Stabilization of Plant Formate Dehydrogenase by Rational Design
A. A. Alekseeva1,2,3,4, S. S. Savin1,3, S. Yu. Kleimenov1,5, I. V. Uporov2,3, E. V. Pometun4, and V. I. Tishkov1,2,3*
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33/2, 117234 Moscow, Russia; fax: (495) 954-2732; E-mail: vitishkov@gmail.com2Chemistry Department, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-2742
3Innovations and High Technologies MSU Ltd., ul. Tsimlyanskaya 16, office 96, 109559 Moscow, Russia; fax: (495) 939-3208
4POMALEX Ltd, Kooperativnaya ul. 2, 140180 Zhukovsky, Moscow Region, Russia
5Koltzov Institute of Developmental Biology, Russian Academy of Sciences, ul. Vavilova 26, 119334 Moscow, Russia; fax: (499) 135-8012
* To whom correspondence should be addressed.
Received July 3, 2012; Revision received July 9, 2012
Recombinant formate dehydrogenase (FDH, EC 1.2.1.2) from soy Glycine max (SoyFDH) has the lowest values of Michaelis constants for formate and NAD+ among all studied formate dehydrogenases from different sources. Nevertheless, it also has the lower thermal stability compared to enzymes from bacteria and yeasts. The alignment of full sequences of FDHs from different sources as well as structure of apo- and holo-forms of SoyFDH has been analyzed. Ten mutant forms of SoyFDH were obtained by site-directed mutagenesis. All of them were purified to homogeneity and their thermal stability and substrate specificity were studied. Thermal stability was investigated by studying the inactivation kinetics at different temperatures and by differential scanning calorimetry (DSC). As a result, single-point (Ala267Met) and double mutants (Ala267Met/Ile272Val) were found to be more stable than the wild-type enzyme at high temperatures. The stabilization effect depends on temperature, and at 52°C it was 3.6- and 11-fold, respectively. These mutants also showed higher melting temperatures in DSC experiments – the differences in maxima of the melting curves (Tm) for the single and double mutants were 2.7 and 4.6°C, respectively. For mutations Leu24Asp and Val127Arg, the thermal stability at 52°C decreased 5- and 2.5-fold, respectively, and the Tm decreased by 3.5 and 1.7°C, respectively. There were no differences in thermal stability of six mutant forms of SoyFDH — Gly18Ala, Lys23Thr, Lys109Pro, Asn247Glu, Val281Ile, and Ser354Pro. Analysis of kinetic data showed that for the enzymes with mutations Val127Arg and Ala267Met the catalytic efficiency increased 1.7- and 2.3-fold, respectively.
KEY WORDS: formate dehydrogenase, thermal stability, rational design, catalytic efficiencyDOI: 10.1134/S0006297912100124
Formate dehydrogenases (EC 1.2.1.2) catalyze the oxidation of formate to carbon dioxide coupled with reduction of NAD+ to NADH. These enzymes are widespread in nature, and there are several types of them. NAD+-dependent FDHs are very simple. They consist of two identical subunits and contain no metal ion or prosthetic groups. Genes encoding FDHs have been found in bacteria, yeasts, fungi, and plants [1-3].
Recently, systematic research has been done for the enzymes from bacteria and yeasts. The FDHs from plants have been less studied. The results of sequence analysis show that there is at least one gene encoding FDH in plants [1, 2]. Several genes (up to six [2, 3]) were found in some plants, so several different isoenzymes can be synthesized in plant cells. FDH is an enzyme of stress in plants, and synthesis of FDH sharply increases during such exposures as drought, thermal discontinuity, hard ultraviolet [4-6], hypoxia [7], and pathogen infection [3, 8]. The type of FDH isoform depends on the stress exposure [3].
One of the most important characteristic features of genes encoding FDHs is the presence of special nucleotide sequence encoding signal peptide on the N-terminus of the enzyme. This peptide provides transport of the enzyme from cytoplasm to mitochondria [1, 9]. First experiments of expression the gene of FDH from potato, using plasmid with full cDNA sequence were not successful, because inclusion bodies were formed [9]. For expression of gene of FDH from rice, the sequence, encoded the signal peptide, was deleted. The content of the active enzyme was less than 0.02% of all soluble enzymes [10]. In the middle 2000s in our laboratory expression plasmids with modified cDNA sequences of gens, encoded FDHs from plant Arabidopsis thaliana and soybean Glycine max (AthFDH and SoyFDH, respectively) were constructed and recombinant formate dehydrogenases were expressed in E. coli cells as active and soluble enzymes (initial plasmids with full cDNA of AthFDH and SoyFDH genes were courteous gifts of Prof. J. Markwell, University of Nebraska-Lincoln, USA and Prof. N. Labrou, Agricultural University of Athens, Greece). The conditions of cultivation were optimized and the yield of active enzyme was more than 500 mg per liter of culture medium [11, 12]. Later, in 2009 FDH from Lotus japonicus (LjaFDH) was successfully expressed in E. coli cells [7]. The obtaining of FDHs from A. thaliana (AthFDH) and G. max (SoyFDH) resulted in systematic study of properties of these enzymes [11-16], crystallization, and determination of three-dimensional structure of the enzyme [17]. Due to the best values of Michaelis constants for formate and NAD+ [2, 11, 12], SoyFDH can be potentially useful for practical application. Unfortunately, SoyFDH has lower thermal stability compared to FDHs from bacteria and yeasts [2, 13, 18] and lower value of catalytic constant [19].
Rational design is one of the most effective ways of improving the properties of enzymes and obtaining biocatalysts with expected characteristics. In present work several positions were chosen, using this approach. Ten mutant forms of SoyFDH were obtained and their properties were studied. As a result, mutant forms with high thermal stability and improved catalytic properties were obtained.
