Boronated Derivatives of Chlorin e6 and Fluoride-Containing Porphyrins as Penetrating Anions: a Study Using Bilayer Lipid Membranes
T. I. Rokitskaya1*, A. V. Zaitsev2, V. A. Ol’shevskaya2, V. N. Kalinin2, M. M. Moisenovich3, I. I. Agapov3, and Y. N. Antonenko1
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: rokitskaya@genebee.msu.ru2Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, 119234 Moscow, Russia
3Biological Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received March 22, 2012; Revision received May 23, 2012
Boronated derivatives of porphyrins are studied extensively as promising compounds for boron-neutron capture therapy and photodynamic therapy. Understanding of the mechanism of their permeation across cell membranes is a key step in screening for the most efficient compounds. In the present work, we studied the ability of boronated derivatives of chlorin e6 and porphyrins, which are mono-, di-, and tetra-anions, to permeate through planar bilayer lipid membranes (BLM). The translocation rate constants through the hydrophobic part of the lipid bilayer were estimated for monocarborane and its conjugate with chlorin e6 by the method of electrical current relaxation. They were similar, 6.6 and 6.8 sec–1, respectively. Conjugates of porphyrins carrying two and four carborane groups were shown to permeate efficiently through a BLM although they carry two charges and four charges, respectively. The rate of permeation of the tetraanion estimated by the BLM current had superlinear dependence on the BLM voltage. Because the resting potential of most mammalian cells is negative inside, it can be concluded that the presence of negatively-charged boronated groups in compounds should hinder the accumulation of the porphyrins in cells.
KEY WORDS: penetrating anions, monocarborane, bilayer lipid membrane, photosensitizer, chlorin e6, porphyrinDOI: 10.1134/S0006297912090039
Abbreviations: BLM, bilayer lipid membrane; BNCT, boron-neutron capture therapy; PDT, photodynamic therapy.
Hydrophobic ions with the charge delocalized through a system of
aromatic bonds belong to a class of molecules capable of penetrating
through the lipid membranes, in contrast to other charged molecules and
inorganic ions [1-3]. In recent
years, penetrating cations have been widely used as “electric
locomotives” for the targeted delivery of antioxidants [4, 5] and other compounds [6, 7] into mitochondria.
Mitochondria are the only intracellular organelles whose interior has
negative electrical potential relative to their surroundings. Thus,
penetrating cations, once in a cell, are accumulated predominantly in
mitochondria. The use of anionic groups capable of penetrating across
membranes seems to be a promising approach for the delivery of
compounds into cells and other cellular compartments. Since cells of
different tissues have different resting potential, anionic groups can
provide selective accumulation of charged compounds in such different
animal cells.
Polyhedral carboranes are used in the synthesis of compounds for boron-neutron capture therapy (BNCT) of cancer. In the early works on penetrating ions, one carborane derivative was shown to penetrate through lipid membranes [8, 9]. Nontraditional three-center two-electron bonds form the basis of carboranes, which are electron-deficient structures and can be regarded as three-dimensional delocalized aromatic systems [10, 11]. This is probably the reason for their ability to permeate lipid membranes [12]. The number of boron atoms in the molecule had to be increased so as to increase the thermal neutrons capture cross section and thus increase the efficiency of compounds in BNCT. It is because of this that porphyrin compounds with different number of carboranes [13-15] or dodecaborates [16] in the molecule are being studied now. These compounds are selectively accumulated in cancer cells and can be used both for BNCT and for photodynamic therapy (PDT) of cancer.
