR to the genes is the main postulate of models of LCR action including a scanning stage [72]. Note that the models of LCR action postulating the scanning stage and the models postulating direct contact of the LCR with promoters of activated genes are not incompatible. The scanning model can better explain the mechanism of formation of an active chromatin domain. This process can occur during differentiation stages that precede the beginning of the active transcription of the globin genes.
Subdomain organization of the domain of β-globin genes. Analysis of relative levels of DNase I sensitivity of different segments of the domain of human β-globin genes revealed three subdomains inside this domain: the LCR-involving regulatory subdomain; the embryonic-fetal subdomain involving genes ε, Gγ, and Aγ; and the adult subdomain involving genes σ and β [68]. Although the whole domain of the β-globin genes in erythroid cells is characterized by an increased sensitivity to DNase I, the sensitivities of the embryonic-fetal and adult subdomains are significantly higher in cells where the corresponding genes are transcribed. The LCR-involving subdomain is characterized by very high sensitivity to DNase I in all erythroid cells, including those at the differentiation stage preceding the start of the transcription of the globin genes [68]. These subdomains can also be discriminated by analyzing the histone acetylation levels in different types of erythroid cells [21, 73]. It is reasonable to think that the differential activation of the embryonic-fetal and adult subdomains is essential for expression switching of globin genes during development. The subdomain status was shown to be controlled through so-called intergenic transcription [68]. The deletion of the promoter controlling the transcription of the “adult” subdomain of the domain of human β-globin genes resulted in prevention of transcription of the σ and b genes, although the promoters of these genes and the LCR were not affected. Therefore, it was supposed that the active status of the adult subdomain should be determined by a processive acetylation of histones within boundaries of this subdomain and be mediated through low-level transcription with RNA polymerase II [71].
DOMAIN OF ALPHA-GLOBIN GENES
General characterization. Similarly to domains of β-globin genes, domains of human, mouse, and chicken α-globin genes are best studied. Genes encoding the a-subunit of hemoglobin are located on the 16th chromosome in humans and on the 14th and 11th chromosomes in mouse and chicken, respectively. The domain of α-globin genes has a similar structure in all these species: one embryonic gene (ζ, π), at least one α-globin gene of the adult type (a), and one or two minor α-globin genes of adult type (aD, θ, μ) [74] (Fig. 4). The domains of α-globin genes are open-type domains with corresponding characteristic features: they are situated in gene-enriched regions of chromosomes, are preferentially sensitive to nucleases in all cell types, and are replicated at the beginning of the S-phase of the cell cycle [15, 75, 76]. The cluster of α-globin genes is flanked by housekeeping genes, which are actively transcribed in all known types of cells [16, 77].
Fig. 4. Domains of α-globin genes of human, mouse, and chicken. a) Region of gene synteny flanking clusters of α-globin genes from the 5′-end. Vertical arrows indicate DHSs co-localized with MCS-R1-MCS-R4 (see text). Numerals above the arrows indicate the distance in kb between DHSs and the α-globin gene cluster. In the schemes, traditional names of the α-globin genes are used. The horizontal arrows indicate the transcription direction: “globin” – the genes above the line, “anti-globin” – the genes under the line. b) Structure of activator complex of the human α-globin gene domain and experiments demonstrating the key role of α-MRE in the activator complex assemblage (according to [96]). The plots show summarized results of experiments analyzing the spatial interaction of DHSs of the domain with the α2- and α1-globin gene promoters that have been obtained using the 3C approach; under the plots the 3C data interpretation is presented.
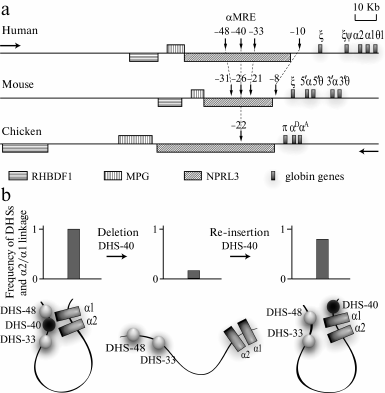
Comparative analysis of positions of the genes and conserved regions of the DNA sequence around the cluster of α-globin genes of human, mouse, chicken, and zebra fish has revealed a synteny region that includes the whole cluster of α-globin genes and the extended 5′-terminal region (Fig. 4) [16, 75]. In all known genomes of vertebrates, directly before the cluster of α-globin genes a gene is situated that is transcribed in the direction opposite to the direction of the globin gene transcription. For a long time this gene was represented in different databases under the name of C16orf35, but recently it was renamed as NPRL3 (NPR3 like) [78]. NPRL3 is transcribed in cells differentiated by various pathways (i.e. it is a housekeeping gene). In the fifth intron of NPRL3, a major regulatory element of the α-globin gene domain is located that has some features of the locus control region [16, 79]. The elimination of this element as a result of a spontaneous deletion of a 16-kb DNA fragment leads to a severe thalassemia. The transcription level of the α gene in the affected chromosome is lower than 1% of the normal level [80].
The eliminated regulatory element was first identified in the domain of human α-globin genes and was called DHS40 because it was found in one of DNase I-sensitive sites at a distance of 40 kb before the first gene of the domain [79]. Later it was named a-MRE (Major Regulatory Element) [81, 82]. DHS40 is a powerful erythroid-specific enhancer [83], and its core element representing a fragment with length of 350 bp contains several binding sites of erythroid-specific transcriptional factors, in particular GATA1/2 and NF-E2 [84]. The most important elements of a-MRE in all known organisms are two NF-E2 binding sites separated by a linker of 21 bp [16]. In the literature, these two sites are frequently called Maf-binding sites (YGCTGASTCAY; Maf recognition sequences, MARE) after the name of the small unit of NF-E2 [85]. The pair of MARE elements is flanked by binding sites of the erythroid-specific transcription factor GATA.
In addition to a-MRE, domains of α-globin genes of different vertebrates also contain other regulatory elements. In particular, this is indicated by the presence in these domains of a number of conserved DNase I hypersensitivity sites and of so-called multispecies conserved sequences (MCS) of the genome. MCSs co-localized with promoters (MCS-P) and regulatory elements (MCS-R) are the most conserved [74]. For our discussion, MCS-R1-MCS-R4 located in the 5′-flanking region of the cluster of α-globin genes are the most interesting. In the human genome they are co-localized with DHS-48 (MCS-R1), DHS-40 (MCS-R2), DHS-33 (MCS-R3), and DHS-10 (MCS-R4); a-MRE is located in MCS-R2 (Fig. 4a) [74].
