REVIEW: Role of Nucleotide Excision Repair Proteins in Oxidative DNA Damage Repair: an Updating
B. Pascucci1,2, M. D’Errico2, E. Parlanti2, S. Giovannini2, and E. Dogliotti2*
1Istituto di Cristallografia, Consiglio Nazionale delle Ricerche, Via Salaria Km 29.300, 00016 Monterotondo Stazione, Rome, Italy; fax: +3(906)9067-2630; E-mail: barbara.pascucci@mlib.ic.cnr.it2Department of Environment and Primary Prevention, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy; fax: +3(906)4990-3650; E-mail: mariarosaria.derrico@iss.it; eleonora.parlanti@iss.it; giovannini.sara@hotmail.it; eugenia.dogliotti@iss.it
* To whom correspondence should be addressed.
Received September 15, 2010; Revision received October 8, 2010
DNA repair is a crucial factor in maintaining a low steady-state level of oxidative DNA damage. Base excision repair (BER) has an important role in preventing the deleterious effects of oxidative DNA damage, but recent evidence points to the involvement of several repair pathways in this process. Oxidative damage may arise from endogenous and exogenous sources and may target nuclear and mitochondrial DNA as well as RNA and proteins. The importance of preventing mutations associated with oxidative damage is shown by a direct association between defects in BER (i.e. MYH DNA glycosylase) and colorectal cancer, but it is becoming increasingly evident that damage by highly reactive oxygen species plays also central roles in aging and neurodegeneration. Mutations in genes of the nucleotide excision repair (NER) pathway are associated with diseases, such as xeroderma pigmentosum and Cockayne syndrome, that involve increased skin cancer and/or developmental and neurological symptoms. In this review we will provide an updating of the current evidence on the involvement of NER factors in the control of oxidative DNA damage and will attempt to address the issue of whether this unexpected role may unlock the difficult puzzle of the pathogenesis of these syndromes.
KEY WORDS: oxidative damage, DNA repair, oxidative metabolism, xeroderma pigmentosum, Cockayne syndromeDOI: 10.1134/S0006297911010032
Abbreviations: Abl-1, Abelson murine leukemia kinase; AAF, acetylaminofluorene; AOA1, ataxia oculomotor apraxia type 1; APE1, apurinic/apyrimidinic endonuclease 1; ATR, ataxia telangiectasia and Rad 3 related; BER, base excision repair; BPDE, benzo(a)pyrene diol epoxide; CS, Cockayne syndrome; CSN, COP9 signalosome; cyPudN, 8,5′-cyclopurine 2′-deoxynucleosides; DDB, DNA damage-binding protein; DRC, damage repair capacity; DSBs, double-strand DNA breaks; ES, embryonic stem; FapyA, formamide pyrimidine adenine; FapyG, formamide pyrimidine guanine; FPG, formamidopyrimidine DNA glycosylase; GGR, global genome repair; HCR, host cell reactivation; HMGN1, high-mobility group nucleosome binding domain 1; HNE, 4-hydroxy-2-nonenal-modified protein; MEF, mouse embryo fibroblasts; mtSSBP-1, mitochondrial single stranded DNA binding protein; NEIL1, nei endonuclease VIII-like 1; NER, nucleotide excision repair; OGG1, 8-oxoguanine DNA glycosylase; 8-oxoA, 8-OH-adenine; 8-oxoG, 8-OH-guanine; PARP-1, poly(ADP-ribose) polymerase 1; PCNA, proliferating cell nuclear antigen; PGBD3, PiggyBac transposable element-derived protein 3; RNAPII, RNA polymerase II; ROS, reactive oxygen species; RPA, replication protein A; SCAN1, spinocerebellar ataxia with axonal neuropathy; SIRT1, sirtuin 1; SMUG1, single-strand-specific monofunctional uracil-DNA glycosylases; SNP, single nucleotide polymorphisms; SOD, superoxide dismutase; SSBs, single-strand DNA breaks; TCR, transcription-coupled repair; TDG, thymine DNA glycosylase; TFIIH, transcription factor H-II; TFIIS, transcription factor S-II; Tg, thymine glycol; TGD, transglutaminase-homology domain; UVsS, UV sensitive syndrome; XAB2, XPA binding protein 2; XP, xeroderma pigmentosum.
In 1968 Jim Cleaver reported that a disorder characterized by high
incidence of skin cancer upon sunlight exposure, xeroderma pigmentosum
(XP), is caused by a defect in the repair of UV lesions. Since then all
the eight genes that cause XP (XPA-XPG and XPV or
variant) have been cloned. All of them, with the exception of
XPV, work in different steps of the same biochemical pathway,
the nucleotide excision repair (NER). The complex biochemistry of this
pathway has been clarified by the joint effort of several groups. We
know today that NER operates by two distinct pathways: global genome
repair (GGR) that removes lesions from the genome overall and
transcription-coupled repair (TCR) that repairs transcriptionally
active domains. The step of damage recognition involves different
factors in the two pathways. In GGR, XPC–HR23B/centrin-2 and XPE
(UV-DDB) protein complexes, and in TCR the RNA polymerase II (RNAPII)
stalled at a lesion on the transcribed strand, play a role in the
recognition step. Transcription arrest is increased by CSA and CSB
proteins that are required for ubiquitylation of the carboxy-terminal
domain of RNAPII. The repair process follows then the same path
involving the binding of the ten-component basal transcription factor
H-II (TFIIH) via interaction with either XPC or the arrested
transcription apparatus. Two helicases, XPB and XPD, initiate the
opening around the lesion, and the DNA around the damaged site is
cleaved by the XPG 3′ nuclease and the XPF-ERCC1 5′
nuclease. Once the damaged oligonucleotide is removed resynthesis
occurs by proliferating cell nuclear antigen (PCNA), DNA polymerase
δ, DNA polymerase κ, and DNA ligase.
The complex biochemistry of NER has been established by using UV-induced photoproducts as model lesions and similar chemically induced products that distort DNA are recognized and repaired by the same factors. However, the clinical heterogeneity in disorders with NER mutations opens the question of whether defects in this pathway are solely due to impaired repair of helix-distorting DNA lesions. XP patients with also defects in TCR (XP-A, XP-B, XP-D, and XP-G) present, besides increased skin cancer risk, accelerated neurodegeneration. Patients with Cockayne syndrome (CS) show also severe developmental and neurological symptoms but do not show skin cancer despite the presence of photosensitivity. Neuronal death might be due to accumulated endogenous damage, and indeed a growing body of evidence indicates that NER proteins participate in the processing of oxidative DNA lesions that are produced by the normal cell metabolism. The role of NER proteins in different pathways might explain the heterogeneity in disorders with NER mutations.