The mechanism of penetration of hydrophobic anions across lipid membranes has been thoroughly studied for tetraphenylborate, dipicrylamine, and analogs of these molecules [17, 18]. Kinetic models have been developed [17, 19] that make it possible to describe the translocation of hydrophobic ions through a BLM. These models assume the existence of three main stages of the process of penetration ion X: adsorption on the membrane surface (constants k and γk), ion transfer across the hydrophobic layer to the other membrane surface (constant ki), and desorption to the water phase (with the same constants k and γk):
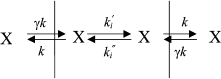
and
where c is the concentration of hydrophobic ions in a solution, z is the valence of the ion, γ is the ratio of surface and bulk concentrations (adsorption constant), β is proportion of the applied potential that affects the ion motion, R is the universal gas constant, F is the Faraday constant, and T is absolute temperature. The ratio of the initial and steady-state current is determined by:
The exponential curve of current relaxation after the application of potential can be explained by the redistribution of hydrophobic anions between the two membrane surfaces. This model is equally applicable to both positively and negatively charged hydrophilic ions. Having determined the dependence of τ and I(0)/I() on the applied potential, we can calculate the constants of ion penetration rates. When studying the penetration of tetraphenylborate through the membrane, it was shown that the initial current I0 was considerably higher than the stationary I, and hence, ki >> k (this can be concluded on the basis of Eq. (3)). The energy barrier of the membrane is relatively low for hydrophobic anions, and ki (the rate constant of transfer from one membrane surface to the other) is significantly higher than k (the rate constant of transfer from the membrane surface to the water phase).
It was shown earlier that, in contrast to the monocation tetraphenyl phosphonium, the triphenyl phosphonium dication could hardly penetrate the BLM at low concentrations and causes leakage of the membrane and its destabilization at high concentrations [20]. Thus, the question arises whether borated derivatives of photosensitizers with several charged groups will penetrate lipid membranes without disrupting their integrity. Results presented in this paper give a positive answer to this question. Having measured the kinetics of current relaxation in the presence of porphyrin compounds with different numbers of monocarboranes, we studied the penetration of compounds with different numbers of negative charges across the membrane. The rates of penetration of monocarborane and chlorin e6 conjugate with one monocarborane molecule across the BLM were found to be 6.6 and 6.8 sec–1, respectively.
MATERIALS AND METHODS
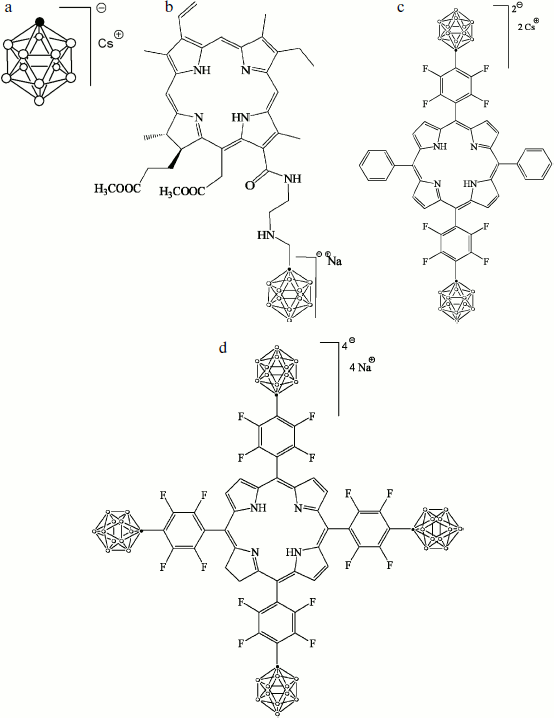
The cesium salt of 1-carba-closo-dodecaborate (monocarborane), the sodium salt of 13(1)-N-{2-[N-(1-carba-closo-dodecaborane-1-yl)methyl]aminoethyl}amide-15(2),17(3)-dimethyl ester of chlorin e6 (compound 1), the cesium salt of {5,15-diphenyl-10,20-bis[4-(1-carba-closo-dodecaborane-1-yl)tetrafluorophenyl]porphyrin)} (compound 2), and the sodium salt of {5,10,15,20-tetrakis[4-(1-carba-closo-dodecaborane-1-yl)tetrafluorophenyl]-17,18-dihydroporphyrin)} (compound 3) were synthesized and described earlier [21]. The structural formulas of these compounds are presented in Fig. 1.