Locations of different regulatory elements are well studied in the domain of chicken α-globin genes. In addition to α-MRE, this domain also contains two erythroid-specific enhancers, one of which located directly after the αA gene [86] activates the expression of adult globin genes, whereas the other located in the intron of the TMEM8 gene is responsible for integration of this gene into the regulatory network of the domain of α-globin genes [87]. Before the cluster of chicken α-globin genes, there is a CpG island harboring the starting site of DNA replication [88], gene NPRL3 promoter [89], and CTCF-dependent silencer, which stabilizes the level of the NPRL3 gene expression in erythroid cells [89]. In non-erythroid cells, a part of this CpG island is methylated, and, according to one of the models, this initiates the inactivation of the α-globin gene cluster [90]. It is interesting that between α-MRE and the chicken α-globin gene cluster there is a full-value insulator possessing both the enhancer-blocking [91] and the barrier [92] activities. The biological role of this insulator is unclear. In erythroid cells, a-MRE undoubtedly stimulates the transcription of the α-globin genes. Thus, the enhancer-blocking element must be inactivated at least in these cells. Also, it cannot be excluded that this element with pronounced activity inside constructs bearing the reporter gene [91] is unable to work in the genomic context. And the barrier element function in the α-globin gene domain is even more unclear because this genomic segment possesses the active chromatin configuration in cells differentiated by different pathways (see above). In this connection, we can note the model proposed by Dillon and Grosveld that postulates that in open-type domains (functional domains) insulators do not perform any function and are retained during evolution in the same positions because they hinder just nothing [18].
Regulation of α-globin gene transcription. It has been said earlier that in the closed-type domains the transcription of tissue-specific genes is activated step-by-step: at first the chromatin domain is activated (its packaging in chromatin changes), then the activity of promoters of one or another gene located in this domain is stimulated [1]. In the open-type domains including domains of α-globin genes, the transcription seems also to be regulated at the level of the chromatin domain, although it is not associated with a cardinal change in the domain packaging inside chromatin. In fact, the domains of α-globin genes possess the open (DNase-sensitive) configuration in both erythroid and non-erythroid cells [15]. Nevertheless, some less cardinal changes in chromatin structure within boundaries of the functional domain of α-globin genes occur in erythroid cells. This is manifested, in particular, by a significant increase in the acetylation level of histones H3 and H4 at some particular positions [8]. Certain data indicate that in the chicken α-globin gene domain this process is controlled by a-MRE through stimulation of low-level transcription of the whole domain of the α-globin genes [93]. In other words, in these cases the same regulatory principles are acting as during the activation of the subdomain of the β-globin gene domain in humans [68].
A crucial role in the regulation of α-globin gene transcription belongs to targeted activation of promoters of individual genes. This is mediated, in particular, by formation of activator chromatin blocks. The step-by-step assembly of such blocks is well studied in chicken erythroid cells. It has been shown that an incompetent activator block containing a-MRE, CpG- island before the α-globin gene cluster, and the αD gene promoter is assembled already in precursors of erythroblasts. In adult type erythroblasts transcribing the αD and αA genes, this block is supplemented with the DNase I hypersensitivity site located at the distance of 9 kb upstream of the cluster of α-globin genes (–9 DHS) and the erythroid-specific enhancer located downstream of the cluster of α-globin genes [94]. Direct interactions of α-MRE and some other erythroid-specific erythroid elements with globin genes were also shown in domains of α-globin genes of human and mouse [95, 96]. Moreover, α-MRE was shown to play a key role in the assembly of the activator complex. The removal of this element resulted in disintegration of the whole complex simultaneously with termination of the transcription of α-globin genes. Re-insertion of α-MRE into another genomic position within the boundaries of the α-globin genes domain ensured the assembly of the normal activator complex and restoration of α-globin gene transcription (Fig. 4b) [96]. Another interesting phenomenon was observed with the mouse model. The activation of the α-globin gene transcription was shown to correlate with attraction of these genes (in the complex with erythroid-specific regulatory elements) to the preexistent transcriptional fabric responsible for transcription of housekeeping genes and located in the genomic synteny region before the cluster of α-globin genes [62]. The same scenario seems to be realized in the domain of chicken α-globin genes. This is evidenced by a direct interaction in erythroblasts of the housekeeping gene NPRL3 promoter with α-MRE and globin genes [94]. Moreover, the promoter of NPRL3 interacts with promoters of other housekeeping genes located near the cluster of α-globin genes [97].
Activation of the expression of α-globin genes during development is under the control of a number of erythroid-specific transcription factors [98]. In multipotent hematopoietic cells, a key role belongs to factor GATA-2, and its plantings are nucleation centers attracting transcription factor NF-E2 and the so-called pentameric complex involving SCL. Starting from the stage of erythroid precursors, GATA-2 is replaced by the related factor GATA-1, which forms a complex with the coregulator FOG-1 [99] and binds with various additional regulatory sites including promoters of globin genes. Similarly to GATA2, GATA-1 also attracts NF-E2 and SCL. The GATA-1-binding sites are co-localized with the majority of earlier described DNase I hypersensitivity sites [98], including the sites which are directly involved in formation of the united activator complex of the α-globin gene domain [95]. Model experiments have shown that the transcription factors are planted independently onto removed regulatory elements and promoters of α-globin genes. However, α-MRE is necessary for attracting RNA polymerase II (Pol II) to promoters of α-globin genes. Pol II is also attracted to α-MRE, and this process does not require the presence of promoters. Therefore, it is likely that Pol II is initially attracted to α-MRE and then somehow transferred onto the promoters of α-globin genes [95].