In this review we concentrate on four NER genes, two involved in DNA damage recognition, XPC and XPA, and two belonging to TCR, CSA and CSB, that have been involved in the response to damage from endogenous sources. The role of XPG in the stimulation of oxidative DNA damage repair has been recently reviewed [1] and will be not covered in this review.
XPC
Biochemical properties and protein structure. The human XPC gene is located on chromosome 3 and encodes a basic protein of 940 amino acids [2] that functions, in concert with XPE, as a damage detector in the first step of GGR. XPC comprises at least four structural domains: a transglutaminase-homology domain (TGD) and three consecutive-hairpin domains (designated BHD1, -2, and -3) (Fig. 1a). Against the conventional dogma that DNA lesions are recognized through direct contacts with modified nucleotides, XPC protein seems to distinguish between damaged DNA and the native double helix by sensing the single-stranded character of non-hydrogen-bonded bases in the undamaged strand [3]. This mode of action is confirmed by structural analysis of the yeast Rad4 homolog that identifies critical chains making contacts with extra-helical nucleotides [4]. In addition, XPC provides a landing platform for TFIIH [5] that, together with XPA and replication protein A (RPA), generates an open repair intermediate in which the DNA around the lesion is melted (over 25-30 nucleotides). XPC is polyubiquitinated by the UV-DDB–Cul4A–Roc1 complex upon DNA damage, a reversible process that does not result in its degradation, but rather increases its affinity for DNA, damaged or not [6]. The human XPC protein in vivo is a heterotrimeric complex including HR23B and centrin-2 proteins [7, 8]. HR23B seems to stabilize XPC, whereas centrin-2 is required to enhance the damage recognition function of XPC [9]. This complex binds to various types of helix-distorting lesions, thus triggering GGR and, unexpectedly, it also stimulates the repair of small base lesions.
The XPC–HR23B complex functionally interacts with 3-methyladenine DNA glycosylase [10] and thymine DNA glycosylase (TDG) [11] that initiate BER of alkylation and deamination products, respectively. XPC–HR23B stimulates TDG activity by promoting the release of TDG following the excision of mismatched T base. In the presence of apurinic/apyrimidinic endonuclease 1 (APE1), XPC–HR23B has an additive effect on TDG turnover without significantly inhibiting the subsequent action of APE. XPC–HR23B complex significantly stimulates also the activity of 8-oxoguanine DNA glycosylase (OGG1) in human cell extracts (Fig. 1b) as well as in a reconstituted repair reaction with purified proteins [12]. OGG1 is known to bind tightly the AP site generated by its glycosylase activity [13], and XPC–HR23B may be required to facilitate its release from the AP site, thereby freeing OGG1 to react with remaining sites. The question of whether XPC operates as an active displacement of the DNA glycosylase or competes at AP sites (it has been shown that XPC complex can bind specifically to AP sites [11]) waits to be clarified. A recent study [14] points to the importance of protein–protein interaction for the stimulation of DNA glycosylases by XPC for AP sites by showing that XPC stimulates the activities of sumoylated TDG and single-strand-specific monofunctional uracil-DNA glycosylases (SMUG1), both of which interact physically with XPC. XPC–HR23B recognizes also 5R-thymine glycol (Tg), formed by exposure to radiation and chemical oxidants, better than the C8-dG acetylaminofluorene (AAF) adduct [15].
Most mutations found in XP-C patients including nonsense mutations, deletions, and splice-site mutations are inactivating null mutations that lead to full loss of function. Nonsense mutations in patients and in vitro mapping studies have allowed the identification of functional regions of the gene that interact with TFIIH, HR23B, and XPA (Fig. 1a). Interestingly, there is a single amino acid substitution in a conserved region (W690) reported in one patient that seems to be essential for NER by stabilizing the binding of XPC to the undamaged strand, thus supporting the mode of action described above [16]. Of relevance for the additional role of XPC in BER is a single amino acid change at position 334 (P334H) in a non-conserved region that has been shown to weaken the interaction with OGG1. Cells from this patient presented low levels of UV-induced unscheduled DNA synthesis and a decreased OGG1 cleavage activity. This patient is also one of the rare XP-C patients who exhibit neurological problems [17].Fig. 1. a) XPC protein structure and functional regions of XPC interacting proteins. b) Cell extracts from two XP-C primary cells (XP26PV and XP28PV) are defective in 8-oxoG cleavage, but addition of purified XPC–HR23B restores normal cleavage activity. The 30-mer duplex oligonucleotides (50 fmol) containing 8-OH-G were incubated with nuclear extracts (5 mg) of XP-C cells at 37°C in the presence of varying concentration of XPC–HR23B as indicated. The 5′ end-labeled oligonucleotide was the 8-oxoG containing strand. The products were separated by denaturing 20% PAGE (modified from [12]).
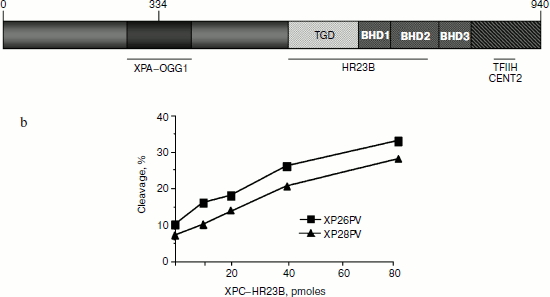
Cell phenotype. It is well known that XP-C primary cells are hypersensitive to UV radiation as a consequence of faulty repair of UV photoproducts. By analyzing the response to oxidizing agents of different types of skin cells we have recently reported that XP-C primary keratinocytes and fibroblasts are hypersensitive also to the killing effects of DNA-oxidizing agents, and this effect is reverted by expression of wild-type XPC [12]. The protective role of XPC from the lethal effects of oxidative stress is supported by data obtained in mouse embryo fibroblasts (MEF) derived from Xpc–/– mice that exhibit a severe decrease in survival when cultured at 20% oxygen compared with 3% oxygen pressure. Even at a low oxygen level of 3%, Xpc-deficient cells seem to be more sensitive as compared to wild-type [18]. Hypersensitivity to oxidative stress might be explained by increased production of reactive oxygen species (ROS) due to altered oxidative metabolism and/or decreased repair of oxidative lesions. Impaired DNA repair is documented by the accumulation of various lesions, such as 8,5′-cyclopurine 2′-deoxynucleosides (cyPudN), 8-OH-guanine (8-oxoG), and 8-OH-adenine (8-oxoA), in XP-C primary fibroblasts upon exposure to oxidizing agents [12]. In addition, host cell reactivation (HCR) of oxidant-treated human adenovirus was reported to be substantially reduced in primary and SV40-trasformed XP-C fibroblasts compared to normal cells [19] supporting the view that XPC is involved in the repair of oxidative DNA damage. It is however of interest to mention that a few reports suggest that the oxidative metabolism of XP-C cells might be altered too. An abnormal low level of catalase activity was reported in XP-C primary fibroblasts, and this defect was corrected upon expression of the wild-type gene [20]. More recently, XPC silencing was shown to cause increased susceptibility to oxidative stress induced by arsenic trioxide in human glioma cells by disturbing redox homeostasis rather than reducing DNA repair [21].