MATERIALS AND METHODS
For genetic engineering experiments we used reagents, marked “molecular biology grade”. In microbiologic experiments, we used bacto-tryptone, yeasts extract, and agar (Difco, USA), glycerol (99.9%), calcium chloride (ultra pure), potassium phosphate dibasic, sodium phosphate monobasic (pure for analysis), and lysozyme (Fluka/BioChemika, Swiss), lactose (analytical grade), ampicillin, and chloramphenicol (Sigma, USA), glucose and sodium chloride (pure for analysis) (Helicon, Russia). For site-directed mutagenesis we used restrictases, T4 DNA ligase and Pfu DNA polymerase, made by Fermentas (Lithuania). DNA gel extraction kit and plasmid mini prep kit (Fermentas) were used for DNA purifying and plasmid extraction, respectively. Oligonucleotides for PCR and sequencing were made by Synthol (Russia). In all experiments we used water, purified by MilliQ (Millipore, USA).
All reactants for protein electrophoresis were made by Bio-Rad (USA). For purifying proteins and studying their properties we used ammonium sulfate (chemically pure; DiaM, Russia), urea (pure for analysis; Reakhim, Russia), NAD+ (99%; AppliChem, Germany), EDTA (pure for analysis; Merck, Germany), sodium formate, and sodium phosphate monobasic (pure for analysis; Reakhim), sodium azide (Serva, Germany).
Site-directed mutagenesis. Replacements in gene encoding SoyFDH were made by two-step polymerase chain reaction (PCR). The initial plasmid pSoyFDH2 was used as a template. For mutagenesis, we used universal primers T7_for and T7_rev for the beginning and the end of gene respectively and forward and reverse primer encoding the necessary mutation in the soyfdh gene. The sequences of the primers are:
T7_For: 5′-TAATACGACTCACTATAGGG-3′
T7_Rev: 5′-GCTAGTTATTGCTCAGCGG-3′
SoyG18A_For: 5′-GTTCTACAAAGCGAATGAGTATGCTAAATTGAACC-3′
SoyG18A_Rev: 5′-GCATACTCATTCGCTTTGTAGAACACCCCCACA-3′
SoyK23T_For: 5′-GAATGAGTATGCTACCTTGAACCCCAATTTCGTGGG-3′
SoyK23T_Rev: 5′-GTTCAAGGTAGCATACTCATTCCCTTTGTAG-3′
SoyL24D_For: 5′-TGAGTATGCTAAAGATAACCCCAATTTCGTGGGGT-3′
SoyL24D_Rev: 5′-ACGAAATTGGGGTTATCTTTAGCATACTCATTCCCTTTGTA-3′
SoyK109P_For: 5′-GATCTCCCGGCTGCAGCTGCTGCTGG-3′
SoyK109P_Rev: 5′-CAGCTGCAGCCGGGAGATCAACGTGATCAGAACC-3′
SoyV127R_For: 5′-AAGCAATGTCAGGTCGGTTGCGGAGGATG-3′
SoyV127R_Rev: 5′-CAACCGACCTGACATTGCTTCCCGTGAC-3′
SoyN247E_For: 5′-GTTTGACAAAGAAAGAATTGCGAAGTGCAAGAAAG-3′
SoyN247E_Rev: 5′-CTTCGCAATTCTTTCTTTGTCAAACAATCCCCTTGTC-3′
SoyA267M_For: 5′-GAGGGGCAATTATGGACACCCAAGCAATTGCA-3′
SoyA267M_Rev: 5′-TTGGGTGTCCATAATTGCCCCTCGAGCATTGTT-3′
SoyI272V_For: 5′-ACCCAAGCAGTTGCAGATGCTTGCTCCAGT-3′
SoyI272V_Rev: 5′-AAGCATCTGCAACTGCTTGGGTGTCCATAATTGC-3′
SoyV281I_For: 5′-AGTGGCCACATTGCAGGTTATAGTGGTGATGTTT-3′
SoyV281I_Rev: 5′-CTATAACCTGCAATGTGGCCACTGGAGCAAG-3′
SoyS354P_For: 5′-TCAACTTGCACCCCAATACCGGTGAAGAATTCG-3′
SoyS354P_Rev: 5′-GGTATTGGGGTGCAAGTTGACCCTCCTTGA-3′
The reaction mixture (total volume 25 µl) consisted of 2.5 µl 10× Pfu buffer (200 mM Tris-HCl, pH 8.8 at 25°C, 100 mM KCl, 100 mM (NH4)2SO4, 20 mM MgSO4, 1% Triton X-100, 1 mg/ml BSA), 2 µl dNTP (dATP, dGTP, dTTP, dCTP, 2.5 mM concentration of each dNTP), 1 µl of DNA template (10 ng/µl), 2 µl forward and reverse primers (10 pmol/µl; Synthol), 0.5 µl Pfu DNA polymerase (2.5 U/µl; Fermentas), and 15 µl deionized water. The PCR was run in a thin-walled tube (volume 0.5 ml; SSI, USA) using a Tercik device (DNA-Technologies, Russia). To avoid evaporation of the reaction mixture, 30 µl of mineral oil were added to each tube. The reaction tube was incubated 5 min at 95°C and then cycled (30 sec at 95°C, 60 sec at 54-58°C, 2 min at 72°C) 25-35 times. After the last cycle, the reaction mixture was additionally incubated for 5 min at 72°C. The temperature during the second step was 3-5°C lower than the melting point of the duplexes formed by the primers.
Two pairs of primers (SoyFor/T7rev and SoyRev/T7for) were used to obtain the fragments of the gene with necessary replacements. The PCR products of the first two reactions were purified using a Gel Extraction Kit (Fermentas) and used as the DNA template in the third PCR with T7_for and T7_rev primers. The PCR product of the third reaction was digested by restriction endonucleases EcoRI and KpnI or NdeI and XhoI (it depends on the position of the mutation). Then, the fragments with necessary mutations were purified and cloned into the initial pSoyFDH2 plasmid digested with the same restrictases. The E. coli Mach1 cell line was used for transformation by the reaction products. Then cells were grown on a Petri dish with agar medium containing 150 μg/ml ampicillin for 16 h at 37°C. For each mutant form, three colonies were used for plasmid extraction. Plasmid DNA was obtained using a Fermentas plasmid extraction kit. Plasmids were sequenced with an Applied Biosystems 3100 Genetic Analyzer to prove the absence of unwanted mutations. Sequence analysis was performed in the Genome Center (Engelhardt Institute of Molecular Biology, Russian Academy of Sciences).