The BLM was formed from 2% solution of diphytanoyl phosphatidylcholine (Avanti Polar Lipids) in decane on a 0.5-mm diameter hole in the barrier that separates the Teflon cell with buffer solution into two compartments [22]. The studied hydrophobic anions were added from concentrated solutions in water or ethanol to the aqueous solution on both sides of the membrane and were thoroughly mixed for 5 min. The aqueous solution contained 100 mM KCl, 10 mM Tris, pH 7.4. All experiments were performed at room temperature (23-25°C).Fig. 1. Structural formulas of hydrophobic anions studied in this paper: a) monocarborane; b-d) compounds 1-3, respectively.
The electric current was recorded under conditions of fixed potential. The potential difference was applied to silver–silver chloride electrodes placed via agar bridges into the Teflon cell. The current was measured using an OES-2 patch-clamp amplifier (OPUS, Russia), digitized using an NI-DAQmx digitizer (National Instruments, USA), and analyzed using the WinWCP Strathclyde Electrophysiology Software developed by J. Dempster (University of Strathclyde, GB). The voltage was applied at the zero time from zero to a certain voltage value, which varied depending on the design of the experiment. Then the current through the membrane decreased from I(0) to I(). Before adding hydrophobic anions to the membrane, we recorded the capacitive response of the unmodified membrane in each experiment (control measurements of current at all used voltage values). Recordings of the current in the presence of hydrophobic anions were analyzed after subtracting the control traces.
RESULTS
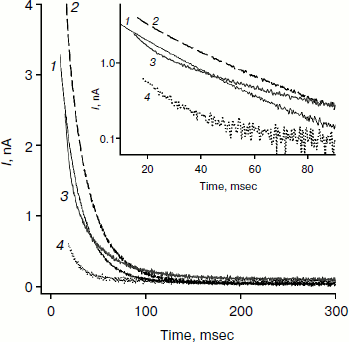
Figure 2 shows recordings of current through a BLM in the presence of monocarborane and its derivatives in response to the application of 125 mV voltage to the membrane. The process of current reduction with time (current relaxation) could be observed after application of the potential difference to the membrane. The inset (Fig. 2) shows the same data presented in semilogarithmic coordinates; this presentation shows that the curves for monocarborane and compound 1 (that contain one anion) are monoexponential functions. Their characteristic relaxation time is 22 msec. The current relaxation curve in the presence of compound 2, which includes two anions, cannot be described by a single exponential and requires at least two components with characteristic times of 6 and 36 msec to be adequately described. In the presence of compound 3, which has four negative charges, the decline in current in response to the application of the voltage has significantly smaller amplitude; the characteristic relaxation time is ~25 msec. (The concentration of compound 3 in solution was five times higher than the monocarborane concentration.)
