REVIEW: Common Themes in Glycoconjugate Assembly Using the Biogenesis of O-Antigen Lipopolysaccharide as a Model System*
M. A. Valvano
Centre for Human Immunology, Department of Microbiology and Immunology and Department of Medicine, University of Western Ontario, London, Ontario N6A 5C1, Canada; fax: +1-519-661-3499; E-mail: mvalvano@uwo.ca* This mini-review is based on a presentation made at the 4th Baltic Conference on Microbial Carbohydrates, Hyytiälä Forestry Field Station, Finland, September 19-22, 2010.
Received February 15, 2011; Revision received February 21, 2011
The biosynthesis of glycoconjugates is remarkably conserved in all types of cells since the biochemical reactions involved exhibit similar characteristics, which can be summarized as follows: (a) the saccharide moiety is formed as a lipid-linked, membrane-associated glycan; (b) the lipid component in most cases is a polyisoprenoid phosphate; (c) the assembly of the lipid-linked saccharide intermediate depends on reactions taking place at both sides of the cell membrane, which requires the obligatory transmembrane movement of amphipathic molecules across the lipid bilayer. These general characteristics are present in the biosynthesis of the O-antigen component of the bacterial lipopolysaccharide, which serves as a model system to investigate the molecular and mechanistic basis of glycoconjugate synthesis, as summarized in this mini-review.
KEY WORDS: lipopolysaccharide, undecaprenyl phosphate, ligase, glycosylphosphate transferase, outer membraneDOI: 10.1134/S0006297911070029
Abbreviations: LPS, lipopolysaccharide; OS, oligosaccharide; PHPT, polyisoprenyl-phosphate hexose-1-phosphate transferases; PNPT, polyisoprenyl-phosphate N-acetylaminosugar-1-phosphate transferases; TM, transmembrane.
A SET OF COMMON PRINCIPLES GOVERNS THE SYNTHESIS
OF GLYCOCONJUGATES IN ALL TYPES OF CELLS
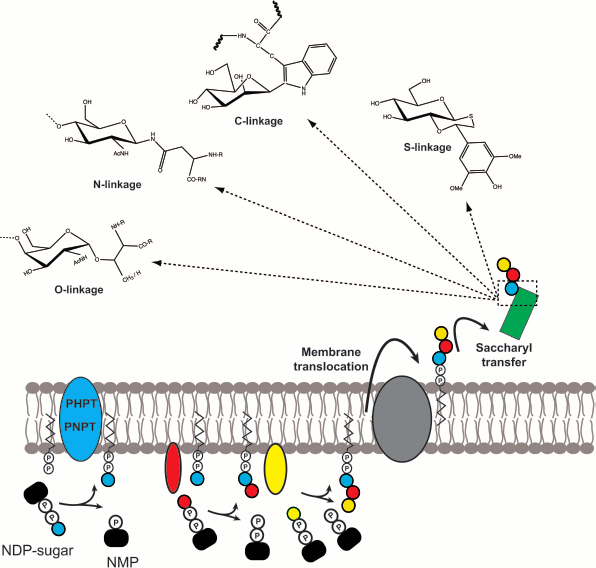
Glycoconjugates are complex carbohydrate molecules covalently linked to other chemical species such as lipids or proteins. These molecules have important structural and functional roles in all types of cells. The biosynthesis of glycoconjugates requires the prior assembly of intermediate components that are independently synthesized and later joined into one macromolecular species. Although there are many classes of glycoconjugates in prokaryotic and eukaryotic cells, their biosynthesis follows some common principles (Fig. 1; see color insert): (i) prior to their transfer to the final molecular species, the saccharide moieties are all formed as lipid-linked glycans; (ii) these lipid-linked saccharides are anchored to cell membranes or the membranes of organelles such as for example the endoplasmic reticulum membrane in eukaryotes; (iii) the most common lipid component of the saccharide intermediates is a polyisoprenoid phosphate; (iv) the majority of the proteins involved in the assembly of polyisoprenoid-linked saccharides are integral membrane proteins; (v) the assembly reactions take place at both sides of the cell membrane, typically commencing at the cytosolic side and continuing at the opposite side, for which the transmembrane movement of the polyisoprenoid-linked saccharides becomes an obligatory step; and (vi) the joining reaction between polyisoprenoid-linked saccharides and acceptor molecules involves a glycosyl transfer reaction to the nucleophilic oxygen of a hydroxyl substituent of the acceptor, but it can also occur to nitrogen (N-linked glycoproteins), sulfur (thioglycosides), and carbon (C-glycosides) nucleophiles.