Mutant mouse models. Mice homozygous for Xpc mutant alleles are viable and display a spectrum of UV exposure-related pathologic skin and eye changes consistent with those found in the human disease [22]. A significantly higher incidence of chemically-induced liver and lung tumors, compared with normal and heterozygous littermates, was also reported in these mice when treated with AAF indicating that, upon environmental exposure, a defect in GGR is associated with internal cancer proneness too [23]. On the other hand the high rate of spontaneous hprt mutations (mainly G>T transversions, the hallmark of 8-oxoG mutagenesis) in T-lymphocytes of Xpc–/– mice [24], and the high frequency of spontaneous tumors and mutation in lung [18] support the view that XPC plays a role in endogenous DNA damage control. It is of note that the neurological phenotype of Csb and Csa knockout mice is greatly increased when the Xpc gene is additionally inactivated (reviewed in [25]). The persistence of cyPudN might be responsible for neuronal death in XP by blocking neuronal gene expression (reviewed in [26]).
XP-C patients and single nucleotide polymorphisms (SNP). XP-C patients exhibit extreme UV sensitivity and present 1000-fold higher risk of skin cancer and 10-fold of internal cancer. XP-C patients commonly do not show neurological alterations, however it should be considered that XP-C patients die in early age and these symptoms could emerge for long-lived patients. As expected, an increased p53 mutation frequency characterizes skin tumors from XP-C patients [27], and the analysis of mutational spectra shows the typical UV signature (tandem CC>TT transitions). Interestingly, in this study three primary internal tumors of young XP children were also analyzed. All of them contained one mutation on the p53 gene, which was different from the ones found in the XP skin tumors and could have resulted from unrepaired lesions caused by oxidative damage. Moreover, mutations which are compatible with 8-oxoG mutagenesis (i.e. G>T transversions) have been reported in the basal layer of human squamous tumors from repair proficient donors [28], indicating that oxidative DNA damage may contribute to skin cancer development too. The function of XPC in BER may thus contribute to increased skin cancer risk and play a major role in internal cancer development in XP-C patients.
An important implication of the newly identified function of XPC in the repair of oxidatively induced DNA lesions is that alterations in the XPC function in the general population (e.g. haploinsufficiency, polymorphisms) might be involved as predisposing factors in cancer development. Functional polymorphisms of the XPC gene and reduced levels of XPC mRNA have been associated with increased cancer risk [29-31]. Two variant alleles of XPC, XPC-PAT+/+ and intron 11 C/A, are characterized by reduced damage repair capacity (DRC) in a HCR [32]. Evidence has been presented that XPC-PAT+/+ subjects are at increasing risk of squamous cell carcinomas of the head and neck [29], bladder cancer [33], and lung cancer [30]. Recently, we reported a borderline association between gastric cancer risk and the XPC-PAT homozygous genotype [34]. All together these observations suggest that the role of XPC in bulky adducts and/or oxidative DNA damage repair may account for increased cancer risk in the general population. The functional characterization of XPC polymorphisms should be addressed by future research.
XPA
Biochemical properties and protein structure. The XPA gene is located on chromosome 9 and encodes a small zinc-finger protein (273 amino acids) that is part of a pre-incision complex with RPA and XPG. XPA was originally thought to be the initial UV damage recognition factor [35]. More recently, it was demonstrated that XPA has a much higher binding affinity for some kinked DNA substrates, such as three-way or four-way junction, than DNA lesions themselves [36]. This suggests that XPA may control the proper assembly of the NER pre-incision complex by probing for appropriately distorted DNA and thereby confirming the existence of the lesion indirectly. DNA binding of XPA is mediated by a positively charged cleft on the protein surface in the C-terminal domain (residues 138-209) [37, 38]. XPA exists as a homodimer either in the free state or as a complex with human RPA [39] by interaction with the zinc-finger domain [37]. It binds to the classical NER substrates (e.g. mismatched DNA bubble substrates and bulky DNA adducts) but also to oxidative DNA lesions, such as Tg paired with adenine. This lesion is an even better substrate in comparison to the C8-dG adduct of AAF [15]. XPA interacts with several proteins (RPA, ERCC1, TFIIH, XPC) [40], and specific interaction domains have been identified by deletion studies. XPA also interacts with the checkpoint ataxia telangiectasia and Rad 3 related (ATR) protein, and this interaction regulates the nuclear import of XPA after UV irradiation [41-43], thus indicating a cross-talk between the DNA damage checkpoint and NER proteins.
More recently, the interaction of XPA with the deacetylase sirtuin 1 (SIRT1) has been reported and shown to be enhanced by UV. SIRT1 deacetylates XPA, and this modification affects UV sensitivity. Moreover, XPA–RPA interaction is enhanced by SIRT1-mediated XPA deacetylation [44]. It is of note that SIRT1 exerts many of the pleiotropic effects of oxidative metabolism and protects cells from oxidative stress [45]. The potential effect of this interaction on the response to oxidative damage should be investigated. Most mutations from XP-A patients are deletions and splice site mutations whereas missense mutations are rare. Mutations in the DNA-binding region of XPA are typically from patients with the more severe disease often associated with neurologic complications, whereas mutations in the C terminus of the protein, which interacts with TFIIH, characterize patients with milder skin disease only [46].
Cell phenotype. It is well established that XP-A cells are sensitive to UVC-induced cell killing [47, 48] whereas the evidence that this sensitivity might extend to oxidative damage is scanty. A recent report [49] shows that fibroblasts derived from patients belonging to the XP-A, XP-C, or XP-G complementation groups are deficient in oxidative DNA damage repair as measured by HCR of H2O2-modified plasmid, and that mutagenicity of H2O2-induced damage in the supF gene is higher in XP-A fibroblasts that in the normal cells. Moreover, melanocytes have a reduced DRC and show a higher mutation frequency for oxidative DNA damage and UV photoproducts compared to that observed in fibroblasts. This could explain the 1000-fold higher incidence of melanoma in XP patients.