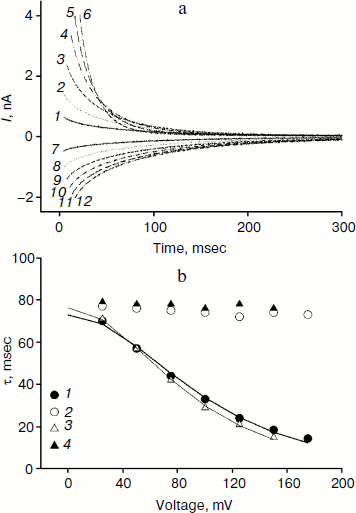
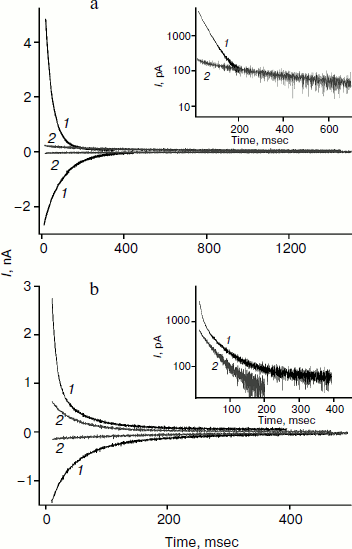
We studied the dependence of the characteristic time of monoexponential current relaxation for monocarborane and compound 1 on the applied potential difference. Figure 3a shows the recordings of current in response application of different voltages (positive current values) and the removal of voltage (negative current values) in the presence of compound 1. In accordance with Eqs. (1) and (2), the current amplitude at the initial time increases and the relaxation accelerates when the applied potential difference is increased. Figure 3b shows the dependence of the characteristic current relaxation time (τon and τoff) in response to the application and removal of voltage for monocarborane and compound 1. The concentrations of the compounds were 20 and 50 nM for compound 1 and monocarborane, respectively; the measured values of I(0) were virtually identical. On the basis of the current relaxation curves, it can be seen that I(0) >> I(). In accordance with Eq. (3), this means that the constant of translocation across the membrane ki substantially exceeds the binding constant k for the studied anions. Approximation of the data for τ (the curves of Fig. 3b) gave ki = 6.6 and 6.8 sec–1 and β = 0.82 and 0.73 for monocarborane and compound 1, respectively.Fig. 2. Recordings of current through a BLM in response to the application of 125 mV voltage in the presence of 50 nM monocarborane (curve 1), 20 nM compound 1 (curve 2), 100 nM compound 2 (curve 3), or 250 nM compound 3 (curve 4). The inset shows the same recordings on a semilogarithmic scale.
Compounds that reduce membrane dipole potential are known to accelerate the transport of hydrophobic cations and slow the transport of anions across a BLM [2, 23, 24]. The amplitude of the monocarborane current relaxation and the rate of its penetration are significantly reduced in the presence of a modifier of the dipole potential of lipid membranes – phloretin (Fig. 4a). Application of U = 100 mV leads to almost 50-fold reduction in I(0) after the addition of 25 μM phloretin, and τon increases 8-fold (Fig. 4a). The kinetics of the current relaxation remains monoexponential (inset in Fig. 4a). The amplitude of the current relaxation for compound 2 is reduced approximately 10-fold in response to addition of phloretin (Fig. 4b). The nature of the current dependence on time becomes monoexponential, as seen in the inset in Fig. 4b.Fig. 3. a) Recordings of the current through a BLM in the presence of 20 nM compound 1 in response to the application and removal of voltage of 25 (curves 1 and 7), 50 (curves 2 and 8), 75 (curves 3 and 9), 100 (curves 4 and 10), 125 (curves 5 and 11), and 150 mV (curves 6 and 12). b) Dependence of the characteristic current relaxation times τon (1) and τoff (2) for compound 1 and τon (3) and τoff (4) for monocarborane. The curves result from data approximation according to Eq. (2) for ki = 6.6 sec–1 and β = 0.82 for monocarborane and ki = 6.8 sec–1 and β = 0.73 for compound 1.
The charge transferred across a BLM after voltage application can be estimated from the area under the relaxation curve. It is reduced 7-fold for monocarborane after addition of phloretin (Fig. 4a). For compounds 1 and 2 this reduction is 1.5-2-fold. Apparently, phloretin adsorption on the BLM surface leads not only to the reduction of the size of the membrane dipole potential, but also to partial displacement of penetrating anion molecules from the membrane to the water phase. Phloretin is a weak acid with pKa = 7.4. Consequently, its adsorption on the membrane surface creates some surface charge. Monocarborane molecules are desorbed from the BLM surface more actively than the molecules of compounds 1 and 2. We can assume this difference is due to the presence of a quite hydrophobic uncharged part – chlorin e6 or porphyrin – in the molecules of these compounds (Fig. 1); these parts increase the affinity of the molecules to the membrane surface or lead to their deeper location. This is confirmed by the differences in the degree of influence of phloretin on the slowing of the characteristic current relaxation time. This slowing is most pronounced for monocarborane (8-fold) and compounds 1 (4-5-fold) and 2 (3-fold). These data correspond to the small potentials when relaxation is close to monoexponential dependence. Thus, the anions of compound 2 are immersed in the bilayer deeper than those of the other compounds and have a lower sensitivity towards the changes in the dipole potential jump.Fig. 4. Recordings of the current through a BLM in the presence of 50 nM monocarborane (a) or 100 nM compound 2 (b) in response to the application and removal of 100 mV voltage (curves 1) and after the addition of 25 μM phloretin (curves 2). The insets show the recordings of current in response to the voltage application on a semilogarithmic scale.