Expression switching of the α-globin genes. Vertebrates have a special embryonic α-type gene (ζ in human and mouse, π in chicken) that is expressed at the yolk bag stage and then is inactivated. Globin genes of both the major and minor adult type are expressed during all stages of development [100, 101]. Thus, the question about mechanisms of expression switching of the α-globin genes is really a question about the inactivation pathways of embryonic genes. Experiments with transfection of various constructs containing the chicken π gene into erythroid cells of the early embryonic and adult types [101], as well as experiments for creating transgenic mice with the genome containing the integrated human ζ gene together with the flanking sequences, have shown that all regulatory elements needed for the inactivation in due time of embryonic α-globin genes are adjacent to these genes. For inactivation of the chicken π gene, DNA sequences are required that are present in the promoter region (~350 bp) of this gene. The inactivation of the π gene correlates with methylation of the promoter region [102]. The inactivation of the human ζ gene upon termination of the early embryonic stage of development is also totally determined by sequences adjacent to this gene [103]. A careful analysis has revealed that these events require both the sequences located in the promoter region (~550 bp before the ζ gene) and the sequences located in the 3′-terminal flanking region of this gene, which is suggested to involve a silencer [100]. It is still unknown whether embryonic genes are fully autonomous, i.e. if they do not need activation with remote regulatory elements. Analysis of the structure of the activator chromatin domain of chicken α-globin genes has revealed that the π gene promoter does not interact either with α-MRE or with an enhancer element located on the 3′-end of the α-globin gene cluster [104]. On the other hand, it has been shown that the location of the ζ gene adjacent to α-MRE is fundamentally important for the expression of this gene during the early embryonic stage. The displacement of the ζ gene into the most removed position through inversion of the whole gene cluster with respect to α-MRE resulted in complete inactivation of the ζ gene during all stages of development [105].
Inactivation of α-globin genes in non-erythroid cells. In closed-type domains, i.e. in the domain of β-globin genes, the inactivation of the genes in non-erythroid cells is mediated through packing the domain into a compact (DNase-resistant) chromatin. In the domain of α-globin genes and other open-type domains, this mechanism is not used. However, the problem of reliable repression of globin genes in non-erythroid cells still remains urgent. This problem could probably be solved by providing for a high tissue-specificity of the α-globin gene promoters. But the human α gene promoter is really not tissue-specific. It is located in the CpG island, and at least in model experiments with promoter-controlled reporter gene transfection it displays a high activity in both erythroid and non-erythroid cells [106]. The mode of inactivation of the α-globin genes in human non-erythroid cells was unclear for a long time. An involvement in this process of the Polycomb group repressor proteins was recently shown [107]. Moreover, nonmethylated CpG island were found to be involved in the attraction of repressor complexes of the Polycomb group to the promoters of human α-globin genes [108].
The Polycomb repressor complexes are known to be responsible for the long-term inactivation of genes expressed during early developmental stages, and they present an important element of the epigenetic system of memory. The involvement of the Polycomb group proteins in repression of tissue-specific α-globin genes was a rather unexpected finding. It will be interesting to find whether these proteins are involved in repression of transcription of other tissue-specific genes situated in open-type domains.
ORIGIN AND EVOLUTION OF DIFFERENT-TYPE DOMAINS
Due to fundamentally different organization of the domains of a- and β-globin genes together with a strictly coordinated expression of these genes, questions concerning the origin and regularities of evolution of different-type domains are especially interesting. Comparative studies on organization of the domains of a- and β-globin genes and their genomic environment in different taxonomic groups represent an approach to this problem. This approach is promising for understanding the level of openness of the two domain types for integration of foreign genes and to answer the question which domain – open (functional) or closed – is primary from the evolutionary standpoint. And finally, this approach allows one to get an idea of coordination of evolution of the a- and β-globin genes and their genomic environment.
Comparative analysis of the genomic environment of the cluster of α-globin genes in different species revealed that in chicken the region adjacent to the α-globin genes from the 3′-end underwent a chromosomal rearrangement that resulted in inverted locations of some orthologous genes adjacent to the cluster of α-globin genes in chicken as compared to human and other vertebrates. The gene TMEM8 in chicken occurred to be located nearer to the cluster of α-globin genes, whereas in humans it is situated at a distance of 170 kb from it (Fig. 5). This relocation correlates with the pronounced change in the expression type and structure of the gene TMEM8. In humans, this gene encodes a transmembrane protein that is expressed in the pancreas, placenta, spleen, and lymphocytes, where it is a marker of their inactive state. In chicken the TMEM8 gene is preferentially expressed in erythroid cells and is not expressed in lymphoid cells. Moreover, induction of erythroid differentiation was found to be accompanied by a significant increase in the transcriptional level of the TMEM8 gene [87]. The significant change in tissue specificity of TMEM8 expression correlates with the acquisition of its own erythroid-specific enhancer. Studies by the 3C approach of the domain configuration revealed that the TMEM8 gene was integrated into the regulatory system of the α-globin genes domain but retained some independence [87]. These observations suggest that in the chicken genome functional boundaries of the α-globin gene domain have been extended as compared to the genome of mammals. Unfortunately, at present one cannot say why this expansion of the functional domain occurred upon the genomic rearrangement, what reasons induced the acquisition by the TMEM8 gene of the erythroid-specific expression type, whether this event was occasional or necessary, primary or secondary relative to the inversion, and whether the selection pressure acted towards the “erythroid specificity” of TMEM8. It seems important to note that exons situated in the end of the gene are much more conserved during evolution than exons situated in the beginning of the gene, in the “dangerous vicinity” of the cluster of α-globin genes. In other words, the evolutionary pressure for changing the TMEM8 gene was the higher, the nearer was the exon to the α-globin genes [87].
Fig. 5. Genomic organization of joint clusters of α- and β-globin genes and of separate clusters of α- and β-globin genes in different vertebrates. The locus containing the MPG-NPRL3-α-β-GBY-LUC7L genes is an ancestral α-β-globin cluster of gnathostomatous vertebrates [118] that is retained during the evolution of amphibians. The complex of MPG-NPRL3-α genes is specific for all contemporary vertebrates. In the genome of bony fishes (zebra fish) and amphibians, the α- and β-globin genes are located closely. In the genome of birds and mammals, the β-globin and α-globin clusters are located on different chromosomes, and in mammals and monotremes this cluster is flanked by genes of the olfactory receptors from both sides, and in the birds’ genome only from one side. The region contacting the α-globin cluster in the birds’ genome is also rearranged from the 3′-end as compared to the genome of mammals: a large chromosomal region flanked with the TMEM8 and LUC7 genes is inverted. In the duckbill’s genome, the α-globin gene cluster contains a β-like gene of ω-globin and the GBY gene homologous to cytoglobin, which was separated structurally and functionally long before the division of the joined α-β-globin domain [117, 118].