Expression of mutant forms of SoyFDH in E. coli cells. Mutant and wild-type SoyFDHs were expressed in E. coli BL21(DE3)CodonPlus/pLysS cells. Competent E. coli cells were transformed by pSoyFDH2 plasmids with the desired mutation, and the cells were grown overnight at 37oC in agar with 2YT medium (16 g/liter bacto-tryptone, 10 g/liter yeast extract, 5 g/liter sodium chloride, pH 7.0) with 100 μg/ml ampicillin and 25 μg/ml chloramphenicol. One colony from a Petri dish was placed in a test tube with 5 ml of 2YT medium supplemented with chloramphenicol and ampicillin (25 and 150 μg/ml, respectively). The cells were cultivated at 30ºC and 180 rpm until absorbance at 600 nm (A600) reached 0.6-0.8. Then the cells were transferred to a 1-liter shaker flask with 200 ml of 2YT medium with 150 μg/ml ampicillin and cultivated at 30ºC and 80-90 rpm. Lactose solution (300 g/liter) was added to the final concentration 20 g/liter when A600 was 0.6-0.8, and the cells were cultivated for 17 h at 20ºC and 120 rpm. Biomass was collected using a Beckman J-21centrifuge (20 min, 7500 rpm, 4ºC). The cells were resuspended in 0.1 M sodium phosphate buffer, pH 8.0 (1 : 4 w/w), and kept at –20°C.
Purification of wild-type and mutant SoyFDHs. The enzymes were purified using a slightly modified procedure developed for purification of recombinant PseFDH [20]. The purification procedure included sonication of the cells at 4°C using a Branson 250 Sonifier (Germany), 30-min centrifugation at 11,000 rpm and 4°C in and Eppendorf 5804 R centrifuge, and overnight precipitation of proteins in ammonium sulfate solution (85% saturation) in 0.1 M sodium phosphate buffer, pH 7.0 (buffer A). Then the precipitate was dissolved in ammonium sulfate solution (45% saturation) in buffer A. Undissolved proteins were removed by centrifugation, and the supernatant was applied on a column with Phenyl-Sepharose Fast Flow (Pharmacia Biotech) equilibrated with 45% ammonium sulfate solution in buffer A. The column was washed with five volumes of 45% ammonium sulfate solution in buffer A, and the formate dehydrogenase was eluted using a 45-0% linear gradient of ammonium sulfate in buffer A. Fractions with active enzyme were collected and desalted by gel filtration through Sephadex G-25 Fine (Pharmacia Biotech). The enzyme preparations were analyzed with standard denaturing sodium dodecyl sulfate electrophoresis in 12% polyacrylamide gel.
Measurement of formate dehydrogenase activity. FDH activity was determined in 0.1 M sodium phosphate buffer, pH 7.0, at 30°C by measuring NADH production at 340 nm (ε340 = 6220 M–1·cm–1) using a Shimadzu UV 1800PC spectrophotometer. The concentrations of sodium formate and NAD+ in the cuvette were 0.6 M and 0.2 mg/ml, respectively.
Determination of Michaelis constants. Michaelis constants for NAD+ and formate were determined from dependences of reaction rate on variable substrate concentration (0.4-6 KM) at fixed saturating (>30 KM) concentration of the second substrate. The concentration of NAD+ was determined by measurement of absorption at 260 nm using the Shimadzu spectrophotometer (ε260 = 17,800 M–1·cm–1). The solution of sodium formate was prepared by dissolving the necessary amount of the substrate in 0.1 M sodium phosphate buffer, pH 7.0. Nonlinear regression (Origin Pro 8.5) was used to analyze the experimental kinetic curves.
Determination of catalytic constants. Several solutions with different concentrations of enzyme were prepared for determination of kcat. For each solution, the activity and active sites concentration were determined. The concentration of the active sites was measured from dependences of fluorescence quenching of FDH–NAD+ complex after addition of azide ion as described in [19]. The measurements were carried out in 0.1 M sodium phosphate buffer, pH 7.0, using a Cary Eclipse spectrofluorimeter (Varian, USA). The catalytic rate constant was calculated from the slope of the linear dependence of maximum velocity on active site concentration using Origin Pro 8.5 software.
Thermal inactivation kinetic study. Thermal stability of the enzyme was studied in sodium phosphate buffer with the concentration range of 0.01-1 M, pH 7.0. Several plastic tubes (0.5 ml) with 100 μl of enzyme (0.2 mg/ml) were prepared for each experiment. The tubes were incubated in a constant temperature bath that was previously preheated to the required temperature (46-60°C, the accuracy of thermostatic control was ±0.1°C). At definite periods of time, some tubes were incubated at 4°C for 5 min, and then the solution was centrifuged for 3 min at 12,000 rpm in an Eppendorf 5415D centrifuge. Residual activity of FDH was measured as described previously. The thermal inactivation rate constant was calculated from the slope of the linear dependence of ln(A/A0) on time using the Origin Pro 8.5 software.
Differential scanning calorimetry. The DSC experiments were carried out in a DASM-4M instrument (NPO Biopribor, Russia) as described in [13, 14]. The operating volume of the cell was 0.48 ml. To avoid evaporation at high temperatures, pressure of 2 atm was maintained in the cell. The device was calibrated using fixed heating in one of the cells (ΔW = 25 µW). The instrumental baseline was determined before the experiment. Buffer was put in the reference cell, and the solution with SoyFDH to be investigated was put in working cell. The concentration of the enzyme was 2.0-3.7 mg/ml and the heating rate was 1°C/min.