Mutant mouse models. Xpa–/– mice share with XP-A patients increased risk of UV-induced skin cancer [50] and spontaneous cancer. In particular, Xpa–/– mice develop spontaneous liver cancer [18, 51, 52]. However, in contrast with humans, no neuropathology was reported in Xpa–/– mice, but only subtle neurological defects [53]. The Csb–/– mouse also presents mild neurological symptoms (see below). However, deleting Xpa in a Csb defective mouse causes the appearance of heavier neurological symptoms (tremors, abnormal gait, poor balance, and progressive ataxia) and neuropathy in the cerebellum (decreased proliferation and increased apoptosis in the early germinal layer) [54]. Whether this indicates that both genes are involved in endogenous damage processing awaits to be clarified.
XP-A patients and SNP. Mutations in XPA are associated with skin cancer, although the risk is stronger when XPC is mutated. On the other hand, mutations in XPA are associated with a higher risk of neurodegeneration [55, 56]. Neurological defects appear between 2 and 8 years of age, with mild cognitive impairment followed by cerebellar alterations and, later, neuropathy. Corticospinal involvement occurs in the third decade, when cognitive impairment becomes severe [57]. Accumulation of oxidative damage has been reported in the brains of autopsied XP-A patients that showed also a reduced neuronal concentration of superoxide dismutase (SOD) [58]. It has been speculated that some forms of oxidative DNA damage that are repaired by XPA as NER core factor, such as cyclopurine adducts [59], or in which XPA is involved, such as Tg [15], might play a role in neurodegeneration.
In line with the involvement of XPA in the control of oxidative DNA damage, in a population-based study, carriers of the A allele of XPA-23G>A polymorphism have been shown to present higher levels of sites sensitive to formamidopyrimidine DNA glycosylase (Fpg) (that reveals oxidative DNA damage) in lymphocytes [60]. Interestingly, the combination of this genotype with the unfavorable XPC-PAT+/+ polymorphism was shown to be associated with significantly higher anti-benzo(a)pyrene diol epoxide (BPDE)-DNA adduct levels in a study conducted on polycyclic aromatic hydrocarbon exposed workers [61], with increased BPDE sensitivity in a twin study [62], and increased gastric cancer risk in a high-risk Italian population [34]. Moreover, the XPA 23GG genotype was associated with a significantly decreased risk of lung cancer when compared with AA and AG genotypes [63-65]. Functional studies on the effects of these SNPs are needed.
CSA AND CSB
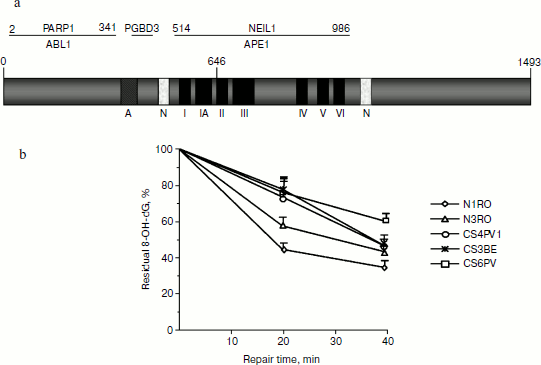
Biochemical properties and structure. The human CSA gene is located on chromosome 5. The CSA gene encodes a 396-amino acid protein that contains a WD repeat (WD40 repeat) domain (reviewed by [66]). The human CSB gene is located on chromosome 10 and encodes a 1493-amino acid protein that contains 7 consecutive domains that are conserved between DNA and RNA helicases. The CSB protein has no functional helicase activity demonstrated in vitro, but possesses a DNA-dependent ATPase activity, which is strongly stimulated by dephosphorylation of CSB upon UV irradiation [67]. CSA and CSB are both required for TCR. In addition, CSB has been shown to interact and stimulate transcriptional protein complexes of all three classes of nuclear RNA polymerases [68-72]. In line with this function, CSB-deficient cells exhibit in vivo defects in transcription initiation and elongation [73].
It has recently been demonstrated that CSB is essential for the re-initiation of transcription after UV irradiation even in undamaged housekeeping genes and that CSB can influence the transcription of specific sets of genes after ligand stimulation or in hypoxic conditions [74, 75]. CSA protein has been shown to interact with CSB and p44, a subunit of the RNA polymerase II basal transcription factor TFIIH [76] but the great surprise was the discovery by Groisman et al. [77] that CSA is a component of a protein complex that contains also the COP9 signalosome (CSN), a known regulator of cullin-based ubiquitin ligases. DNA damage-binding protein 2 (DDB2) is part of a similar but distinct protein complex via interaction with DDB1. CSN was shown to play a key role in NER by differentially regulating ubiquitin ligase activity of the DDB2 and CSA complexes in response to UV irradiation. The analysis of UV-stalled transcription elongation complexes from human cells revealed the nature and order of molecular events that take place during TCR and the different, albeit interconnected, role played by CSA and CSB [78]. CSB is a prerequisite factor for the assembly of NER proteins, histone acetyltransferase p300, and CSA-DDB1 E3-ubiquitin ligase complex with the COP9 signalosome to stalled RNA polymerase II, whereas functional CSA, in cooperation with CSB, is required to recruit the nucleosomal binding proteins high-mobility group nucleosome binding domain 1 (HMGN1), XPA binding protein 2 (XAB2), and transcription factor S-II (TFIIS) [78] but is dispensable for attraction of NER proteins to lesion stalled RNAPII. To complete this picture, CSB has been shown to contain a ubiquitin-binding domain that is required for excision of the lesion (not for assembly of NER factors) [79].
Thus, an integrated model would involve the ubiquitylation of CSB or the RNAPII large subunit by CSA as a signal for CSB to disassemble the initial TCR complex, leaving the NER complex to finish the job. If the roles of CSA and CSB in TCR have been finely dissected, the evidence that these proteins might have an additional function in the removal of oxidative damage in nuclei and in mitochondria is large but still lacks mechanistic insights. Mutations in the ATPase domains V and VI of CSB lead to accumulation of 8-oxoG in the cell genome [80] whereas domain VI appears to be involved in the repair of 8-oxoA [81]. Purified CSB greatly enhances nei endonuclease VIII-like 1 (NEIL1) catalytic activity via stimulation of the strand-incision step in repair of formamide pyrimidine guanine (FapyG) and formamide pyrimidine adenine (FapyA) lesions. CSB and NEIL1 also co-localize in HeLa cells and co-immunoprecipitate from HeLa nuclear extracts [82].