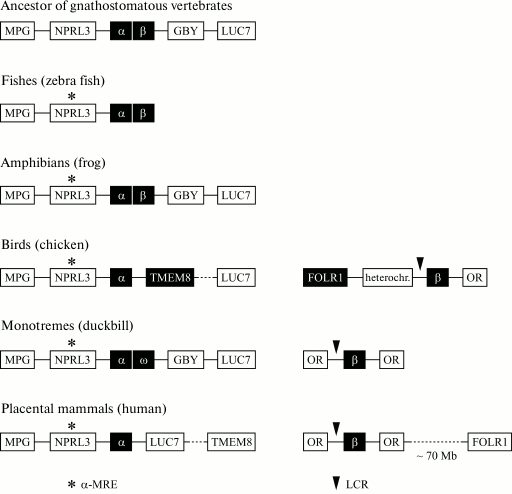
The genomic context of the β-globin gene cluster in mammals and birds is also different (Fig. 5). In mammals, the cluster of β-globin genes is flanked by genes of olfactory receptors (OR). In birds, genes of olfactory receptors flank the β-globin genes from one side, and from the other side the folate receptor gene FOLR1 is situated, which is separated from the cluster of globin genes by a region of condensed heterochromatin [14]. In humans, the FOLR1 gene is situated on the other shoulder of the 11th chromosome, i.e. it is removed from the cluster of β-globin genes. FOLR1 encodes the folic acid receptor also in humans, and according to the UCSC database it is expressed only in tissues of epithelial origin. In chicken, the folate receptor gene is erythroid-specific and is expressed during early stages of erythroid differentiation (the formation of CFU-E) preceding the expression of the globin genes. The cluster of chicken β-globin genes and the folate receptor gene are separated by an insulator, are regulated independently, and are components of different genomic domains [24]. Moreover, a DNA fragment with length of 3 kb situated before the FOLR1 gene includes all regulatory elements required for the correct expression of this gene during the early stage of the erythroid differentiation (CFU-E).
Thus, the translocation-caused rapprochement of the TMEM8 gene and the domain of chicken α-globin genes, on one hand, and the rapprochement of the FOLR1 gene with the domain of chicken β-globin genes, on the other hand, correlated with the gaining by these genes of the erythroid-specific type of expression. And the rapprochement of the TMEM8 gene with the erythroid-specific open domain (the domain of α-globin genes) caused the integration of this gene into the regulatory networks of the domain of α-globin genes, whereas the FOLR1 gene situated beyond the insulator-indicated boundary of the closed-type domain of β-globin genes retained independence and regulatory self-competence.
Which type of domain, open or closed, is evolutionarily more ancient, and is such a question reasonable? It is now generally recognized that the functional complexity and diversity of contemporary genomes appeared as a result of both evolution inside simple genomes of ancestral organisms and multiple vertical and horizontal transfers of genes, genomic fragments, and whole genomes. The ancestor of globin genes is supposed to appear in the genome of ancient unicellular eukaryotes as a result of a horizontal transfer from bacterial genomes concurrently with the appearance of endosymbiotic mitochondria and plastids [109]. A temporal retention of the structural and functional isolation of gene-interventionists seems to be necessary as the only reasonable strategy for survival of a new gene in an aggressive environment of a heterogeneous genome. From this standpoint, closed-type domains isolated from the genomic environment, self-competent and independent of regulation, are similar to such gene-interventionists immediately upon their introduction into the host’s genome.
Two sequential cycles of full-genomic duplication were key stages in the evolution of chordates. A dramatically grown excessiveness of the genome became fuel for evolution that promoted the diversification and specialization of many physiological functions. In particular, specialized systems appeared responsible for oxygen transport and storage (myoglobin, cytoglobin, and hemoglobin). The fourfold excessiveness of the genome in the protogenome of the organism, which appeared as a result of full-genomic duplications, can be also followed in contemporary organisms. The genomic environment of the cluster of α-globin genes, as well as of genes of myoglobin and cytoglobin (paralogon [110]) demonstrates features of a common organization and features of macrosyntheny [110]. Certainly, numerous translocations and duplications during evolution wash away this macrosyntheny. Natural selection corrects the gene complex, and as a result the genes capable of cooperating form a tightly linked group. It is obvious that in addition to the initial functional isolation, some integration processes occur inside the genomes accompanied by gaining certain advantages, alongside with the retention of the initial function. It seems possible that both open-type domains and transcriptional fabrics formed by colinearly situated genes are products of such integration processes. It should be once more reminded that inside the open domain of chicken α-globin genes, inactive insulator-like elements have been detected, which seemed to lose their functions during evolution. In the framework of this logic, the closed-type domains seem to be more ancient.
The reconstructed evolution of α- and β-globin genes, their regulatory elements, and domains as wholes is an inexhaustible source for various hypotheses because the DNA sequence of the globin cluster and its genomic environment is determined for many organisms. Comparing the structure of genes, the composition of α- and β-globin clusters and the corresponding genomic context in different groups of vertebrates, one can follow the behavior during evolution of α- and β-globin genes down to the common ancestor of fishes, amphibians, and higher vertebrates (Fig. 5). In the genome of amphibians (Xenopus laevis, X. tropicalis) and the majority of fishes (Danio rerio, Oryzias latipes, Salmo salar), α- and β-globin genes are not separated but are situated side-by-side in the direction of 5′-α-β-3′ [111-113] and seem to be a joined genomic domain. In the genome of the tropical zebra fish (Danio rerio), genes encoding hemoglobin molecules are located on the 3rd and the 12th chromosomes. On the 12th chromosome, a cluster of embryonic α- and β-globin genes is situated, and products of their expression have been found only at the level of mRNA [114]. On the 3rd chromosome of Danio rerio, the embryonic and adult clusters of α- and β-globin genes are located adjacently [114]. And in both clusters on the 3rd chromosome, the transcription of α- and β-globin genes occurs in opposite directions. The analysis of the genomic environment of the cluster and the search for conserved sequences in different organisms provided mapping of some possible erythroid-specific elements. Upstream of the cluster of α-globin genes, either isolated or joined with the cluster of β-globin genes, the conserved α-MRE-involving housekeeping gene NPRL3 is located. Thus, the linkage of the cluster of globin genes and the NPRL3 gene is retained in all vertebrates and seems to be absolutely necessary (Fig. 5). The regulatory element homologous to human α-MRE in fishes is an enhancer activating both α- and β-genes, which also switches over the expression from the embryonic to the adult type [111].
It is still not known whether the joined domain of α- and β-globin genes of fishes and amphibians is open or closed.