To determine reversibility/irreversibility of the thermal denaturation, the experiment was repeated. The experimental curves were calculated using Mathlab 8.0 software. In the first step, the device baseline was subtracted from the experimental melting curve. After that, the baseline of buffer solution and chemical baseline were also subtracted. Then the values of denaturation heat and maximum position of the melting curves (Tm) were computed. The percentage error was about 5-8% and the experimental error of Tm determination was ±0.2°C.
Computer modeling. The structure of apo-SoyFDH was studied by homology modeling using Insight II (Accelrys) software and the apo-AthFDH structure (3JTM in pdb.org, resolution 1.3 Å). Additional optimization was achieved by molecular mechanic (Discover_3 in Insight II, force field CVFF, 1000 cycles), molecular dynamics (5 psec), and additional molecular mechanics (1000 cycles). The SoyFDH structure was analyzed and the structures of the mutant enzymes were modeled using the Accelrys Discovery Studio 2.1 software package. Pictures of structures were prepared with the Accelrys Studio 2.1 software.
RESULTS AND DISCUSSION
Analysis of SoyFDH structure and selection of promising positions for amino acid replacements. For selection of amino acid replacements, the three-dimensional structures of apo- and holo-forms of SoyFDH and alignment of amino acid sequences of formate dehydrogenases from different sources were analyzed. We obtained crystals and structure with 1.9 Å resolution for the ternary SoyFDH–NAD+–N3– complex (unpublished data). For the apo-form of SoyFDH, computer modeling was made using the apo-form of FDH from Arabidopsis thaliana (3JTM in pdb.org, resolution 1.3 Å) as a template. The RMSD between experimental apo-AthFDH structure and model apo-SoyFDH was only 0.2 Å. Homology between sequences of AthFDH and SoyFDH is 85%. According to [21], the model structure is considered to be valid and the accuracy is sufficient for choosing positions for amino acid replacements.
Stability of protein molecule can be provided by different types of interactions. For this reason, stabilization of protein structure can be achieved by optimizing all types of interactions. For improving thermal stability of SoyFDH, we can use such approaches as creation of additional ion pairs, filling cavities inside the protein globule, increasing of rigidity of the amino acid chain, and optimizing of electrostatic interactions on the surface of the protein globule. All these approaches were successfully applied for improving the stability of FDH from the bacterium Pseudomonas sp. 101 (PseFDH) [1, 18, 20, 22, 23].
Optimization of electrostatic interactions. It was shown by analysis of model apo-SoyFDH structure that in this enzyme there are fewer bonds between two subunits than in FDH from A. thaliana. Therefore, it was decided to create additional ion pairs in the region of subunit contact. Modeling the structure of mutants with additional ion pairs showed that the most promising replacements were Leu24Asp and Val127Arg. These mutants should result in forming ion pairs with Arg164 and Glu169 from the second subunit. The results of modeling Val127Arg are presented in Fig. 1 (a, b) (see color insert).
Fig. 1. Modeling of amino acid replacements A_Val127 (a) for A_Arg127 (b) with formation of an ion pair with B_Glu169 of the second subunit, replacement of Ala267 (c) to Met (d), Asn247 (e) to Glu (f), and Lys109 (g) to Pro (h).
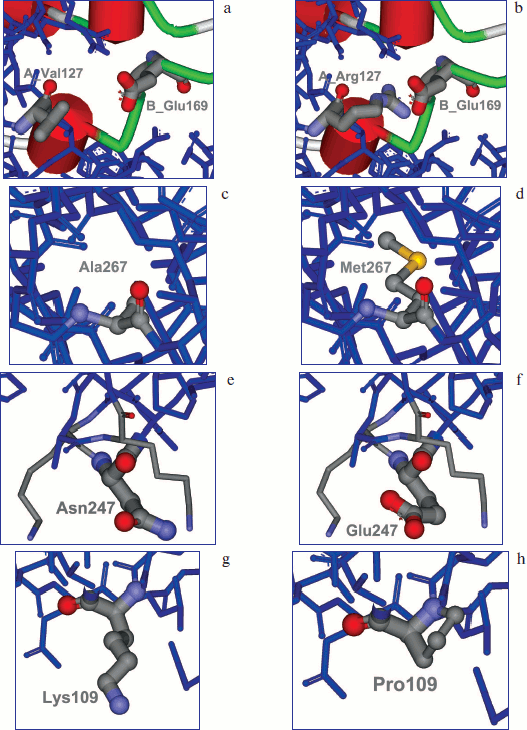
Filling the cavities inside the protein molecule. Cavities filled with water can often be found inside a protein globule. The main disadvantage is that those water molecules are surrounded by hydrophobic residues. Therefore, filling these cavities with large hydrophobic radicals can stabilize the protein structure. Analysis of apo and holo forms of the enzyme amino acid sequence and comparison with structure of more stable FDH from A. thaliana revealed such cavity in the region of the Ala267 residue (Fig. 1c; see color insert). It was decided to change Ala267 to Met (Fig. 1d; see color insert).
Comparison of amino acid sequences. Figure 2 shows aligned amino acid sequences of FDH fragments from different plants in the regions of residues 8-22 and 258-287 (numbers are as in SoyFDH). The chosen residues are shown in gray. The FDHs from plants have a high level of homology in these regions. In position 18, the majority of FDH from plants (except SoyFDH and FDH from rice OsaFDH and sorghum SbiFDH) has an alanine residue. Position 267 of SoyFDH has an alanine residue, while in all other enzymes this position is occupied by methionine. Val272 is semi-conserved (only in SoyFDH this position is occupied by isoleucine). Practically all plant FDHs have an Ile residue in position 281. Preparation of mutant SoyFDHs with replacements Gly18Ala, Ala267Met, Ile272Val, and Val281Ile was suggested. It should be noted that between replacements Ala267Met and Ile272Val structure correlation can be revealed. For that reason, it was decided to obtain the double point mutant form Ala267Met/Ile272Val.