Altogether these results might suggest a mechanism in which CSB would contribute to repair of oxidatively modified bases via interaction with lesion-specific DNA glycosylases; however, CSB resides in a physical and a functional complex with poly(ADP-ribose) polymerase 1 (PARP-1) thus opening as alternative or complementary scenario its role in chromatin remodelling and/or single strand break repair. CSB–PARP1 complex redistributes in the nucleus in response to oxidative DNA damage, and CSB is post-translationally modified by PARP-1 after oxidative stress [83]. A more general function of CSB in modulating BER processes is also suggested by its interaction with APE1 and stimulation of APE1 incision activity at AP sites in an ATP-independent manner [84]. Mitochondria possess an independent BER machinery, the components of which are coded by nuclear genes that thoughtfully control the levels of oxidation of mtDNA. CSA and CSB proteins are present in mitochondria after oxidative stress and directly interact with mtDNA and BER-associated human mtOGG1 and mitochondrial single stranded DNA binding protein (mtSSBP-1) upon oxidative stress [85]. This indicates an additional role of CS proteins in the control of oxidative DNA damage in mitochondria, probably by anchoring the BER machinery at lesion sites. As mentioned above, UV irradiation leads to dephosphorylation of CSB protein and stimulation of its ATPase activity [67]. After treatment with oxidative agents, phosphorylation of CSB by Abelson murine leukemia kinase (Abl-1) has been reported [86].
Further studies should explore the role of post-translational modifications in the control of CSB activity after different types of stress. The functions of CSB have been thoroughly mapped by analysis of mutations in patients [87]. Less information is available for CSA. It is worth mentioning for the purpose of this review the Trp361Cys mutation of CSA that has been recently reported in a case of UV sensitive syndrome (UVSS) and was associated with defective TCR but normal oxidative DNA repair capacities [88]. This mutation could help to discriminate the different functions of CSA in various pathways and explain the different phenotypes associated with CSA mutations. Additional in vitro studies are obviously needed to gain further insight into the molecular mechanisms involving the CS proteins.
Cell phenotype. A hallmark of the CS phenotype is the sensitivity to UV light, but CS-deficient cells are hypersensitive also to several types of oxidative DNA damaging agents. After exposure to IR, CS-B transformed fibroblasts, MEF, embryonic stem (ES) cells, and keratinocytes from Csb knockout mice all showed a marked reduction in survival [47, 80, 89, 90]. Even if IR induces a variety of DNA lesions including single-strand DNA breaks (SSBs), double-strand DNA breaks (DSBs), and oxidative base damage, the observed hypersensitivity has been ascribed to oxidative DNA modifications [47, 89], which normally are repaired by BER. Moreover, Csb knockout MEFs were found to be hypersensitive to the oxidizing chemical paraquat [47, 89].
Reports on human SV-40-transformed CS-B cells did not confirm this hypersensitivity [91], although these cells accumulated oxidative DNA damage (see below). CS-A human primary cells, but not SV-40-transformed cells (CS3BE), were shown to be hypersensitive to oxidative damage-inducing agents such as potassium bromate (KBrO3) [92]. This finding should lead to caution in the extrapolation of data on oxidative stress sensitivity in transformed cells where different cell responses (e.g. p53 response) are defective compared to normal cells. The hypersensitivity to oxidative DNA agents is associated with accumulation of oxidative base modifications, including 8-oxoG, 8-oxoA, and 5-hydroxy-2′-deoxycytidine, in both CS-B and CS-A (Fig. 2b) genomic DNA [80, 91, 92] and impaired HCR of plasmids containing a single 8-oxoG [93]. CS-A primary skin cells showed also impaired repair of 8,5′-cyclopurine-2′-deoxyadenosine [92], a transcription-blocking lesion that has been involved in neurodegeneration [26]. The similar defect in oxidative DNA damage repair in CS-A and CS-B cells suggests that these proteins operate in the same pathway. The acceleration of repair at early repair times mediated by CSB [94] and CSA [92] might reflect a role of CS proteins in the regulation of chromatin structure [77, 95, 96], but this is still a matter of speculation. Elevated levels of DNA breakage after KBrO3 [97] and hypersensitivity to AP site-inducing agents, such as methylmethanesulfonate [84], have been reported in CS-B human cells, thus extending the list of lesions that involve CS proteins to BER intermediates.
Because of the dual role of CSB in transcription and TCR, CS-B human fibroblasts were analyzed by using expression arrays and comparative expression analysis. Newman et al. [96] found that expression of wild-type CSB in CS-B cells induced significant changes in gene expression, even in the absence of external stress. Many of the genes regulated by CSB were also affected by inhibitors of histone deacetylase and DNA methylation, as well as by defects in poly(ADP-ribose) polymerase function and RNA polymerase II elongation, supporting the idea that CSB has a general role in chromatin maintenance and remodeling. Moreover, after oxidative stress, genes encoding proteins involved in the ubiquitin proteasome pathway were induced to a lesser extent in CS-B fibroblasts as compared to wild-type cells [98]. It is of note that the defect in the incision of 8-oxoG as reported in CS-B human cells was associated with a downregulation of human OGG1 gene expression and protein level [81, 99].