It is supposed that α- and β-globin genes were produced as a result of duplication of an ancestral gene encoding the protein responsible for oxygen transport. These genes were situated side-by-side in the common ancestor of higher vertebrates and retained the same positions in fishes and amphibians (Fig. 5). Most likely, the divergence of α- and β-globin genes was caused by the necessity of complicating the hemoglobin molecule. The genes encoding α- and β-globin chains occurred on different chromosomes either as a result of translocations or as a result of chromosomal/genomic and in trans duplications and the subsequent inactivation of the genes [115]. The further evolution of both α- and β-globin genes in higher vertebrates was accompanied by multiple duplications of the genes inside the clusters and their further specialization. According to one hypothesis, the evolution of clusters of α- and β-globin genes in birds and mammals was independent after the duplication of the cluster of their common ancestor [116]. Thus, the chromosome region carrying the α-β-globin cluster is supposed to be duplicated and to produce two α-β-globin clusters on different chromosomes before the divergence of birds and mammals. Then these clusters underwent independent evolution in birds and mammals through inactivation of the α- or β-globin components of the cluster. According to another hypothesis, the β-globin cluster was isolated due to replacement onto another chromosome of a copy of the β-globin gene in the ancestor of higher vertebrates [117]. Surprisingly, contemporary “living fossils” retain archaic features of the organization of the α-globin gene domain. Thus, in the duckbill’s genome the cluster of α-globin genes contains not only the embryonic and adult genes, but also the β-like ω gene of ω-globin, as well as the GBY gene homologous to cytoglobin, which separated structurally and functionally long before the division of the joined α-β-globin domain [117, 118].
In any case, evolution was associated with expansion, functional divergence, and the secondary loss of duplication products: the β-globin genes were replaced onto another chromosome and became flanked with genes of the olfactory receptors, which also possessed pronounced tissue specificity. Now it is impossible to determine what caused the isolation of the β-globin genes and the formation of the closed domain: a translocation of some barrier elements of the ancestral joined domain simultaneously with the β-globin gene or the evolutionary pressure directed to appearance of boundary elements de novo. The latter event seems unlikely. In any case, the disappearance of the barrier elements from the environment of the remaining α-globin genes (if they were present there and the ancestral α-β-globin domain was closed) occurred to be advantageous for evolution. The translocation resulted in formation of the open domain, which is integrated with the genomic environment through erythroid-specific remote regulatory elements producing with this environment a common transcriptional fabric, and it is open for intervention of new genes (TMEM8).
At present, it is not quite clear which domains – of the open- or closed-type – are the most typical in the genome of contemporary vertebrates. There is no doubt that studies on the domain of β-globin genes and on other closed-type domains have played an extremely important role in studies of regulatory mechanisms of transcription in higher eukaryotes. Concurrently, it resulted in a certain re-evaluation of the importance of these domains. Many examples appeared recently that even tissue-specific genes not always are concentrated in closed-type domains. Moreover, some genomic domains, which were earlier considered to be typical closed-type domains (e.g. the domain of chicken lysozyme gene [119]), were shown to contain housekeeping gene(s) that are always expressed in all cell types. Thus, the structural–functional organization of the genome is really much more complicated than the organization described even by the hypothesis of domain organization of the genome, although it is adapted to the existence of the open-type domain. This confirms once more that there are no clear boundaries in Nature. And to comprehend principles of activities of genomic domains, which cannot be assigned to the open- or closed-type but are more complicated, is a problem for future research.
This work was supported by the Ministry of Science and Education of the Russian Federation (contracts 16.740.11.0483, 14.740.12.1344, and 16.740.11.0353), by the Russian Foundation for Basic Research (grants 11-04-00361-a; 11-04-91334-NNIO_a, and 12-04-0036-a), by a grant of the Russian Federation President for young scientists (MK-3813.2012.4), and by the Presidium of the Russian Academy of Sciences (grants from the Program on Molecular and Cellular Biology) and by Dmitri Zimin’s Foundation “Dynasty”.
REFERENCES
1.Razin, S. V., Iarovaia, O. V., Sjakste, N.,
Sjakste, T., Bagdoniene, L., Rynditch, A. V., Eivazova, E. R.,
Lipinski, M., and Vassetzky, Y. S. (2007) J. Mol. Biol.,
369, 597-607.
2.Bodnar, J. W. (1988) J. Theor. Biol.,
132, 479-507.
3.Goldman, M. A. (1988) Bioessays, 9,
50-55.
4.Weintraub, H., and Groudine, M. (1976)
Science, 73, 848-856.
5.Forrester, W. C., Thompson, C., Elder, J. T., and
Groudine, M. (1986) Proc. Natl. Acad. Sci. USA, 83,
1359-1363.
6.Lawson, G. M., Knoll, B. J., March, C. J., Woo, S.
L. C., Tsai, M.-J., and O’Malley, B. W. (1982) J. Biol.
Chem., 257, 1501-1507.
7.Krajewski, W. A., and Becker, P. B. (1998) Proc.
Natl. Acad. Sci. USA, 95, 1540-1545.
8.Anguita, E., Johnson, C. A., Wood, W. G., Turner,
B. M., and Higgs, D. R. (2001) Proc. Natl. Acad. Sci. USA,
98, 12114-12119.
9.Kellum, R., and Schedl, P. (1992) Mol. Cell.
Biol., 12, 2424-2431.
10.Felsenfeld, G., Burgess-Beusse, B., Farrell, C.,
Gaszner, M., Ghirlando, R., Huang, S., Jin, C., Litt, M., Magdinier,
F., Mutskov, V., Nakatani, Y., Tagami, H., West, A., and Yusufzai, T.
(2004) Cold Spring Harb. Symp. Quant. Biol., 69,
245-250.
11.Wallace, J. A., and Felsenfeld, G. (2007)
Curr. Opin. Genet. Dev., 17, 400-407.
12.Forrester, W. C., Epner, E., Driscoll, M. C.,
Enver, T., Brice, M., Papayannopoulou, T., and Groudine, M. (1990)
Genes Dev., 4, 1637-1649.
13.Grosveld, F., van Assandelt, G. B., Greaves, D.
R., and Kollias, B. (1987) Cell, 51, 975-985.
14.Razin, S. V., Farrell, C. M., and Recillas-Targa,
F. (2003) Int. Rev. Cytol., 226, 63-125.
15.Craddock, C. F., Vyas, P., Sharpe, J. A., Ayyub,
H., Wood, W. G., and Higgs, D. R. (1995) EMBO J., 14,
1718-1726.