The remarkable conservation of these basic steps in all types of cells suggests a common mechanism at play and especially, conserved biochemical reactions. I believe that learning the detailed biochemical mechanisms of these key reactions will provide general framework knowledge on the process of glycoconjugate synthesis that can be applicable to the majority of these molecules regardless of their source and cell type.Fig. 1. General model depicting the conserved steps in glycoconjugate biosynthesis found in all cells. The synthesis of the isoprenoid-linked saccharide begins with the formation of a phosphoanhydride bond, a reaction catalyzed by membrane proteins that belong to either the PHPT or the PNPT families. Subsequent extension depends on glycosyltransferases. The assembled glycan is subsequently translocated across the membrane lipid bilayer with the assistance of proteins. Depending on the specific system, this process may or may not require ATP hydrolysis. Once on the other side of the membrane the glycan may be further modified and ultimately transferred to an acceptor molecule by a glycosyl transfer reaction that may result in the formation of O-, N-, C-, and/or S-linkages.
LIPOPOLYSACCHARIDE, A MODEL BACTERIAL GLYCOCONJUGATE
Lipopolysaccharide (LPS) is a surface glycoconjugate unique to Gram-negative bacteria and a key elicitor of innate immune responses, ranging from local inflammation to disseminated sepsis. Gram-negative bacteria have two membrane layers separated by a periplasmic space: an inner or plasma membrane and the outer membrane. LPS is a major component of the outer leaflet of the outer membrane [1] and consists of lipid A, core oligosaccharide (OS), and O-specific polysaccharide or O antigen [1, 2]. The O antigen, which is the most surface-exposed LPS moiety, mediates pathogenicity by protecting infecting bacteria from serum complement killing and phagocytosis [2-5]. O antigens are polymers of OS repeating units. The chemical composition, structure, and antigenicity of the O antigens vary widely among Gram-negative bacteria, giving rise to a large number of O-serotypes [6]. LPS biosynthesis involves a large number of enzymes and assembly proteins encoded by more than 40 genes, recently reviewed in references [7-9]. It begins at the cytosolic or inner membrane, followed by the transit of the molecule to the outer leaflet of the outer membrane where it becomes surface exposed. The O antigen is synthesized as a lipid-linked glycan intermediate by a process that is remarkably similar to the biogenesis of lipid-linked OSs for protein N-glycosylation [10]. The lipid carrier in bacteria is undecaprenyl phosphate (Und-P), while eukaryotic cells and Archaea utilize dolichyl phosphate (Dol-P).
The core OS, made of hexoses, glycero-manno-heptose, and 3-deoxy-D-manno-oct-2-ulosonic acid [11], is assembled on preformed lipid A by the sequential transfer of sugar components. The complete lipid A–core OS unit is translocated to the periplasmic face of the inner membrane by the MsbA transporter [12], which is a member of the glyco ATP-binding cassette (ABC) transporters superfamily requiring ATP hydrolysis [13]. In a separate pathway, the O antigen is assembled as an Und-P-P-linked glycan. Depending on the specific system the assembled O-antigen precursors are translocated to the periplasmic side of the inner membrane by ATP-dependent or ATP-independent mechanisms. The O antigen and lipid A–core OS are joined by a ligation reaction that results in the formation of a novel glycosyl bond with the concomitant release of Und-P-P [7, 9, 11, 14]. Und-P-P is recycled into Und-P by a poorly characterized pathway that involves the hydrolysis of the terminal phosphate and is also conserved for the recycling of Dol-P-P in eukaryotic cells [9, 15-17]. The complete LPS molecule is translocated to the outer leaflet of the outer membrane by the Lpt system, a multiprotein complex that spans the periplasmic space, connecting inner and outer membranes [18, 19].
LIPID-LINKED SACCHARIDE FORMATION: THE INITIATION REACTION
FOR O-ANTIGEN SYNTHESIS
The O-unit synthesis starts at the cytosolic face of the plasma membrane by the formation of a phosphoanhydride linkage between Und-P and the first sugar 1-phosphate of the O-antigen unit transferred from a sugar nucleoside diphosphate. This reaction results in the release of nucleoside monophosphate. In the literature, including our own early work [20], the formation of the Und-P-P–sugar bond is often referred incorrectly to as a phosphodiester bond. In a phosphodiester bond, one phosphate group joins adjacent carbons through ester linkages. This is the typical bonding between sugars and phosphates of the backbone of nucleic acids. On the contrary, the phosphoanhydride bond is a high-energy linkage found in sugar nucleoside diphosphates and polyisoprenoid-P-P-saccharides (Fig. 2).