Mutant mouse models. Two mouse models for the TC-NER disorder CS are available. In Csb-deficient mice, a truncation mutation in the CSB gene of a CS-B patient [100] was mimicked, while in Csa–/– mice the Csa gene was knocked out by interrupting exon 2 [101]. Csb–/– as well as Csa–/– mice are viable and exhibit all of the CS repair characteristics: UV sensitivity, inactivation of TC-NER, unaffected GG-NER, and inability to resume RNA synthesis after UV exposure. However, in contrast with CS patients, these mouse models present a mild form of growth failure, neurological dysfunction, and skin cancer susceptibility although modest and apparent only after chronic exposure to daily doses of UV light [100, 102]. The Csb–/– mice present some age-related pathological features such as renal karyomegaly and retinal atrophy. The Csb–/– mouse retina is hypersensitive to ionizing radiation, which suggests that oxidative DNA lesions are at the basis of this premature-aging phenotype [103]. CSB deficiency makes retinal photoreceptors more sensitive to apoptosis, resulting in progressive spontaneous photoreceptor loss with age. Moreover, it was recently demonstrated that mice deficient in the Csb gene are more susceptible to enhanced fetal oxidative DNA damage and neurodevelopmental deficits resulting from in utero exposure to xenobiotics, like methamphetamine, that enhance the fetal formation of ROS. These results provide the first evidence that, in addition to OGG1 [104], CSB protects the fetus from xenobiotic-enhanced DNA oxidation and post-natal functional deficits [105]. In agreement with a cross-talk between OGG1 and CSB, a more pronounced accumulation of Fpg-sensitive sites and increased spontaneous mutation frequency was reported in the livers of Csbm/m/Ogg1–/– mice as compared to Ogg1–/– mice, whereas the basal levels of these lesions were not significantly affected in Csbm/m mice [94, 106].Fig. 2. a) CSB protein structure and functional regions of interacting proteins. b) Repair kinetics of 8-oxoG in primary fibroblasts from two normal (N1RO, N3RO) and three CS-A donors (CS4PV, CS6PV, and CS3BE) after exposure to 40 mM KBrO3 (30 min). DNA was isolated at the indicated times, and the kinetics of 8-oxoG removal was measured by HPLC-ED (modified from [91]).
The role of CS proteins in maintaining the mitochondrial membrane integrity is supported by reports on partially disassembled complexes of the inner mitochondrial membrane in CSB null mice together with hypersensitivity to bioenergetic inhibitors, impaired ability to recover from cellular ATP depletion [107] and reduced 8-oxoG, uracil, 5-hydroxy-uracil, and AP site-incision activities in defective mitochondrial extracts [108].
CS patients and SNP. In contrast with CS mice that are cancer prone, CS patients are cancer-free. The cardinal clinical features of CS are pre- or post-natal growth failure, leading to a characteristic appearance of so-called cachectic dwarfism and progressive neurological dysfunction. Associated clinical features are gait defects, progressive pigmentary retinopathy, and other ocular anomalies such as cataracts and optic disc atrophy, sensorineural hearing loss, impaired sexual development, skeletal abnormalities, dental caries, and cutaneous photosensitivity. The severity of the symptoms can be quite variable depending on the complementation group and on the nature of the mutation. Another disease with mutation in CSA and CSB is UVsS, which resembles to CS but presents mild skin abnormalities and normal growth and mental development [88, 109].
The analysis of oxidative products, such as nitrotyrosine, glycation end product, and 4-hydroxy-2-nonenal-modified protein (HNE), conducted in the brains of autopsied patients, showed a higher level of these oxidative products in the globus pallidus of CS patients compared to XPA patients [110] and in both cases higher than in normal brains. The involvement of CS proteins in oxidative DNA damage repair opens the question of whether this new function might account for some of the neurological abnormalities of these patients. The link between altered processing of oxidative DNA lesions and neurological disorders is supported by the growing body of evidence of defects in the repair of DNA SSBs in central nervous system disorders, e.g. spinocerebellar ataxia with axonal neuropathy (SCAN1) and ataxia oculomotor apraxia type 1 (AOA1). Certain aspects of the clinical features of CSB overlap with the phenotypes associated with mitochondrial dysfunction, including severe neurological deficiencies, dysfunction in skeletal muscle and heart, and premature aging symptoms. Thus, it was speculated that CS-B cells accumulate mutations in mtDNA and develop mitochondrial dysfunction that contributes to the phenotype and progression of disease in CS-B patients [111]. Several polymorphisms have been documented in the CSB-coding sequence alone, mostly in the C-terminal third of the protein (Ensembl and NCBI SNP databases) [112]. Recently, the combined variant genotypes of four loci (rs2228526, rs4253160, rs12571445, and rs3793784) of the CSB gene have been reported to associate with a significantly increased lung cancer risk in a Chinese population [113]. More studies should address whether SNP in these genes are associated with health effects in the general population.
In this review we present an overview of the growing body of evidence that supports the role of NER proteins in the control of endogenous/oxidative DNA damage. The mechanistic basis is still unknown and the precise source of internal damage has to be identified. However, what emerges is a complex picture where the participation of NER factors is likely to involve not only repair capacity but also ROS production and more in general the cellular oxidative metabolism (table). Alterations of catalase and SOD activity, mitochondrial dysfunction, and hypersensitivity to bioenergetic inhibitors have been reported in XP-C, XP-A, and CS-B cells. How NER proteins might determine alteration in the cell metabolism is an open question. In the case of the most studied NER factor, CSB, several scenarios have been envisaged. It has been proposed that the elevated levels of p21 that characterize CS cells might be responsible for the high intracellular ROS level [114], or p21 itself may transcriptionally regulate key metabolic enzymes. As alternative mechanism a direct interaction of CSB with mitochondrial enzymes is suggested by its interaction with 3-hydroxyisobutyryl-coenzyme A hydrolase that belongs to the valine catabolic pathway [108] and by its function as anchorage of BER complexes associated with the inner mitochondrial membrane [107]. In addition, an effect on transcription of a set of genes involved in oxidative metabolism cannot be ruled out, as suggested by genome-wide transcription profiling of CSB mutant mouse models [115]. The range of oxidative lesions that involve NER factors in their repair is large (table), and although interactions of XPC and CSB proteins with specific DNA glycosylases/AP endonucleases have been described, the variety of lesions that are potential substrates suggest that a more general function of these factors might be involved too. The role of CSB in chromatin remodeling [96] has been invoked as a plausible mechanism. The recent discovery of the recruitment of NER factors, such as XPC, XPA, XPG, and XPF-ERCC1, to active promoters to facilitate RNA polymerase II transcription [116] and their requirement for DNA demethylation and histone post-translational modifications opens the question of whether this “double life” of NER factors in transcription and repair might impact on the accessibility of the repair machinery to endogenous damage. Further studies have to be engaged to define the mechanistic basis of NER involvement in oxidative DNA damage control and to address whether the severity of DNA damage from endogenous sources might be a factor in the clinical variations that characterize NER disorders.
Multifaceted role of NER factors in control of oxidative damage

We acknowledge grant support from Associazione Italiana per la Ricerca sul Cancro, Fondazione Cariplo, Programma Malattie Rare (Institute of Public Health), and Progetto Integrato Oncologia (Ministry of Public Health).
REFERENCES
1.Scharer, O. D. (2008) Adv. Exp. Med. Biol.,
637, 83-92.
2.Masutani, C., Sugasawa, K., Yanagisawa, J.,
Sonoyama, T., Ui, M., Enomoto, T., Takio, K., Tanaka, K., van der Spek,
P. J., Bootsma, D., et al. (1994) EMBO J., 13,
1831-1843.