16.Flint, J., Tufarelli, C., Peden, J., Clark, K.,
Daniels, R. J., Hardison, R., Miller, W., Philipsen, S., Tan-Un, K. C.,
McMorrow, T., Frampton, J., Alter, B. P., Frischauf, A. M., and Higgs,
D. R. (2001) Hum. Mol. Genet., 10, 371-382.
17.Makalowska, I., Lin, C. F., and Makalowski, W.
(2005) Comput. Biol. Chem., 29, 1-12.
18.Dillon, N., and Sabbatini, P. (2000)
Bioessays, 22, 657-665.
19.Forget, B. G. (1998) Ann. N. Y. Acad.
Sci., 850, 38-44.
20.Bulger, M., Bender, M., Farrell, C., Felsenfeld,
G., Wertman, B., Groudine, M., and Hardison, R. (2000) Blood Cells
Mol. Dis., 26, 490.
21.Schubeler, D., Francastel, C., Cimbora, D. M.,
Reik, A., Martin, D. I. K., and Groudine, M. (2000) Genes Dev.,
14, 940-950.
22.Hanscombe, O., Whyatt, D., Fraser, P.,
Yannoutsos, N., Greaves, D., Dillon, N., and Grosveld, F. (1991)
Genes Dev., 5, 1387-1394.
23.Hebbes, T. R., Clayton, A. L., Thorne, A. W., and
Crane-Robinson, C. (1994) EMBO J., 13, 1823-1830.
24.Prioleau, M.-N., Nony, P., Simpson, M., and
Felsenfeld, G. (1999) EMBO J., 18, 4035-4048.
25.Guerrero, G., Delgado-Olguin, P.,
Escamilla-Del-Arenal, M., Furlan-Magaril, M., Rebollar, E., De La
Rosa-Velazquez, I. A., Soto-Reyes, E., Rincon-Arano, H.,
Valdes-Quezada, C., Valadez-Graham, V., and Recillas-Targa, F. (2007)
Comp. Biochem. Physiol. A. Mol. Integr. Physiol., 147,
750-760.
26.Geyer, P. K. (1997) Curr. Opin. Genet.
Dev., 7, 242-248.
27.West, A. G., Gaszner, M., and Felsenfeld, G.
(2002) Genes Dev., 16, 271-288.
28.Chung, J. H., Whiteley, M., and Felsenfeld, G.
(1993) Cell, 74, 505-514.
29.Recillas-Targa, F., Pikaart, M. J.,
Burgess-Beusse, B., Bell, A. C., Litt, M. D., West, A. G., Gaszner, M.,
and Felsenfeld, G. (2002) Proc. Natl. Acad. Sci. USA, 99,
6883-6888.
30.Chung, J. H., Bell, A. C., and Felsenfeld, G.
(1997) Proc. Natl. Acad. Sci. USA, 94, 575-580.
31.Bell, A. C., West, A. G., and Felsenfeld, G.
(1999) Cell, 98, 387-396.
32.Wendt, K. S., Yoshida, K., Itoh, T., Bando, M.,
Koch, B., Schirghuber, E., Tsutsumi, S., Nagae, G., Ishihara, K.,
Mishiro, T., Yahata, K., Imamoto, F., Aburatani, H., Nakao, M.,
Imamoto, N., Maeshima, K., Shirahige, K., and Peters, J. M. (2008)
Nature, 451, 796-801.
33.Xiao, T., Wallace, J., and Felsenfeld, G. (2011)
Mol. Cell. Biol., 31, 2174-2183.
34.Huang, S., Li, X., Yusufzai, T. M., Qiu, Y., and
Felsenfeld, G. (2007) Mol. Cell. Biol., 27,
7991-8002.
35.Li, X., Wang, S., Li, Y., Deng, C., Steiner, L.
A., Xiao, H., Wu, C., Bungert, J., Gallagher, P. G., Felsenfeld, G.,
Qiu, Y., and Huang, S. (2011) Blood, 118, 1386-1394.
36.Saitoh, N., Bell, A. C., Recillas-Targa, F.,
West, A. G., Simpson, M., Pikaart, M., and Felsenfeld, G. (2000)
EMBO J., 19, 2315-2322.
37.Farrell, C. M., West, A. G., and Felsenfeld, G.
(2002) Mol. Cell. Biol., 22, 3820-3831.
38.Li, Q., and Stamatoyannopoulos, G. (1994)
Blood, 84, 1399-1401.
39.Tanimoto, K., Liu, Q., Bungert, J., and Engel, J.
D. (1999) Nature, 398, 344-348.
40.Li, Q., Zhang, M., Han, H., Rohde, A., and
Stamatoyannopoulos, G. (2002) Nucleic Acids Res., 30,
2484-2491.
41.Talbot, D., Collis, P., Antoniou, M., Vidal, M.,
Grosveld, F., and Greaves, D. R. (1989) Nature, 338,
352-355.
42.Li, G., Lim, K. C., Engel, J. D., and Bungert, J.
(1998) Genes Cells, 3, 415-429.
43.Cimbora, D. M., Schubeler, D., Reik, A.,
Hamilton, J., Francastel, C., Epner, E. M., and Groudine, M. (2000)
Mol. Cell. Biol., 20, 5581-5591.
44.Philipsen, S., Talbot, D., Fraser, P., and
Grosveld, F. (1990) EMBO J., 9, 2159-2167.
45.Ney, P. A., Sorrentino, B. P., McDonagh, K. T.,
and Nienhuis, A. W. (1990) Genes Dev., 4, 993-1006.
46.Strauss, E. C., and Orkin, S. H. (1992) Proc.
Natl. Acad. Sci. USA, 89, 5809-5813.
47.Lowrey, C. H., Bodine, D. M., ana Nienhuis, A. W.
(1992) Proc. Natl. Acad. Sci. USA, 89, 1143-1147.
48.Stamatoyannopoulos, G., Goodwin, A., Joyce, T.,
and Lowrey, C. H. (1995) EMBO J., 14, 106-116.
49.Li, Q., Zhou, B., Powers, P., Enver, T., and
Stamatoyannopoulos, G. (1990) Proc. Natl. Acad. Sci. USA,
87, 8207-8211.