Two different families of integral membrane proteins catalyze the initiation reaction [9]. They are referred to as polyisoprenyl-phosphate N-acetylaminosugar-1-phosphate transferases (PNPT) and polyisoprenyl-phosphate hexose-1-phosphate transferases (PHPT). PNPT proteins are found in both prokaryotes and eukaryotes. The eukaryotic members are UDP-GlcNAc:Dol-P GlcNAc-1-P transferases that reside in the rough endoplasmic reticulum membrane. The transfer reaction initiates the formation of Dol-P-P-linked OS, an essential step for protein N-glycosylation in eukaryotes [21], as well as C- and O-mannosylation, and the biosynthesis of glycosylphosphatidylinositol anchors [22]. Bacterial prototypes of this family are WecA and MraY. The former is a UDP-GlcNAc:Und-P GlcNAc-1-P transferase [23-26], while the latter is a UDP-N-acetylmuramyl (MurNAc)-pentapeptide:Und-P MurNAc-pentapeptide-1-P transferase that initiates the synthesis of cell wall peptidoglycan.Fig. 2. Phosphoanhydride bonds joining Und-P-P-GlcNAc and Dol-P-P-GlcNAc. The difference in the saturation of the α-isoprene unit of Und and Dol is shown.
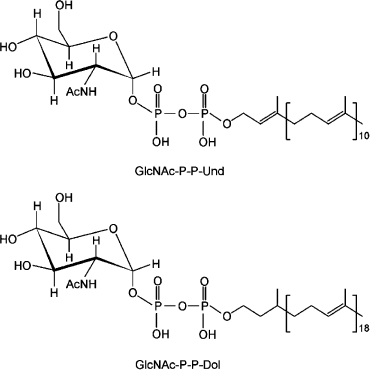
Two major differences exist in the eukaryotic vs. prokaryotic PNPTs. First, the eukaryotic members exclusively function with UDP-GlcNAc, while the bacterial enzymes can handle different diphospho N-acetylaminosugar nucleoside substrates [10]. Second, the eukaryotic PNPTs are specific for Dol-P [27], while the bacterial enzymes function only with Und-P [28]. Und-P contains 11 isoprene units, all of which are unsaturated, while Dol-P can be made of 15 to 19 isoprene units that have a saturated α-isoprene (Fig. 2) [29]. The α-isoprene is the phosphorylated end of the molecule, which participates in the phosphoanhydride bond formation with the N-acetylaminosugar 1-phosphate. Given that the active site of these enzymes is on the cytosolic face of the membrane, it is reasonable to assume that recognition of the α-isoprene-P region of the isoprenoid acceptor is determined by residues located near or at the boundaries of the membrane/solvent interface whereby the phosphate can be placed in proximity to the nucleotide sugar substrate.
These enzymes have multiple transmembrane (TM) helices, and several studies suggest that all the cytosolic loops contribute residues to form a putative catalytic site [26, 30]. Early enzyme kinetics studies on MraY have suggested that the reaction involves the formation of an enzyme-phospho-N-acetylmuramyl-pentapeptide intermediate prior to its transfer to Und-P [31]. However, recent experiments using purified MraY proteins suggests a one-step catalytic mechanism in which a base contributed by the side chain of a conserved aspartic acid residue would permit the deprotonation of a hydroxyl of the Und-P terminal phosphate. This deprotonated hydroxyl group would be involved in the nucleophilic attack on the β-phosphate of the UDP-MurNAc-pentapeptide [30].
Enzymes of the PHTP family are also integral membrane proteins, but they occur only in bacteria. PHPTs catalyze the synthesis of O antigen, exopolysaccharide (EPS), capsular polysaccharide (CPS), and/or glycans for surface layer (S-layer) protein glycosylation and also general protein glycosylation [9, 32, 33]. The most common substrates are UDP-glucose or UDP-galactose, but some members of the family can utilize UDP-2,4-diacetamido-2,4,6-trideoxyglucose (bacillosamine) [34] and UDP-2(4)-acetamido-4(2)-glyceramido-2,4,6-trideoxyhexose [35, 36].
The predicted topology of PHPT proteins is very different than that of the PNPTs. The distinctive feature of this family is a large cytosolic C-terminal tail. Experiments involving deletion and mutagenesis demonstrate that the enzymatic activity resides in this region of the protein [32, 37, 38]. The functional significance of the other regions in these proteins is unclear. In fact, some members of the family only have the C-terminal tail and one predicted TM helix that holds the protein to the membrane. A central soluble region is also characteristic of some of the proteins in this family. Studies in the Streptococcus pneumoniae CpsE suggest that this region plays a regulatory role of the enzymatic activity, probably exerting an allosteric effect on the initiation reaction that controls the rate of polymer synthesis [39]. The reminder of the protein, largely corresponding to an N-terminal domain containing several TM domains may serve as a scaffold [38], but it is the least conserved portion of the protein.