Fig. 2. Fragments of alignment of amino acid sequences of FDHs from different plants in the regions of residues chosen for site-directed mutagenesis (such residues are marked gray). The residues that are conservative for all FDHs are marked with asterisk (*). Residues conservative for plant FDHs only are marked with symbol ^. SoyFDH to be investigated is marked gray. The FDHs from various species are as follows: HvuFDH1, barley Hordeum vulgare; TaeFDH1, wheat Triticum aestivum; ZmaFDH, maize Zea mays; OsaFDH1, rice Oryza sativa; SbiFDH2, sorghum Sorghum bicolor; LjaFDH1, legume Lotus japonicus; LesFDH1, tomato Lycopersicon esculentum; StuFDH, potato Solanum tuberosum; ZofFDH, ginger Zingiber officinale; QroFDH1, oak Quercus robur; RcoFDH, castor Ricinus communis; BnaFDH2, raps Brassica napus; BolFDH1, cabbage Brassica oleracea; MdoFDH, apple Malus domestica; AthFDH, Arabidopsis thaliana.

Modification of electrostatic interactions on the protein surface. Surface amino acid residues play a very important role in stability of the protein globule. Analysis of three-dimensional structure of the SoyFDH–NAD+–N3– ternary complex revealed two such residues – Lys23 and Asn247. It was decided to change them to Thr and Glu, respectively (Fig. 1, e and f; see color insert).
Increasing rigidity of the amino acid chain. Using this approach, some residues in protein molecule were changed to Pro, which increase the rigidity of the amino acid chain. In the case of SoyFDH, analysis of protein structure and comparison of amino acid sequences allowed us to change residues Lys109 and Ser354 to Pro. Modeling of the Lys109Pro replacement is shown in Fig. 1, g and h (see color insert).
Therefore, base on the results of structure analysis and comparison of amino acid sequences of SoyFDH and other FDHs from plants, 10 replacements were chosen. Taking into consideration that in case of Ala267 and Ile272 double point mutation should be more optimal, it was decided to prepare the mutant form of SoyFDH with both replacements Ala267Met and Ile272Val. As a result, we obtain nine single point mutant forms of SoyFDH and one double point mutant form.
Preparation of mutant SoyFDH. Nucleotide replacements providing the necessary amino acid mutations were made using the polymerase chain reaction. Three plasmids were obtained for each of the ten mutant forms. Sequence analysis showed that all plasmids contained the soyfdh gene with the desired mutations and lacking other nucleotide replacements. Plasmids containing replacements encoding Gly18Ala, Lys23Thr, Leu24Asp, Lys109Pro, Val127Arg, Asn247Glu, Ala267Met, Ala267Met/Ile272Val, Val281Ile, and Ser354Pro were transformed in E. coli strain BL21(DE3)Codon Plus/pLysS. The resulting recombinant strains were cultivated according to the technique described in “Materials and Methods”. All 10 mutant forms of SoyFDH were expressed in active and soluble form. They were purified as described in “Materials and Methods”. Analytical electrophoresis in polyacrylamide gel in the presence of sodium dodecyl sulfate showed that the purity of the resulting enzymes was at least 95% (Fig. 3, a and b).
Fig. 3. Protein electrophoresis of enzymatic preparations. a: 1) SoyFDH Asn247Glu; 2) SoyFDH Gly18Ala; 3) SoyFDH Lys23Tyr; 4) SoyFDH Lys109Pro; 5) SoyFDH Ala267Met; 6) SoyFDH Ala267Met/Ile272Val; 7) SoyFDH Ser354Pro; 8) wt-SoyFDH. b: 1) SoyFDH Val127Arg; 2) SoyFDH Leu24Asp; 3) SoyFDH Val281Ile; 4) wt-SoyFDH; M, markers.

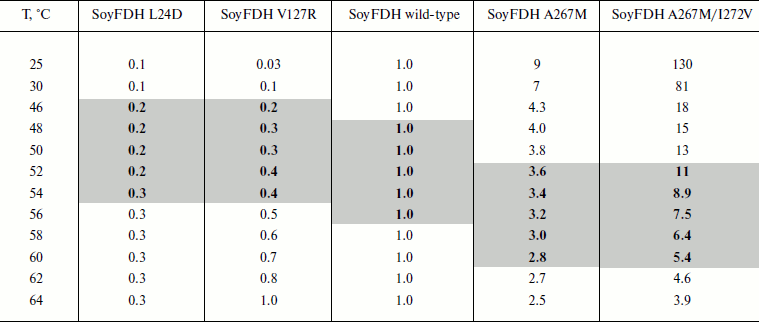
Thermal stability. Study of thermal stability from the kinetics of thermal inactivation. The differences in thermal stability of the mutant forms of SoyFDH prompted study of the kinetics of inactivation at different temperatures. In the case of less stable mutant forms Leu24Asp and Val127Arg, we studied inactivation kinetics in the temperature range 46-54°C. Wild-type SoyFDH and the mutant forms with replacements Gly18Ala, Lys23Thr, Lys109Pro, Asn247Glu, Val281Ile, Ser354Pro were studied in the 48-56°C range, and the more stable enzymes with replacements Ala267Met and Ala267Met/Ile272Val were studied at temperatures of 52-60°C. For all of the mutant forms and wt-SoyFDH the dependences of residual activity (A/A0) on time were exponential and thus linear in coordinates ln(A/A0) – t. Inactivation rate constant (kin) can be calculated from the slope of these dependences. Change in protein concentration over a wide range (0.1-2.5 mg/ml) had no influence on the value of kin, i.e. inactivation of the wt-SoyFDH and mutant forms is a monomolecular process. Consequently, the data show that the amino acid replacements did not change the mechanism of thermal inactivation.