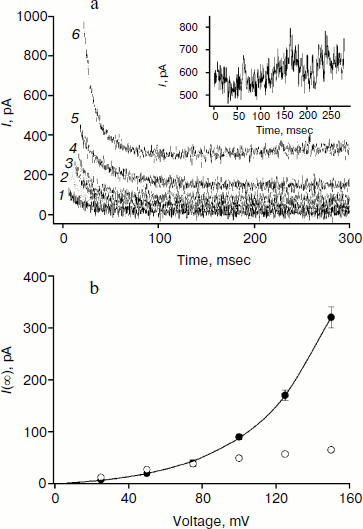
The amplitude of the kinetics of current relaxation in response to the applied potential differences in the presence of compound 3, which has four monocarborane groups, is significantly smaller than the amplitudes for the other studied compounds. Moreover, its concentration was an order of magnitude higher than the concentrations of the other compounds (Fig. 2). Figure 5a shows the curves of current relaxation for different potentials in the presence of 400 nM compound 3 added to the membrane. Accurate measurement of the dependence of τon on potential is rather difficult because of the presence of small currents and current fluctuations; nevertheless, it can be noted that a significant steady-state current through the membrane is observed in the presence of this compound, and this current increases nonlinearly with the increase in the applied voltage (Fig. 5b). We assume the steady-state current to be determined by the flow of compound 3 from the water phase on one side of the membrane to the water phase on the other side. Exponential dependence of the steady-state current on the applied voltage can be explained by the presence of four charges in the compound 3 molecule. In the presence of high concentrations of compound 1, steady-state current is linearly dependent on voltage and approaches saturation at high potentials (Fig. 5b).
The application of a high voltage (150 mV) to BLM in the presence of significant concentrations of compound 3 (>250 nM) for a long time (>5 sec) often leads to the appearance of current fluctuations (Fig. 5a, inset) associated with the destabilization of the membrane, often followed by its rupture. Therefore, the increasing the concentration of compound 3 in the aqueous solution or increasing applied voltage was difficult.Fig. 5. a) Recordings of current through a BLM in the presence of 400 nM compound 3 in response to the application of voltage of 25 (curve 1), 50 (curve 2), 75 (curve 3), 100 (curve 4) 125 (curve 5), and 150 mV (curve 6). The inset shows the current recording at 150 mV. b) Dependences of the steady-state current through the BLM at 1.5 sec after the voltage application in the presence of 100 nM compound 1 (light circles) or 400 nM compound 3 (dark circles). The data for compound 3 were approximated by the exponential curve I(U) = aebU + I(0), where a = 8.3, b = 0.025, and I(0) = –9.
The potential difference on the BLM was also measured in an open circuit when creating a concentration gradient of the studied penetrating anions. A 10-fold monocarborane concentration gradient caused potential difference of 57 ± 1.5 mV on the BLM. This value is virtually identical to the theoretical limit value of the potential on the membrane that has ideal selectivity for this ion (Nernst equation, 58 mV at room temperature). For a ten-fold concentration gradient of compound 3, the potential difference was 20 ± 2 mV, which was higher than expected (14.5 mV) from the Nernst equation taking into account the presence of four negative charges in one molecule. For 5-fold gradient of compound 2, the potential difference across the BLM was 32 ± 1 mV, which again was somewhat higher than the theoretical value (20.5 mV) for a penetrating compound with the molecule charge of two. The experimentally measured potential values for compounds 2 and 3 were shown to be higher than the theoretical values. The reason for this excess is unclear and requires further study. One possible explanation is a nonlinear dependence of the parameter γ on the bulk concentration of the compound, which leads to a difference between the membrane- and the bulk gradients of the compound.