An interesting aspect of the initiation reaction for both PNPT and PHPT enzymes is that these are the only true integral membrane proteins involved in the synthesis of the O-antigen units. It is tempting to speculate that these proteins have also a structural role to localize the biosynthesis of O antigen by establishing contacts with other integral membrane proteins involved in the subsequent steps including the translocation of the Und-P-P-linked saccharide across the membrane. Some evidence supporting this notion exists in the case of WecA, as demonstrated by a punctate pattern of fluorescence at the bacterial cell membrane, as well as by fractionation studies [26] suggesting that WecA molecules are localized in membrane microdomains. After the initiation reaction, the O-antigen synthesis progresses by the activities of other sugar transferases that sequentially add additional sugars to make the O-antigen unit or polysaccharide. In contrast to the initiating transferases, these glycosyltransferases catalyze glycoside bond formation and are outside the scope of this review.
TRANSMEMBRANE MOVEMENT OF POLYISOPRENOID-LINKED SACCHARIDES:
DIFFERENT SOLUTIONS TO THE SAME PROBLEM
The remainder of the assembly of the O antigen after the initiation reaction varies depending on the pathways used. Four assembly pathways are known to date: Wzy/Wzx-, Wzm/Wzt-, synthase-, and Wzk-dependent pathways recently reviewed in ref. [40]. From these, the Wzy (polymerase)- and synthase-dependent pathways do not require ATP hydrolysis for the membrane translocation step, while the other two employ different classes of ABC transporters. In the synthase, Wzt/Wzm, and Wzk pathways the polymeric O antigen is formed on the cytoplasmic side of the plasma membrane prior to its export (reviewed in refs. [7, 9]). Wzm and Wzt are the permease and ATPase components of an ABC transporter. They have been recently reviewed [13] and will not be further discussed. The Wzk pathway has been recently discovered in Helicobacter pylori and involves an ABC transporter similar to that used in bacterial protein N-glycosylation [41]. The synthase pathway employs a protein that has similarities to the hyaluronan synthase family of glycosaminoglycan glycosyltransferases [42]. These proteins catalyze a vectorial polymerization reaction by a processive mechanism resulting in the extension of the polysaccharide chain with the simultaneous extrusion of the nascent polymer across the plasma membrane [42, 43].
In the Wzy-dependent pathway, O-units are synthesized at the cytosolic side of the plasma membrane. Each unit is subsequently translocated across the membrane in an ATP hydrolysis-independent manner mediated by the protein Wzx [7, 9, 44, 45]. On the periplasmic side of the plasma membrane, translocated units polymerize by the concerted functions of Wzy (O-antigen polymerase) and Wzz (O-antigen chain length regulator) to a certain length distribution that is unique to each O antigen. Finally, the polysaccharide is ligated “en bloc” to the lipid A–core OS [46-48]. The Wzy-dependent pathway coordinates the synthesis of many O antigens [49] and is remarkably similar to the pathway operating in eukaryotes for N-glycosylation of proteins. In this case, a Dol-P-P-linked saccharide is built at the cytosolic side of the endoplasmic reticulum membrane and translocated across the membrane by the Rft1 protein [50]. Once in the endoplasmic reticulum lumen, the saccharide is further modified and finally transferred to proteins targeted for glycosylation [15, 51].
Wzx and Rft1 are both integral membrane proteins with multiple TM domains, and although they carry similar functions they share no similarity in their amino acid sequences. Also, both lack any distinctive motifs that could provide clues to their function. In the case of Wzx proteins the lack of distinctive regions is even more puzzling as these proteins display very low conservation in their primary amino acid sequences. The variability in the amino acid sequence of Wzx proteins could be due to the fact that they may recognize very different O-unit structures. However, our laboratory discovered that an incomplete O-antigen unit could be translocated across the cytoplasmic membrane in a Wzx-dependent fashion and incorporated into the lipid A–core OS of Escherichia coli K-12 [52]. Experiments leading to progressive truncation of the O-antigen unit unequivocally revealed that only the first sugar bound to Und-P-P is the minimal substrate for translocation [52]. Furthermore, Wzx proteins from different bacteria could restore O-antigen synthesis in an E. coli K-12 Δwzx mutant [53]. Importantly, the ability of a given Wzx protein to restore O-antigen synthesis is clearly associated to the nature of the initiating sugar, as Wzx proteins from O-antigen systems that use Und-P-P-GlcNAc or Und-P-P-GalNAc for the initiation of the biosynthesis of the O-antigen unit can fully complement the formation of E. coli O16 LPS, while complementation with the Wzx protein from systems using Und-P-P-Gal was only possible under high levels of protein expression [53]. These observations suggest that Wzx proteins, like the initiating Und-P-P-sugar transferases, occur in at least two functional classes that can distinguish among Und-P-P-linked N-acetylamino sugars or hexoses. The genetic reconstitution of O-antigen synthesis in E. coli K-12 as well as the heterologous expression of the O antigen of E. coli O7 provided strong evidence that cognate Wzy, Wzz, and Wzx work in concert as a functional multiprotein complex [54].