3.Camenisch, U., Trautlein, D., Clement, F. C., Fei,
J., Leitenstorfer, A., Ferrando-May, E., and Naegeli, H. (2009) EMBO
J., 28, 2387-2399.
4.Min, J. H., and Pavletich, N. P. (2007)
Nature, 449, 570-575.
5.Yokoi, T., Ohmichi, M., Tasaka, K., Kimura, A.,
Kanda, Y., Hayakawa, J., Tahara, M., Hisamoto, K., Kurachi, H., and
Murata, Y. (2000) J. Biol. Chem., 275, 21639-21647.
6.Sugasawa, K., Okuda, Y., Saijo, M., Nishi, R.,
Matsuda, N., Chu, G., Mori, T., Iwai, S., Tanaka, K., and Hanaoka, F.
(2005) Cell, 121, 387-400.
7.Araki, M., Masutani, C., Takemura, M., Uchida, A.,
Sugasawa, K., Kondoh, J., Ohkuma, Y., and Hanaoka, F. (2001) J.
Biol. Chem., 276, 18665-18672.
8.Charbonnier, J. B., Renaud, E., Miron, S., Le Du,
M. H., Blouquit, Y., Duchambon, P., Christova, P., Shosheva, A., Rose,
T., Angulo, J. F., and Craescu, C. T. (2007) J. Mol. Biol.,
373, 1032-1046.
9.Popescu, A., Miron, S., Blouquit, Y., Duchambon,
P., Christova, P., and Craescu, C. T. (2003) J. Biol. Chem.,
278, 40252-40261.
10.Miao, F., Bouziane, M., Dammann, R., Masutani,
C., Hanaoka, F., Pfeifer, G., and O’Connor, T. R. (2000) J.
Biol. Chem., 275, 28433-28438.
11.Shimizu, Y., Iwai, S., Hanaoka, F., and Sugasawa,
K. (2003) EMBO J., 22, 164-173.
12.D’Errico, M., Parlanti, E., Teson, M., de
Jesus, B. M., Degan, P., Calcagnile, A., Jaruga, P., Bjoras, M.,
Crescenzi, M., Pedrini, A. M., Egly, J. M., Zambruno, G., Stefanini,
M., Dizdaroglu, M., and Dogliotti, E. (2006) EMBO J., 25,
4305-4315.
13.Hill, J. W., Hazra, T. K., Izumi, T., and Mitra,
S. (2001) Nucleic Acids Res., 29, 430-438.
14.Shimizu, Y., Uchimura, Y., Dohmae, N., Saitoh,
H., Hanaoka, F., and Sugasawa, K. (2010) J. Nucleic Acids, ID
805698.
15.Brown, K. L., Roginskaya, M., Zou, Y.,
Altamirano, A., Basu, A. K., and Stone, M. P. (2010) Nucleic Acids
Res., 38, 428-440.
16.Yasuda, G., Nishi, R., Watanabe, E., Mori, T.,
Iwai, S., Orioli, D., Stefanini, M., Hanaoka, F., and Sugasawa, K.
(2007) Mol. Cell Biol., 27, 6606-6614.
17.Bernardes de Jesus, B. M., Bjoras, M., Coin, F.,
and Egly, J. M. (2008) Mol. Cell Biol., 28,
7225-7235.
18.Melis, J. P., Wijnhoven, S. W., Beems, R. B.,
Roodbergen, M., van den Berg, J., Moon, H., Friedberg, E., van der
Horst, G. T., Hoeijmakers, J. H., Vijg, J., and van Steeg, H. (2008)
Cancer Res., 68, 1347-1353.
19.Kassam, S. N., and Rainbow, A. J. (2007)
Biochem. Biophys. Res. Commun., 359, 1004-1009.
20.Quilliet, X., Chevallier-Lagente, O., Zeng, L.,
Calvayrac, R., Mezzina, M., Sarasin, A., and Vuillaume, M. (1997)
Mutat. Res., 385, 235-242.
21.Liu, S. Y., Wen, C. Y., Lee, Y. J., and Lee, T.
C. (2010) Toxicol. Sci., 116, 183-193.
22.Sands, A. T., Abuin, A., Sanchez, A., Conti, C.
J., and Bradley, A. (1995) Nature, 377, 162-165.
23.Hoogervorst, E. M., van Oostrom, C. T., Beems, R.
B., van Benthem, J., van den Berg, J., van Kreijl, C. F., Vos, J. G.,
de Vries, A., and van Steeg, H. (2005) DNA Repair (Amst.),
4, 3-9.
24.Wijnhoven, S. W., Kool, H. J., Mullenders, L. H.,
van Zeeland, A. A., Friedberg, E. C., van der Horst, G. T., van Steeg,
H., and Vrieling, H. (2000) Oncogene, 19, 5034-5037.
25.Friedberg, E. C. (2004) DNA Repair
(Amst.), 3, 183-195.
26.Brooks, P. J. (2008) DNA Repair (Amst.),
7, 1168-1179.
27.Giglia, G., Dumaz, N., Drougard, C., Avril,
M.-F., Daya-Grosjean, L., and Sarasin, A. (1998) Cancer Res.,
58, 4402-4409.
28.Agar, N. S., Halliday, G. M., Barnetson, R. S.,
Ananthaswamy, H. N., Wheeler, M., and Jones, A. M. (2004) Proc.
Natl. Acad. Sci. USA, 101, 4954-4959.
29.Shen, H., Sturgis, E. M., Khan, S. G., Qiao, Y.,
Shahlavi, T., Eicher, S. A., Xu, Y., Wang, X., Strom, S. S., Spitz, M.
R., Kraemer, K. H., and Wei, Q. (2001) Cancer Res., 61,
3321-3325.
30.Marin, M. S., Lopez-Cima, M. F., Garcia-Castro,
L., Pascual, T., Marron, M. G., and Tardon, A. (2004) Cancer
Epidemiol. Biomarkers Prev., 13, 1788-1793.
31.Shen, M. R., Hsu, Y. M., Hsu, K. F., Chen, Y. F.,
Tang, M. J., and Chou, C. Y. (2006) Carcinogenesis, 27,
962-971.
32.Qiao, Y., Spitz, M. R., Shen, H., Guo, Z., Shete,
S., Hedayati, M., Grossman, L., Mohrenweiser, H., and Wei, Q. (2002)
Carcinogenesis, 23, 295-299.