DISCUSSION
Overall, our data suggest all the studied compounds including monocarborane and the porphyrin derivatives with different numbers of monocarboranes in the molecule penetrate through the BLM. The following data support this suggestion: generation of electric potentials caused by concentration gradients of these compounds; current induction in their presence in response to voltage jump on the membrane; increase in the steady-state membrane conduction caused by their addition to the aqueous solution.
As discussed earlier, the current relaxation is caused by the redistribution of hydrophobic anions between the two membrane surfaces under the influence of the potential difference. The characteristic current relaxation times in the presence of monocarborane and compound 1 (with one monocarborane) are very similar in the entire range of applied voltages, and for voltages close to zero τ0 = 77 and 74 msec for monocarborane and compound 1, respectively. It would be interesting to compare the translocation times of monocarborane and the well-studied tetraphenylborate. Characteristic current relaxation time in the presence of penetrating ions is known to depend on the lipid composition of the membrane. For example, in case of dioleoyl lecithin τ0 = 55 (for current relaxation in the presence of tetraphenylborate [17]), 2 (for membranes from bacterial phosphatidylcholine [19]), and 83 msec (for diphytanoyl phosphatidylcholine [25]). Since the membrane was formed of diphytanoyl phosphatidylcholine in this work, the relaxation times for monocarborane can be compared to those for tetraphenylborate given in [25]. We conclude that tetraphenylborate and monocarborane are translocated across the BLM with similar rates.
The rate constants of transmembrane transport calculated according to Eq. (2) from the dependence of characteristic time on applied voltage were found to be 6.6 and 6.8 sec–1 for monocarborane and compound 1, respectively. The β values, which characterize the depth of the molecule location on the membrane surface and the portion of the applied potential affecting the anion movement, were found to be 0.82 and 0.73, respectively. These data suggest the penetration rate of compound 1 is determined mainly by the properties of monocarborane; the covalently bound group of chlorin e6 does not make anion translocation across the lipid membrane more difficult. The change in β values indicates a deeper location of compound 1 in relation to the surface of the lipid membrane as compared to that of monocarborane, the phenomenon being apparently connected to the high binding constant of chlorin e6 with the lipid membrane [26]. The higher concentration of monocarborane (when compared to compound 1 concentration) necessary for the registration of the same initial current value also indicates a higher affinity of compound 1 to the membrane.
Boronated porphyrin compounds have been actively studied recently because of the prospects of their use in boron-neutron capture therapy of cancer, combined with photodynamic therapy. Up to four molecules of anionic boron hydrides are bound to photosensitizer so as to increase the efficiency of BNCT. Dodecaborate [27, 28] and monocarborane [29] have the greatest prospects in terms of new drug development, for they are di- and monoanions and provide water solubility. Boronated porphyrin [30], chlorin [31], and hemin [32] were found to be more effective photosensitizers both in vitro and in vivo than their predecessors. Compound 1 was previously shown to accumulate in intracellular organelles such as the Golgi apparatus, endoplasmic reticulum, and other membrane compartments [31]. In addition, this compound has a higher light-induced cytotoxic activity compared to non-boronated chlorin e6. These differences can be attributed to the ability of compound 1 to penetrate effectively the bilayer lipid membrane (in contrast to unmodified chlorin e6).