The initial demonstration of Wzx could be a flippase was obtained by determining the accumulation of radioactive O-antigen precursors in the membrane of Salmonella enterica [45]. Biochemical in vitro evidence for membrane translocation was obtained using a soluble analogue of Und-P-P-GlcNAc in sealed membrane vesicles of E. coli K-12 [44]. This assay provided important clues such as demonstrating that the flipping process does not require ATP. However, it does not appear to function with all flippases. One complication of in vitro assays is the requirement of detergents to prepare membrane extracts, which can introduce bias in the assay by providing continuity between the leaflets of the membranes [55-57].
Wzx proteins contain 12 predicted TM helices as determined by experimental topological studies in Wzx proteins of S. enterica [58], Rhizobium leguminosarum (PssL) [59], Pseudomonas aeruginosa [60], and E. coli O157 [61]. The common lesson learned from these studies is that Wzx proteins have two or more TM domains containing charged amino acids. The role of the charged amino acids was directly investigated in the Wzx flippase from E. coli O157, resulting in the identification of four charged residues that were critical for the restoration of O-antigen synthesis when replaced by alanine [61]. The topological distribution of these residues was surprising: two were located in TM helices, and each of the other two in internal and external soluble loops. Conservative replacements at these positions demonstrated that the charge, but not the nature of the targeted amino acid is critical to retain the ability of Wzx to support O-antigen production. Charged residues in TM helices suggest possible contacts with substrates directly or via water molecules. Therefore, it is tempting to speculate that some of the helices in Wzx may interact with each other and locate further away from the lipid bilayer, similar to the arrangement of helices in the LacY permease [62]. Thus, charged residues at both sides of the membrane and in two TM helices could contribute to create an electrostatic cavity [62, 63] and perhaps even electrostatic interactions with the phosphate groups of Und-P-P-linked sugars, which may allow localized perturbation of the lipid bilayer to facilitate the movement of the Und-PP-linked saccharide substrate across the membrane.
An important player in the flipping process, usually overlooked, is the polyisoprenoid lipid carrier. Using an energy-minimized molecular modeling approach, Zhou and Troy (reviewed in ref. [64]) determined that the long axis of Dol-P and Und-P in phosphatidylcholine membranes is oriented almost perpendicular to the plane of the bilayer. Molecular modeling calculations revealed that hydrophobic peptides corresponding to putative polyisoprenoid recognition sequences could bind cooperatively to a single Dol-P molecule, and this structure could have the potential to form a membrane channel. The biophysical properties of Und-P and Dol-P in membranes and in the context of TM helices of membrane proteins such as Wzx and Rft1 could facilitate the translocation of polyisoprenoid-P-P-saccharides across membranes with minimal energetic requirements. This could also explain why polyisoprenoids are evolutionarily conserved as the preferred carrier lipid for glycoconjugate synthesis in all types of cells.
THE OLIGOSACCHARYL TRANSFER REACTION MEDIATED BY THE O-ANTIGEN
LIGASE, A “GLORIFIED” GLYCOSYLTRANSFERASE
The so-called ligation reaction, which results in the covalent linkage between lipid A–core OS and O antigen, is in effect a glycosyl transfer reaction that is required for the formation of any glycoconjugate molecule. Acceptors for “ligation” reactions are proteins, lipids, and other carbohydrate structures. Therefore, this reaction would be best defined as saccharyl transfer reaction. The ligase protein, called WaaL, is also an integral membrane protein with multiple TM helices and has a characteristic large periplasmic segment.