50.Mason, M. M., Lee, E., Westphal, H., and Reitman,
M. (1995) Mol. Cell. Biol., 15, 407-414.
51.Hardison, R., Slightom, J. L., Gumicio, D. L.,
Goodman, M., Stojanovic, N., and Miller, W. (1997) Gene,
205, 73-94.
52.Bulger, M., and Groudine, M. (1999) Genes
Dev., 13, 2465-2477.
53.Peterson, K. R., and Stamatoyannopoulos, G.
(1993) Mol. Cell. Biol., 13, 4836-4843.
54.Dillon, N., Trimborn, T., Stronboulis, J.,
Fraser, P., and Grosveld, F. (1997) Mol. Cell, 1,
131-139.
55.Wijgerde, M., Grosveld, F., and Fraser, P. (1995)
Nature, 377, 209-213.
56.Gribnau, J., de Boer, E., Trimborn, T., Wijgerde,
M., Milot, E., Grosveld, F., and Fraser, P. (1998) EMBO J.,
17, 6020-6027.
57.Dekker, J., Rippe, K., Dekker, M., and Kleckner,
N. (2002) Science, 295, 1306-1311.
58.De Laat, W., and Grosveld, F. (2003)
Chromosome Res., 11, 447-459.
59.Tolhuis, B., Palstra, R. J., Splinter, E.,
Grosveld, F., and de Laat, W. (2002) Mol. Cell, 10,
1453-1465.
60.West, A. G., and Fraser, P. (2005) Hum. Mol.
Genet., 14, 101-111.
61.De Laat, W., Klous, P., Kooren, J., Noordermeer,
D., Palstra, R. J., Simonis, M., Splinter, E., and Grosveld, F. (2008)
Curr. Top. Dev. Biol., 82, 117-139.
62.Zhou, G. L., Xin, L., Song, W., Di, L. J., Liu,
G., Wu, X. S., Liu, D. P., and Liang, C. C. (2006) Mol. Cell.
Biol., 26, 5096-5105.
63.Kooren, J., Palstra, R. J., Klous, P., Splinter,
E., von Lindern, M., Grosveld, F., and de Laat, W. (2007) J. Biol.
Chem., 282, 16544-16552.
64.Armstrong, J. A., Bieker, J. J., and Chen, X.
(1998) Cell, 95, 93-104.
65.Blobel, G. (2000) Blood, 95,
745-755.
66.Bresnick, E. H., and Tze, L. (1997) Proc.
Natl. Acad. Sci. USA, 94, 4566-4571.
67.Forsberg, E. C., Johnson, K., Zaboikina, T. N.,
Mosser, E. A., and Bresnick, E. H. (1999) J. Biol. Chem.,
274, 26850-26859.
68.Gribnau, G., Diderich, K., Pruzina, S.,
Calzolari, R., and Frazer, P. (2000) Mol. Cell, 5,
377-386.
69.Wilson, C. J., Chao, D. M., Imbalzano, A. N.,
Schnitzler, G. R., Kingston, R. E., and Yong, R. A. (1996) Cell,
84, 235-244.
70.Cho, H., Orphanides, G., Sun, X., Yang, X. J.,
Ogryzko, V., Lees, E., Nakatani, Y., and Reinberg, D. (1998) Mol.
Cell. Biol., 18, 5355-5363.
71.Travers, A. (1999) Proc. Natl. Acad. Sci.
USA, 96, 13634-13637.
72.Herendeen, D. R., Kassavetis, G. A., and
Geiduschek, E. P. (1992) Science, 256, 1298-1303.
73.Forsberg, E. C., and Bresnick, E. H. (2001)
Bioessays, 23, 820-830.
74.Hughes, J. R., Cheng, J. F., Ventress, N.,
Prabhakar, S., Clark, K., Anguita, E., De Gobbi, M., de Jong, P.,
Rubin, E., and Higgs, D. R. (2005) Proc. Natl. Acad. Sci. USA,
102, 9830-9835.
75.Tufarelli, C., Hardison, R., Miller, W., Hughes,
J., Clark, K., Ventress, N., Frischauf, A. M., and Higgs, D. R. (2004)
Genome Res., 14, 623-630.
76.Klochkov, D. B., Gavrilov, A. A., Vassetzky, Y.
S., and Razin, S. V. (2009) Genomics, 93, 481-486.
77.Tufarelli, C., Frischauf, A. M., Hardison, R.,
Flint, J., and Higgs, D. R. (2001) Genomics, 71,
307-314.
78.Neklesa, T. K., and Davis, R. W. (2009) PLoS
Genet., 5, e1000515.
79.Higgs, D. R., Wood, W. G., Jarman, A. P., Sharpe,
J., Lida, J., Pretorius, I.-M., and Ayyub, H. (1990) Genes Dev.,
4, 1588-1601.
80.Viprakasit, V., Harteveld, C. L., Ayyub, H.,
Stanley, J. S., Giordano, P. C., Wood, W. G., and Higgs, D. R. (2006)
Blood, 107, 3811-3812.
81.Jarman, A. P., Wood, W. G., Sharpe, J. A.,
Gourdon, G., Ayyub, H., Higgs, and D. R. (1991) Mol. Cell.
Biol., 11, 4679-4689.
82.Anguita, E., Sharpe, J. A., Sloane-Stanley, J.
A., Tufarelli, C., Higgs, D. R., and Wood, W. G. (2002) Blood,
100, 3450-3456.
83.Chen, H., Lowrey, C. H., and Stamatoyannopoulos,
G. (1997) Nucleic Acids Res., 25, 2917-2922.
84.De Gobbi, M., Anguita, E., Hughes, J.,
Sloane-Stanley, J. A., Sharpe, J. A., Koch, C. M., Dunham, I., Gibbons,
R. J., Wood, W. G., and Higgs, D. R. (2007) Blood, 110,
4503-4510.
85.Igarashi, K., Itoh, K., Hayashi, N., Nishizawa,
M., and Yamamoto, M. (1995) Proc. Natl. Acad. Sci. USA,
92, 7445-7449.
86.Knezetic, J., and Felsenfeld, G. (1989) Mol.
Cell. Biol., 9, 893-901.
87.Philonenko, E. S., Klochkov, D. B., Borunova, V.
V., Gavrilov, A. A., Razin, S. V., and Iarovaia, O. V. (2009)
Nucleic Acids Res., 37, 7394-7406.
88.Razin, S. V., Kekelidze, M. G., Lukanidin, E. M.,
Scherrer, K., and Georgiev, G. P. (1986) Nucleic Acids Res.,
14, 8189-8207.