Thus, the increase in the number of carboranes in the photosensitizer does not preclude the ability of compounds to penetrate through a BLM. The concentration of charged molecules within the cells is determined not only by their permeability across the plasma membrane, but also by the presence of potential on this membrane. Since the resting potential of the majority of animal cells is negative inside, the internal concentration of anionic porphyrins should be lower than the outer one. This is especially true of compounds bearing two or more charges, since, according to the Nernst equation, the equilibrium ratio of concentrations inside and outside the membrane for charged molecules depends exponentially on the potential value multiplied by the number of charges in the molecule. It should be noted that cancer cells have lower resting potentials [33], which can be attributed to differences in the level of expression of ion channels [34]. This difference in resting potential can determine the selectivity in the accumulation of charged molecules in different tissues. Our work has also shown that polyanionic porphyrins to have lesser affinity to the lipid membrane, which should also reduce the accumulation of porphyrins in the lipid-containing structures of animal cells.
We express our gratitude to E. A. Kotova and A. A. Shtil for fruitful discussion of the results.
This work was financially supported by the Russian Foundation for Basic Research (grant 12-04-00199), the Ministry of Education and Science of the Russian Federation under the federal target program “Scientific and Scientific-Pedagogical Personnel of Innovative Russia for 2009-2013” (state contract P 407 from 12.05.2010), and the Ministry of Industry and Trade of the Russian Federation under the federal target program “Development of Pharmaceutical and Medical Industry of the Russian Federation to 2020 and Beyond” (state contract 11411.1008700.13.085 from 13.09.2011).
REFERENCES
1.Liberman, E. A., Topaly, V. P., Tsofina, L. M.,
Jasaitis, A. A., and Skulachev, V. P. (1969) Nature, 222,
1076-1078.
2.Liberman, E. A., and Topaly, V. P. (1969)
Biofizika, 14, 452-461.
3.LeBlanc, O. H. (1969) Biochim. Biophys.
Acta, 193, 350-360.
4.Kelso, G. F., Porteous, C. M., Hughes, G.,
Ledgerwood, E. C., Gane, A. M., Smith, R. A., and Murphy, M. P. (2002)
Ann. N. Y. Acad. Sci., 959, 263-274.
5.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L. E.,
Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Y.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Y., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Y., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1273-1287.
6.Filipovska, A., Kelso, G. F., Brown, S. E., Beer,
S. M., Smith, R. A., and Murphy, M. P. (2005) J. Biol. Chem.,
280, 24113-24126.
7.Stoyanovsky, D. A., Vlasova, I. I., Belikova, N.
A., Kapralov, A., Tyurin, V., Greenberger, J. S., and Kagan, V. E.
(2008) FEBS Lett., 582, 725-728.
8.Grinius, L. L., Jasaitis, A. A., Kadziauskas, Y.
P., Liberman, E. A., Skulachev, V. P., Topali, V. P., Tsofina, L. M.,
and Vladimirova, M. A. (1970) Biochim. Biophys. Acta,
216, 1-12.
9.Bakeeva, L. E., Grinius, L. L., Jasaitis, A. A.,
Kuliene, V. V., Levitsky, D. O., Liberman, E. A., Severina, I. I., and
Skulachev, V. P. (1970) Biochim. Biophys. Acta, 216,
13-21.
10.Wade, K. (1976) Inorg. Chem. Radiochem.,
18, 1-66.
11.King, R. B. (1993) Izv. RAN. Ser. Khim.,
8, 1353-1360.
12.Liberman, E. A. (1970) Biofizika,
15, 278-297.
13.El Zaria, M. E., Ban, H. S., and Nakamura, H.
(2010) Chemistry, 16, 1543-1552.
14.Ol’shevskaya, V. A., Zaytsev, A. V.,
Savchenko, A. N., Shtil, A. A., Cheong, C. S., and Kalinin, V. N.
(2007) Bull. Korean Chem. Soc., 28, 1910-1916.
15.Friso, E., Roncucci, G., Dei, D., Soncin, M.,
Fabris, C., Chiti, G., Colautti, P., Esposito, J., De Nardo, L.,
Riccardo, R. C., Nitti, D., Giuntini, F., Borsetto, L., and Jori, G.