The ligation reaction has two distinctive features. First, although WaaL catalyzes the formation of a glycosidic bond, these proteins share no similarities with any of the glycosyltransferases that use sugar nucleotides. Second, the specificity of the reaction is driven by the requirement for a specific lipid A–core OS acceptor structure [65-68] but not Und-P-P-linked substrate. Indeed, donor Und-P-P-linked glycans originating from various biosynthesis pathways can be substrates for ligation, such as for example, colanic acid, a cell surface capsular material that is usually loosely associated with the bacterial cell [69]. The topology of the O-antigen ligases is reasonably well established [60, 65, 67, 68, 70]. However, it is still unclear how WaaL recognizes the Und-PP-linked O antigen and in particular, which part of the donor molecule participates in the enzymatic reaction. The low sequence conservation among O-antigen ligases makes comparative analyses difficult [71], and therefore it is not straightforward to identify residues involved in ligase activity. Conceivably, WaaL activity involves amino acids exposed to the periplasmic space where they could interact with the donor and acceptor molecules. A tri-dimensional structural model of the WaaL large periplasmic loop was proposed, which consists of two pairs of almost perpendicular α-helices. In this model, all the conserved residues in other WaaL proteins cluster within a putative catalytic region, as demonstrated by site-directed amino acid replacements [72]. The model predicts that arginine-288 and histidine-337, two residues that are critical for WaaL function, face each other and are exposed to the solvent in a spatial arrangement that suggests interactions with substrate molecules. This critical histidine is also found in oligosaccharyl-peptide transferases that ligate Und-PP-linked O-antigen precursors to pilin [73]. In addition, a conserved arginine, also critical for WaaL function, is invariably present in the short periplasmic loop preceding the large loop [72].
Given that the ligase catalyzes a glycosyl transfer reaction and the product released is Und-P-P, it is reasonable to assume the cleavage takes place between the distal phosphate group of the Und-P-P-saccharide and the sugar. This would be a situation analogous to the cleavage of a nucleoside diphosphate sugar by any classical glycosyltransferase. Therefore, it is unlikely that the ligase requires ATP hydrolysis for the ligation reaction, as reported for the P. aeruginosa enzyme [74]. These authors incorrectly assumed the ligation reaction is similar to the reaction involving DNA and RNA ligation, which require ATP. However, ligation of nucleic acid molecules is very different chemically than glycosyl bond formation. Consistent with this, the requirement for ATP in the ligation reaction could not be confirmed in other WaaL proteins [41], and extensive mutagenesis of amino acid motifs that could putatively be involved in ATP binding or hydrolysis did not afford ligation-defective proteins [72].
Research in the author’s laboratory is supported by grants from the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada, the Mizutani Foundation, and Cystic Fibrosis Canada. The author holds a Canada Research Chair in Infectious Diseases and Microbial Pathogenesis.
REFERENCES
1.Nikaido, H. (1996) in Escherichia coli and
Salmonella: Cellular and Molecular Biology (Neidhardt, F. C.,
Curtiss, R., Ingraham, J. L., Lin, E. C. C., Low, K. B., Magasanik, B.,
Reznikoff, W. S., Riley, M., Schaechter, M., and Umbarger, H. E., eds.)
ASM Press, Washington, DC, pp. 29-47.
2.Whitfield, C., and Valvano, M. A. (1993) Adv.
Microb. Physiol., 35, 135-246.
3.Pluschke, G., Mercer, A., Kusecek, B., Pohl, A.,
and Achtman, M. (1983) Infect. Immun., 39,
599-608.
4.Pluschke, G., and Achtman, M. (1984) Infect.
Immun., 43, 684-692.
5.Joiner, K. A. (1988) Ann. Rev.
Microbiol., 42, 201-230.
6.Jansson, P.-E. (1999) in Endotoxin in Health and
Disease (Brade, H., Morrison, D. C., Vogel, S., and Opal, S., eds.)
Marcel Dekker, New York, pp. 155-178.
7.Raetz, C. R. H., and Whitfield, C. (2002) Annu.
Rev. Biochem., 71, 635-700.
8.Samuel, S., and Reeves, P. (2003) Carbohydr.
Res., 338, 2503-2519.
9.Valvano, M. A. (2003) Front. Biosci.,
8, s452-471.
10.Valvano, M. A. (2010) in Comprehensive Natural
Products Chemistry II. Vol. 6. Carbohydrates, Nucleosides and
Nucleic Acids (Mander, L. N., and Liu, H.-W., eds.) Elsevier,
Oxford, pp. 297-314.
11.Heinrichs, D. E., Valvano, M. A., and Whitfield,
C. (1999) in Endotoxin in Health and Disease (Brade, H.,
Morrison, D. C., Vogel, S., and Opal, S., eds.) Marcel Dekker, New
York, pp. 305-330.
12.Doerrler, W. T., and Raetz, C. R. H. (2002) J.
Biol. Chem., 277, 36697-36705.
13.Cuthbertson, L., Kos, V., and Whitfield, C.
(2010) Microbiol. Mol. Biol. Rev., 74,
341-362.
14.Heinrichs, D. E., Yethon, J. A., and Whitfield,
C. (1998) Mol. Microbiol., 30, 221-232.
15.Burda, P., and Aebi, M. (1999) Biochim.
Biophys. Acta, 1426, 239-257.
16.Fernandez, F., Rush, J. S., Toke, D. A., Han, G.,
Quinn, J. E., Carman, G. M., Choi, J.-Y., Voelker, D. R., Aebi, M., and
Waechter, C. J. (2001) J. Biol. Chem., 276,
41455-41464.