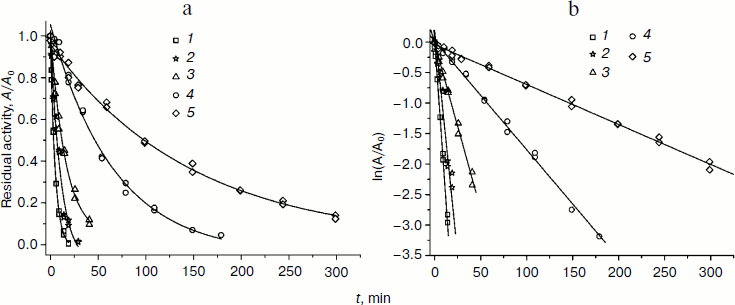
Dependences of residual activity on time in common and semilogarithmic coordinates at 54°C for several mutant forms and wt-SoyFDH are shown in Fig. 4 (a and b). The mutant forms with replacements Ala267Met and Ala267Met/Ile272Val are more stable than wt-SoyFDH, but mutants Val127Arg and Leu24Asp are less stable. Replacements Gly18Ala, Lys23Thr, Lys109Pro, Asn247Glu, Val281Ile and Ser354Pro did not cause any change in the thermal stability of SoyFDH, so we do not provide any data for these mutant forms.
Fig. 4. Dependence of residual activity on time in common (a) and semilogarithmic (b) coordinates for four mutant forms of SoyFDH and the wild-type enzyme, 0.1 M sodium-phosphate buffer, 0.01 M EDTA, pH 7.0, 54°C. 1) SoyFDH Leu24Asp; 2) SoyFDH Val127Arg; 3) wt-SoyFDH; 4) SoyFDH Ala267Met; 5) SoyFDH Ala267Met/Ile272Val.
The monomolecular mechanism of inactivation was observed for all mutant forms and wt-SoyFDH over a wide range of temperatures. In this case, transition state theory can be applied to analyze the process. The theoretical equation for the dependence of the first order constant kin on temperature T can be linearized using the equation:
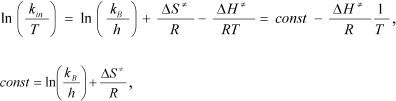
where kB and h are the Boltzmann and Plank constants, respectively, R is the universal thermodynamic constant, and ΔH ≠ and ΔS ≠ are the activating parameters for the thermal inactivation of the enzyme.
Dependences of inactivation rate constants on temperature were studied for the wild-type enzyme and the mutant forms of SoyFDH that had the strongest stabilization (Ala267Met and Ala267Met/Ile272Val) and destabilization (Leu24Asp and Val127Arg) effects. The dependences are presented in Fig. 5. The dependences in coordinates ln(kin/T) – 1/T are linear. The linearity of these dependences for the wild-type SoyFDH and its mutants (Fig. 5) confirmed the applicability of the transition state theory to describe the process of thermal inactivation of SoyFDH. From the slope the enthalpy ΔH ≠ and from the intercept the entropy ΔS ≠ can be calculated. The values of activation parameters for four chosen mutants and wt-SoyFDH are presented in Table 1. The difference in the activation parameters is greater than the measuring error. This means that the kin for all the mutants depends differently on temperature. The higher value of activation enthalpy ΔH ≠ of mutant forms SoyFDH A267M and SoyFDH A267M/I272V means that by decreasing the temperature the value of kin decreases much faster than for the wt-SoyFDH. So, when the temperature is decreased, a stabilization effect increases. We used the values of activation parameters ΔH ≠ and ΔS ≠ calculated from the experimental data to estimate the stabilization effect. The stabilization effect was calculated as the ratio of kin for wt-SoyFDH to kin for a mutant form at the same temperature. In Table 2, the values of the stabilization effect are presented. The experimental values are indicated with gray and bold. The table shows that when temperature is decreased the stabilization effect for mutant forms SoyFDH A267M and SoyFDH A267M/I272V increases. At room temperature the values are 9- and 130-fold, respectively.
Fig. 5. Dependence of inactivation rate constant of mutants and wt-SoyFDH on temperature in coordinates ln(kin/T) – 1/T (0.1 M sodium phosphate buffer, 0.01 M EDTA, pH 7.0). 1) wt-SoyFDH; 2) SoyFDH Leu24Asp; 3) SoyFDH Val127Arg; 4) SoyFDH Ala267Met; 5) SoyFDH Ala267Met/Ile272Val.
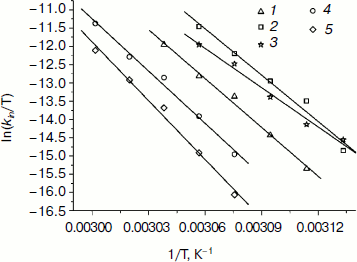
Table 1. Activation parameters for mutant
forms of SoyFDH
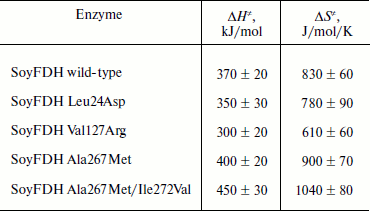
Table 2. The values of stabilization effect at different
temperatures (0.1 M sodium phosphate buffer, pH 7.0).
Experimental data are marked bold
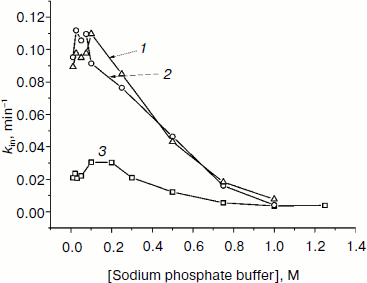
We also studied the dependence of kin on buffer concentration for the most stable mutants. Figure 6 shows such plots for wt-SoyFDH and mutants Ala267Met and Ala267Met/Ile272Val. These mutant forms have different stability; therefore, experiments were carried out at different temperatures. Figure 6 shows that for mutant forms A267M and A267M/I272V the dependences of kin on buffer concentration is similar. For the double mutant the stabilization effect is lower than for the single mutant. When the concentration of phosphate is increased from 0.01 to 0.1 M, the thermal stability becomes lower, but above 0.1 M the stabilization effect increases and reaches the value of 14-fold. The maximum of the plot (the lowest stability) is located at 0.1 M phosphate buffer, and the minimum is located at high concentrations of phosphate of about 1 M. The destabilization effect at lower ion strength may be due to increase in dielectric penetration. This can affect electrostatic interactions on the surface of the protein globule. Increase in stability at high concentrations of phosphate ion may be due to strengthening of hydrophobic interactions in the protein globule (the effect of strengthening of hydrophobic interactions at high ionic strength is a basis for a protein purification method; we used this method for purifying our enzymes).