(2006) Photochem. Photobiol. Sci., 5, 39-50.
16.Efremenko, A. V., Ignatova, A. A., Borsheva, A.
A., Grin, M. A., Bregadze, V. I., Sivaev, I. B., Mironov, A. F., and
Feofanov, A. V. (2012) Photochem. Photobiol. Sci., 11,
645-652.
17.Ketterer, B., Neumcke, B., and Lauger, P. (1971)
J. Membr. Biol., 5, 225-245.
18.Benz, R. (1988) Biophys. J., 54,
25-33.
19.Andersen, O. S., and Fuchs, M. (1975) Biophys.
J., 15, 795-830.
20.Severina, I. I., Vyssokikh, M. Y., Pustovidko, A.
V., Simonyan, R. A., Rokitskaya, T. I., and Skulachev, V. P. (2007)
Biochim. Biophys. Acta, 1767, 1164-1168.
21.Olshevskaya, V. A., Zaitsev, A. V., Sigan, A. L.,
Kononova, E. G., Petrovsky, P. V., Chkanikov, N. D., and Kalinin, V. N.
(2010) Dokl. AN. Khimiya, 435, 755-759.
22.Mueller, P., Rudin, D. O., Tien, H. T., and
Wescott, W. C. (1963) J. Phys. Chem., 67, 534-535.
23.Andersen, O. S., Finkelstein, A., and Katz, I.
(1976) J. Gen. Physiol., 67, 749-771.
24.Pohl, P., Rokitskaya, T. I., Pohl, E. E., and
Saparov, S. M. (1997) Biochim. Biophys. Acta, 1323,
163-172.
25.Erukova, V. Y., Krylova, O. O., Antonenko, Y. N.,
and Melik-Nubarov, N. S. (2000) Biochim. Biophys. Acta,
1468, 73-86.
26.Kepczynski, M., Pandian, R. P., Smith, K. M., and
Ehrenberg, B. (2002) Photochem. Photobiol., 76,
127-134.
27.Longuet-Higgins, H. C., and Roberts, M. (1955)
Proc. Roy. Soc. (London) A, 230, 110-119.
28.Pitochelli, A. R., and Hawthorne, M. F. (1960)
J. Am. Chem. Soc., 82, 3228-3229.
29.Korbe, S., Michl, J., and Schreiber, P. J. (2006)
Chem. Rev., 106, 5208-5249.
30.Hill, J. S., Kahl, S. B., Stylli, S. S.,
Nakamura, Y., Koo, M. S., and Kaye, A. H. (1995) Proc. Nat. Acad.
Sci. USA, 92, 12126-12130.
31.Moisenovich, M. M., Ol’shevskaya, V. A.,
Rokitskaya, T. I., Ramonova, A. A., Nikitina, R. G., Savchenko, A. N.,
Tatarskiy, V. V., Jr., Kaplan, M. A., Kalinin, V. N., Kotova, E. A.,
Uvarov, O. V., Agapov, I. I., Antonenko, Y. N., and Shtil, A. A. (2010)
PLoS One, 5, e12717.
32.Ol’shevskaya, V. A., Nikitina, R. G.,
Zaitsev, A. V., Luzgina, V. N., Kononova, E. G., Morozova, T. G.,
Drozhzhina, V. V., Ivanov, O. G., Kaplan, M. A., Kalinin, V. N., and
Shtil, A. A. (2006) Org. Biomol. Chem., 4, 3815-3821.
33.Binggeli, R., and Cameron, I. L. (1980) Cancer
Res., 40, 1830-1835.
34.Cherubini, A., Taddei, G. L., Crociani, O.,
Paglierani, M., Buccoliero, A. M., Fontana, L., Noci, I., Borri, P.,
Borrani, E., Giachi, M., Becchetti, A., Rosati, B., Wanke, E.,
Olivotto, M., and Arcangeli, A. (2000) Br. J. Cancer, 83,
1722-1729.