17.Tatar, L. D., Marolda, C. L., Polischuk, A. N.,
van Leeuwen, D., and Valvano, M. A. (2007) Microbiology,
153, 2518-2529.
18.Sperandeo, P., Deho, G., and Polissi, A. (2009)
Biochim. Biophys. Acta, 1791, 594-602.
19.Ruiz, N., Kahne, D., and Silhavy, T. J. (2009)
Nat. Rev. Micro., 7, 677-683.
20.Amer, A. O., and Valvano, M. A. (2002)
Microbiology, 148, 571-582.
21.Lehrman, M. A. (1994) Glycobiology,
4, 768-771.
22.Schenk, B., Fernandez, F., and Waechter, C. J.
(2001) Glycobiology, 11, 61R-70R.
23.Rick, P. D., and Silver, R. P. (1996) in
Escherichia coli and Salmonella: Cellular and Molecular Biology
(Neidhardt, F. C., Curtiss III, R., Ingraham, J. L., Lin, E. C. C.,
Low, K. B., Magasanik, B., Reznikoff, W. S., Riley, M., Schaechter, M.,
and Umbarger, H. E., eds.) ASM Press, Washington, DC, pp. 104-122.
24.Klena, J. D., and Schnaitman, C. A. (1993)
Mol. Microbiol., 9, 393-402.
25.Alexander, D. C., and Valvano, M. A. (1994) J.
Bacteriol., 176, 7079-7084.
26.Lehrer, J., Vigeant, K. A., Tatar, L. D., and
Valvano, M. A. (2007) J. Bacteriol., 189,
2618-2628.
27.Mankowski, T., Sasak, W., and Chojnacki, T.
(1975) Biochem. Biophys. Res. Commun., 65,
1292-1297.
28.Rush, J. S., Rick, P. D., and Waechter, C. J.
(1997) Glycobiology, 7, 315-322.
29.Pennock, J. F., Hemming, F. W., and Morton, R. A.
(1960) Nature, 186, 470-472.
30.Al-Dabbagh, B., Henry, X., El Ghachi, M., Auger,
G., Blanot, D., Parquet, C., Mengin-Lecreulx, D., and Bouhss, A. (2008)
Biochemistry, 47, 8919-8928.
31.Heydanek, M. G., Struve, W. G., and Neuhaus, F.
C. (1969) Biochemistry, 8, 1214-1221.
32.Wang, L., Liu, D., and Reeves, P. R. (1996) J.
Bacteriol., 178, 2598-2604.
33.Steiner, K., Novotny, R., Patel, K., Vinogradov,
E., Whitfield, C., Valvano, M. A., Messner, P., and Schaffer, C. (2007)
J. Bacteriol., 189, 2590-2598.
34.Glover, K. J., Weerapana, E., Chen, M. M., and
Imperiali, B. (2006) Biochemistry, 45, 5343-5350.
35.Power, P. M., Roddam, L. F., Dieckelmann, M.,
Srikhanta, Y. N., Tan, Y. C., Berrington, A. W., and Jennings, M. P.
(2000) Microbiology, 146, 967-979.
36.Chamot-Rooke, J., Rousseau, B., Lanternier, F.,
Mikaty, G., Mairey, E., Malosse, C., Bouchoux, G., Pelicic, V., Camoin,
L., Nassif, X., and Dumenil, G. (2007) Proc. Natl. Acad. Sci.
USA, 104, 14783-14838.
37.Patel, K. B., Furlong, S. E., and Valvano, M. A.
(2010) Glycobiology, 20, 1389-1401.
38.Saldias, M. S., Patel, K., Marolda, C. L.,
Bittner, M., Contreras, I., and Valvano, M. A. (2008)
Microbiology, 154, 440-453.
39.Xayarath, B., and Yother, J. (2007) J.
Bacteriol., 189, 3369-3381.
40.Hug, I., and Feldman, M. F. (2011)
Glycobiology, 21, 138-151.
41.Hug, I., Couturier, M. R., Rooker, M. M., Taylor,
D. E., Stein, M., and Feldman, M. F. (2010) PLoS Path.,
6, e1000819.
42.DeAngelis, P. L. (2002) Glycobiology,
12, 9R-16R.
43.DeAngelis, P. L. (1999) Cell. Mol. Life
Sci., 56, 670-682.
44.Rick, P. D., Barr, K., Sankaran, K., Kajimura,
J., Rush, J. S., and Waechter, C. J. (2003) J. Biol. Chem.,
278, 16534-16542.
45.Liu, D., Cole, R. A., and Reeves, P. R. (1996)
J. Bacteriol., 178, 2102-2107.
46.Marino, P. A., McGrath, B. C., and Osborn, M. J.