Fig. 6. Dependences of inactivation rate constant on phosphate buffer concentration (0.01-1 M, pH 7.0). 1) SoyFDH Ala267Met/Ile272Val at 60°C; 2) SoyFDH Ala267Met at 58°C; 3) wt-SoyFDH at 52°C.
Study of thermal stability by differential scanning calorimetry. The thermal stability of the mutant forms of SoyFDH was also studied by DSC. The DSC melting curves are presented in Fig. 7. For the Ala267Met and Ala267Met/Ile272Val mutants the position of the maximum (Tm) of the melting curves is higher (stabilization effect). For the mutant forms with replacements Leu24Asp and Val127Arg, the values of Tm are lower (destabilization effect).
Fig. 7. DSC melting curves of mutant SoyFDHs and wt-SoyFDH (0.1 M sodium phosphate buffer, 0.01 M EDTA, pH 7.0). Enzyme concentration, 2 mg/ml. 1) SoyFDH Ala267Met/Ile272Val; 2) SoyFDH Ala267Met; 3) wt-SoyFDH; 4) SoyFDH Val127Arg; 5) SoyFDH Leu24Asp.
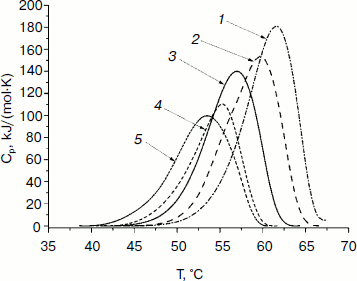
Parameters of the denaturation process for the mutant forms of SoyFDH and wt-FDH from different sources are presented in Table 3. The most stable FDH is from the bacterium Pseudomonas sp. 101 (PseFDH), and its mutant form PseFDH GAV has the highest values of ΔHcal and Tm. Amino acid replacements in SoyFDH that stabilized the enzyme (Ala267Met and Ala267Met/Ile272Val) also increased the values of ΔHcal and Tm. For the mutants with destabilization effect, these parameters decreased. The half-width of the plot (T1/2) is a factor of cooperativity of the denaturation process. Data from Table 3 show that for SoyFDH this parameter is lower than for the FDHs from other sources. There is no correlation between the values of T1/2 and the stabilization effect.
Table 3. DSC parameters for mutant SoyFDHs and wt-FDHs from
different sources
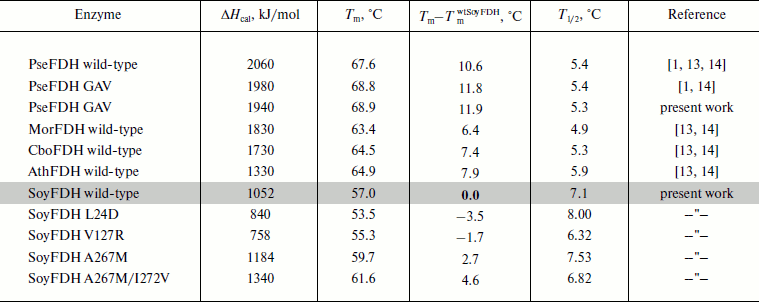
In conclusion, the DSC data for the mutant SoyFDHs are in good correspondence with data obtained from analysis of inactivation kinetics.
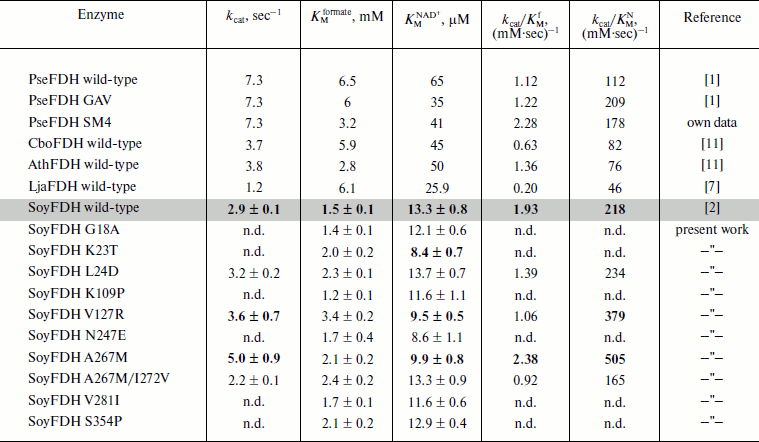
Kinetic parameters of the mutant enzymes. Michaelis constants for formate and NAD+ calculated for all mutant forms as well as catalytic constants for four mutant SoyFDHs are presented in Table 4. Also, data for FDHs from the bacterium Pseudomonas sp. 101 (PseFDH), the yeast C. boidinii (CboFDH), and the plants A. thaliana (AthFDH) and L. japonicus (LjaFDH) and two mutant forms of PseFDH with highest catalytic parameters and thermal stability are presented in Table 4. For some mutants the Michaelis constant for formate slightly increased (maximum 2.2-fold for Val127Arg). But all values of KMformate are less than that for the FDHs from the bacterium and yeast. Such increase in KMformate for the mutant SoyFDHs is not very important for practical application in chiral synthesis with an NADH regeneration system because high concentrations of formate (1-3 M) are used in such processes. These values are much higher than KMformate for SoyFDH. For practical application, KMNAD+ is much more important. In the case of SoyFDH with amino acid replacements Lys23Thr, Val127Arg, and Ala267Met, the KMNAD+ are lower (better).
Table 4. Catalytic parameters of SoyFDH mutants and FDHs
from other sources
Note: n.d., not determined.
One of the most important enzyme characteristics is the catalytic constant. For mutants Leu24Asp and Val127Arg, the kcat value increased by 10 and 25%, respectively (Table 4). Mutation Ala267Met in SoyFDH increased kcat by 72%. However, the double mutation A267M/Ile272Val decreased kcat. For SoyFDH, the forming of the ternary complex is accompanied by a change in the conformation of the enzyme [1]. Therefore, this suggests that the second mutation Ile272Val causing high stabilization effect would also decrease mobility of the protein molecule. However, it should be noted that for the most stable SoyFDH A267M/I272V the KMNAD+ value did not change.