89.Klochkov, D., Rincon-Arano, H., Ioudinkova, E.
S., Valadez-Graham, V., Gavrilov, A., Recillas-Targa, F., and Razin, S.
V. (2006) Mol. Cell. Biol., 26, 1589-1597.
90.Razin, S. V., Ioudinkova, E. S., and Scherrer, K.
(2000) J. Mol. Biol., 209, 845-852.
91.Valadez-Graham, V., Razin, S. V., and
Recillas-Targa, F. (2004) Nucleic Acids Res., 32,
1354-1362.
92.Furlan-Magaril, M., Rebollar, E., Guerrero, G.,
Fernandez, A., Molto, E., Gonzalez-Buendia, E., Cantero, M., Montoliu,
L., and Recillas-Targa, F. (2011) Nucleic Acids Res., 39,
89-103.
93.Razin, S. V., Rynditch, A., Borunova, V.,
Ioudinkova, E., Smalko, V., and Scherrer, K. (2004) J. Cell.
Biochem., 92, 445-457.
94.Gavrilov, A. A., and Razin, S. V. (2008)
Nucleic Acids Res., 36, 4629-4640.
95.Vernimmen, D., De Gobbi, M., Sloane-Stanley, J.
A., Wood, W. G., and Higgs, D. R. (2007) EMBO J., 26,
2041-2051.
96.Vernimmen, D., Marques-Kranc, F., Sharpe, J. A.,
Sloane-Stanley, J. A., Wood, W. G., Wallace, H. A., Smith, A. J., and
Higgs, D. R. (2009) Blood, 114, 4253-4260.
97.Gavrilov, A. A., Zukher, I. S., Philonenko, E.
S., Razin, S. V., and Iarovaia, O. V. (2010) Nucleic Acids Res.,
38, 8051-8060.
98.Anguita, E., Hughes, J., Heyworth, C., Blobel, G.
A., Wood, W. G., and Higgs, D. R. (2004) EMBO J., 23,
2841-2852.
99.Pal, S., Cantor, A. B., Johnson, K. D., Moran, T.
B., Boyer, M. E., Orkin, S. H., and Bresnick, E. H. (2004) Proc.
Natl. Acad. Sci. USA, 101, 980-985.
100.Liebhaber, S. A., Wang, Z., Cash, F. E., Monks,
B., and Russell, J. E. (1996) Mol. Cell. Biol., 16,
2637-2646.
101.Knezetic, J. A., and Felsenfeld, G. (1993)
Mol. Cell. Biol., 13, 4632-4639.
102.Singal, R., vanWert, J. M., and Ferdinand, L.,
Jr. (2002) Blood, 100, 4217-4222.
103.Sabath, D. E., Spangler, E. A., Rubin, E. M.,
and Stamatoyannopoulos, G. (1993) Blood, 82,
2899-2905.
104.Ioudinkova, E. S., Ulianov, S. V., Bunina, D.,
Iarovaia, O. V., Gavrilov, A. A., and Razin, S. V. (2011)
Epigenetics, 6, 1481-1488.
105.Tang, Y., Wang, Z., Huang, Y., Liu, D. P., Liu,
G., Shen, W., Tang, X., Feng, D., and Liang, C. C. (2006) Genes
Cells, 11, 123-131.
106.Whitelaw, E., Hogben, P., Hanscombe, O., and
Proudfoot, N. J. (1989) Mol. Cell. Biol., 9, 241-251.
107.Garrick, D., De Gobbi, M., Samara, V., Rugless,
M., Holland, M., Ayyub, H., Lower, K., Sloane-Stanley, J., Gray, N.,
Koch, C., Dunham, I., and Higgs, D. R. (2008) Blood, 112,
3889-3899.
108.Lynch, M. D., Smith, A. J., De Gobbi, M.,
Flenley, M., Hughes, J. R., Vernimmen, D., Ayyub, H., Sharpe, J. A.,
Sloane-Stanley, J. A., Sutherland, L., Meek, S., Burdon, T., Gibbons,
R. J., Garrick, D., and Higgs, D. R. (2011) EMBO J., Nov 4. doi:
10.1038/emboj.2011.399.
109.Vinogradov, S. N., Hoogewijs, D., Bailly, X.,
Arredondo-Peter, R., Gough, J., Dewilde, S., Moens, L., and
Vanfleteren, J. R. (2006) BMC Evol. Biol., 6, 31.
110.Hoffmann, F. G., Opazo, J. C., and Storz, J. F.
(2012) Mol. Biol. Evol., in press.
111.Maruyama, K., Yasumasu, S., Naruse, K., Mitani,
H., Shima, A., and Iuchi, I. (2004) Gene, 335,
89-100.
112.Fuchs, C., Burmester, T., and Hankeln, T.
(2006) Cytogenet. Genome Res., 112, 296-306.
113.Chan, F. Y., Robinson, J., Brownlie, A.,
Shivdasani, R. A., Donovan, A., Brugnara, C., Kim, J., Lau, B. C.,
Witkowska, H. E., and Zon, L. I. (1997) Blood, 89,
688-700.
114.Brownlie, A., Hersey, C., Oates, A. C., Paw, B.
H., Falick, A. M., Witkowska, H. E., Flint, J., Higgs, D., Jessen, J.,
Bahary, N., Zhu, H., Lin, S., and Zon, L. (2003) Dev. Biol.,
255, 48-61.
115.Jeffreys, A. J., Wilson, V., Wood, D., Simons,
J. P., Kay, R. M., and Williams, J. G. (1980) Cell, 21,
555-564.
116.Wheeler, D., Hope, R. M., Cooper, S. J.,
Gooley, A. A., and Holland, R. A. (2004) J. Mol. Evol.,
58, 642-652.
117.Wheeler, D., Hope, R., Cooper, S. B., Dolman,
G., Webb, G. C., Bottema, C. D., Gooley, A. A., Goodman, M., and
Holland, R. A. (2001) Proc. Natl. Acad. Sci. USA, 98,
1101-1106.
118.Patel, V. S., Cooper, S. J., Deakin, J. E.,
Fulton, B., Graves, T., Warren, W. C., Wilson, R. K., and Graves, J. A.
(2008) BMC Biol., 6, 34.
119.Chong, S., Riggs, A. D., and Bonifer, C. (2002)
Nucleic Acids Res., 30, 463-467.