(1991) J. Bacteriol., 173, 3128-3133.
47.McGrath, B. C., and Osborn, M. J. (1991) J.
Bacteriol., 173, 649-654.
48.Mulford, C. A., and Osborn, M. J. (1983) Proc.
Natl. Acad. Sci. USA, 80, 1159-1163.
49.Keenleyside, W. J., and Whitfield, C. (1999) in
Endotoxin in Health and Disease (Brade, H., Morrison, D. C.,
Vogel, S., and Opal, S., eds.) Marcel Dekker, New York, pp.
331-358.
50.Helenius, J., Ng, D. T., Marolda, C. L., Walter,
P., Valvano, M. A., and Aebi, M. (2002) Nature, 415,
447-450.
51.Helenius, J., and Aebi, M. (2002) Semin. Cell.
Dev. Biol., 13, 171-178.
52.Feldman, M. F., Marolda, C. L., Monteiro, M. A.,
Perry, M. B., Parodi, A. J., and Valvano, M. A. (1999) J. Biol.
Chem., 274, 35129-35138.
53.Marolda, C. L., Vicarioli, J., and Valvano, M. A.
(2004) Microbiology, 150, 4095-4105.
54.Marolda, C. L., Tatar, L. D., Alaimo, C., Aebi,
M., and Valvano, M. A. (2006) J. Bacteriol., 188,
5124-5135.
55.Kol, M. A., de Kroon, A. I., Rijkers, D. T.,
Killian, J. A., and de Kruijff, B. (2001) Biochemistry,
40, 10500-10506.
56.Kol, M. A., van Dalen, A., de Kroon, A. I., and
de Kruijff, B. (2003) J. Biol. Chem., 278,
24586-24593.
57.Kol, M. A., van Laak, A. N., Rijkers, D. T.,
Killian, J. A., de Kroon, A. I., and de Kruijff, B. (2003)
Biochemistry, 42, 231-237.
58.Cunneen, M. M., and Reeves, P. R. (2008) FEMS
Microbiol. Lett., 287, 76-84.
59.Mazur, A., Marczak, M., Krol, J. E., and
Skorupska, A. (2005) Arch. Microbiol., 184,
1-10.
60.Islam, S. T., Taylor, V. L., Qi, M., and Lam, J.
S. (2010) MBio, 1, e00189-10.
61.Marolda, C. L., Li, B., Lung, M., Yang, M.,
Hanuszkiewicz, A., Roa Rosales, A., and Valvano, M. A. (2010) J.
Bacteriol., 192, 6160-6171.
62.Sorgen, P. L., Hu, Y., Guan, L., Kaback, H. R.,
and Girvin, M. E. (2002) Proc. Natl. Acad. Sci. USA,
99, 14037-14040.
63.Abramson, J., Smirnova, I., Kasho, V., Verner,
G., Kaback, H. R., and Iwata, S. (2003) Science, 301,
610-615.
64.Zhou, G. P., and Troy, F. A. (2005)
Glycobiology, 15, 347-359.
65.Abeyrathne, P., Daniels, C., Poon, K. K.,
Matewish, M. J., and Lam, J. (2005) J. Bacteriol.,
187, 3002-3012.
66.Heinrichs, D. E., Yethon, J. A., Amor, P. A., and
Whitfield, C. (1998) J. Biol. Chem., 273,
29497-29505.
67.Heinrichs, D. E., Monteiro, M. A., Perry, M. B.,
and Whitfield, C. (1998) J. Biol. Chem., 273,
8849-8859.
68.Schild, S., Lamprecht, A. K., and Reidl, J.
(2005) J. Biol. Chem., 280, 25936-25947.
69.Meredith, T. C., Mamat, U., Kaczynski, Z.,
Lindner, B., Holst, O., and Woodard, R. W. (2007) J. Biol.
Chem., 282, 7790-7798.
70.Nesper, J., Kraiss, A., Schild, S., Blass, J.,
Klose, K. E., Bockemuhl, J., and Reidl, J. (2002) Infect.
Immun., 70, 2419-2433.
71.Raetz, C. R., Reynolds, C. M., Trent, M. S., and
Bishop, R. E. (2007) Annu. Rev. Biochem., 76,
295-329.
72.Perez, J. M., McGarry, M. A., Marolda, C. L., and
Valvano, M. A. (2008) Mol. Microbiol., 70,
1424-1440.
73.Qutyan, M., Paliotti, M., and Castric, P. (2007)
Mol. Microbiol., 66, 1444-1458.
74.Abeyrathne, P., and Lam, J. (2007) Mol.
Microbiol., 65, 1345-1359.