The catalytic efficiency kcat/KM is also used for comparison of enzyme catalytic parameters. For the SoyFDH A267M mutant we got very good results – kcat/KM increased both for NAD+ and for formate, 2.3- and 1.2-fold, respectively. This mutant has the best catalytic characteristics among all the enzymes listed in Table 4. In case of kcat/KMNAD+, the A267M mutant is two times better than the best mutant of bacterial FDH PseFDH GAV and is practically the same as PseFDH SM4.
So, the SoyFDH A267M mutant is the most interesting of the 10 mutants described in this work. It is an effective biocatalyst with high thermal stability and excellent catalytic and kinetic characteristics.
Positive results in thermal stability and catalytic parameters were found using only one approach. This approach is based on filling cavities inside the protein globule. Negative effects using other approaches do not mean that they could not be applied for this enzyme in the future. It is more important to find positions in amino acid sequences that are responsible for catalytic parameters and thermal stability. In this work, one such position was found. It is Ala267. Experiments using rational design of SoyFDH proceed in our laboratory. The analysis of protein structure helped us to find several positions that may be responsible for the most important enzyme properties. The data of preliminary experiments show that approaches used in this work should have some positive results.
This work was supported by the Ministry of Education and Science of Russian Federation (contract 16.512.11.2148) and Russian Foundation for Basic Research (grant 11-04-00920).
REFERENCES
1.Tishkov, V. I., and Popov, V. O. (2004)
Biochemistry (Moscow), 69, 1252-1267.
2.Alekseeva, A. A., Savin, S. S., and Tishkov, V. I.
(2011) Acta Naturae, 3, 38-54.
3.David, P., des Francs-Small, C. C., Sevignac, M.,
Thareau, V., Macadre, C., Langin, T., and Geffroy, V. (2010) Theor.
Appl Genet., 121, 87-103.
4.Hourton-Cabassa, C., Ambard-Bretteville, F.,
Moreau, F., Davy de, V., Remy, R., and des Francs-Small, C. C. (1998)
Plant Physiol., 116, 627-635.
5.Suzuki, K., Itai, R., Suzuki, K., Nakanishi, H.,
Nishizawa, N. K., Yoshimura, E., and Mori, S. (1998) Plant
Physiol., 116, 725-732.
6.Thompson, P., Bowsher, C. G., and Tobin, A. K.
(1998) Plant Physiol., 118, 1089-1099.
7.Andreadeli, A., Flemetakis, E., Axarli, I., Dimou,
M., Udvardi, M. K., Katinakis, P., and Labrou, N. E. (2009) Biochim.
Biophys. Acta, 1794, 976-984.
8.Wojtasik, W., Kulma, A., Kostyn, K., and Szopa, J.
(2011) Plant Physiol. Biochem., 49, 862-872.
9.Colas des Francs-Small, C., Ambard-Bretteville, F.,
Small, I. D., and Remy, R. (1993) Plant Physiol., 102,
1171-1177.
10.Shiraishi, T., Fukusaki, E., and Kobayashi, A.
(2000) J. Biosci. Bioeng., 89, 241-246.
11.Sadykhov, E. G., Serov, A. E., Yasnyi, I. E.,
Voinova, N. S., Alexeeva, A. A., Petrov, A. S., and Tishkov, V. I.
(2006) Vestnik Mosk. Univ. Ser. 2. Khim., 47, 31-34.
12.Alekseeva, A. A. (2011) Recombinant Formate
Dehydrogenase from Soya Glycine max: Protein Engineering and Structure
Analysis: Ph.D thesis [in Russian], Lomonosov Moscow State
University.
13.Sadykhov, E. G., Serov, A. E., Voinova, N. S.,
Uglanova, S. V., Petrov, A. S., Alexeeva, A. A., Kleimenov, S. Yu.,
Popov, V. O., and Tishkov, V. I. (2006) Appl. Biochem.
Microbiol., 42, 236-240.
14.Sadykhov, E. G. (2007) Obtaining and Structure
Analysis of Formate Dehydrogenases from Different Sources: Ph.D
thesis [in Russian], Bach Institute of Biochemistry, Russian Academy of
Sciences.
15.Voinova, N. S., Savin, S. S., Alekseeva, A. A.,
Skirgello, O. E., and Tishkov, V. I. (2008) Moscow Univ. Chem.
Bull., 63, 60-62.
16.Savin, S. S., and Tishkov, V. I. (2010) Acta
Naturae, 2, 97-102.
17.Shabalin, I. G., Serov, A. E., Skirgello, O. E.,
Timofeev, V. I., Samygina, V. R., Popov, V. O., Tishkov, V. I., and
Kuranova, I. P. (2010) Crystallogr. Rep., 55,
806-810.
18.Tishkov, V. I., and Popov, V. O. (2006)
Biomol. Eng., 23, 89-110.
19.Romanova, E. G., Alekseeva, A. A., Pometun, E.
V., and Tishkov, V. I. (2010) Moscow Univ. Chem. Bull.,
65, 127-130.
20.Rojkova, A. M., Galkin, A. G., Kulakova, L. B.,
Serov, A. E., Savitsky, P. A., Fedorchuk, V. V., and Tishkov, V. I.
(1999) FEBS Lett., 445, 183-188.
21.Baker, D., and Sali, A. (2001)
Science, 295, 93-96.
22.Fedorchuk, V. V., Galkin, A. G., Yasny, I. E.,
Kulakova, L. B., Rojkova, A. M., Filippova, A. A., and Tishkov, V. I.
(2002) Biochemistry (Moscow), 67, 1145-1151.
23.Serov, A. E., Odintzeva, E. R., Uporov, I. V.,
and Tishkov, V. I. (2005) Biochemistry (Moscow), 70,
804-808.