33.Sanyal, S., Festa, F., Sakano, S., Zhang, Z.,
Steineck, G., Norming, U., Wijkstrom, H., Larsson, P., Kumar, R., and
Hemminki, K. (2004) Carcinogenesis, 25, 729-734.
34.Palli, D., Polidoro, S., D’Errico, M.,
Saieva, C., Guarrera, S., Calcagnile, A. S., Sera, F., Allione, A.,
Gemma, S., Zanna, I., Filomena, A., Testai, E., Caini, S., Moretti, R.,
Gomez-Miguel, M. J., Nesi, G., Luzzi, I., Ottini, L., Masala, G.,
Matullo, G., and Dogliotti, E. (2010) Mutagenesis, Epub ahead of
print.
35.Kuraoka, I., Morita, E. H., Saijo, M., Matsuda,
T., Morikawa, K., Shirakawa, M., and Tanaka, K. (1996) Mutat.
Res., 362, 87-95.
36.Missura, M., Buterin, T., Hindges, R., Hubscher,
U., Kasparkova, J., Brabec, V., and Naegeli, H. (2001) EMBO J.,
20, 3554-3564.
37.Ikegami, T., Kuraoka, I., Saijo, M., Kodo, N.,
Kyogoku, Y., Morikawa, K., Tanaka, K., and Shirakawa, M. (1998) Nat.
Struct. Biol., 5, 701-706.
38.Buchko, G. W., Daughdrill, G. W., de Lorimier,
R., Rao, B. K., Isern, N. G., Lingbeck, J. M., Taylor, J. S., Wold, M.
S., Gochin, M., Spicer, L. D., Lowry, D. F., and Kennedy, M. A. (1999)
Biochemistry, 38, 15116-15128.
39.Yang, Z. G., Liu, Y., Mao, L. Y., Zhang, J. T.,
and Zou, Y. (2002) Biochemistry, 41, 13012-13020.
40.Gillet, L. C., and Scharer, O. D. (2006) Chem.
Rev., 106, 253-276.
41.Wu, X., Shell, S. M., Yang, Z., and Zou, Y.
(2006) Cancer Res., 66, 2997-3005.
42.Wu, X., Shell, S. M., Liu, Y., and Zou, Y. (2007)
Oncogene, 26, 757-764.
43.Shell, S. M., Li, Z., Shkriabai, N.,
Kvaratskhelia, M., Brosey, C., Serrano, M. A., Chazin, W. J., Musich,
P. R., and Zou, Y. (2009) J. Biol. Chem., 284,
24213-24222.
44.Fan, W., and Luo, J. (2010) Mol. Cell,
39, 247-258.
45.He, W., Wang, Y., Zhang, M. Z., You, L., Davis,
L. S., Fan, H., Yang, H. C., Fogo, A. B., Zent, R., Harris, R. C.,
Breyer, M. D., and Hao, C. M. (2010) J. Clin. Invest.,
120, 1056-1068.
46.States, J. C., McDuffie, E. R., Myrand, S. P.,
McDowell, M., and Cleaver, J. E. (1998) Hum. Mutat., 12,
103-113.
47.De Waard, H., de Wit, J., Gorgels, T. G., van den
Aardweg, G., Andressoo, J. O., Vermeij, M., van Steeg, H., Hoeijmakers,
J. H., and van der Horst, G. T. (2003) DNA Repair (Amst.),
2, 13-25.
48.De Waard, H., Sonneveld, E., de Wit, J.,
Esveldt-van Lange, R., Hoeijmakers, J. H., Vrieling, H., and van der
Horst, G. T. (2008) DNA Repair (Amst.), 7, 1659-1669.
49.Wang, H. T., Choi, B., and Tang, M. S. (2010)
Proc. Natl. Acad. Sci. USA, 107, 12180-12185.
50.De Vries, A., van Oostrom, C. T., Hofhuis, F. M.,
Dortant, P. M., Berg, R. J., de Gruijl, F. R., Wester, P. W., van
Kreijl, C. F., Capel, P. J., van Steeg, H., et al. (1995)
Nature, 377, 169-173.
51.De Vries, A., Dolle, M. E., Broekhof, J. L.,
Muller, J. J., Kroese, E. D., van Kreijl, C. F., Capel, P. J., Vijg,
J., and van Steeg, H. (1997) Carcinogenesis, 18,
2327-2332.
52.Takahashi, Y., Nakatsuru, Y., Zhang, S., Shimizu,
Y., Kume, H., Tanaka, K., Ide, F., and Ishikawa, T. (2002)
Carcinogenesis, 23, 627-633.
53.Wijnhoven, S. W., Hoogervorst, E. M., de Waard,
H., van der Horst, G. T., and van Steeg, H. (2007) Mutat. Res.,
614, 77-94.
54.Murai, M., Enokido, Y., Inamura, N., Yoshino, M.,
Nakatsu, Y., van der Horst, G. T., Hoeijmakers, J. H., Tanaka, K., and
Hatanaka, H. (2001) Proc. Natl. Acad. Sci. USA, 98,
13379-13384.
55.Andrews, A. D., Barrett, S. F., and Robbins, J.
H. (1978) Proc. Natl. Acad. Sci. USA, 75, 1984-1988.
56.Kraemer, K. H., Lee, M. M., and Scotto, J. (1987)
Arch. Dermatol., 123, 241-249.
57.Anttinen, A., Koulu, L., Nikoskelainen, E.,
Portin, R., Kurki, T., Erkinjuntti, M., Jaspers, N. G., Raams, A.,
Green, M. H., Lehmann, A. R., Wing, J. F., Arlett, C. F., and Marttila,
R. J. (2008) Brain, 131, 1979-1989.
58.Hayashi, M., Araki, S., Kohyama, J., Shioda, K.,
and Fukatsu, R. (2005) Brain Dev., 27, 34-38.
59.Brooks, P. J. (2007) Neuroscience,
145, 1407-1417.
60.Dusinska, M., Dzupinkova, Z., Wsolova, L.,
Harrington, V., and Collins, A. R. (2006) Mutagenesis,
21, 205-211.
61.Pavanello, S., Pulliero, A., Siwinska, E.,
Mielzynska, D., and Clonfero, E. (2005) Carcinogenesis,
26, 169-175.
62.Lin, J., Swan, G. E., Shields, P. G., Benowitz,
N. L., Gu, J., Amos, C. I., de Andrade, M., Spitz, M. R., and Wu, X.
(2007) Cancer Epidemiol. Biomarkers Prev., 16,
2065-2071.
63.Park, J. Y., Park, S. H., Choi, J. E., Lee, S.
Y., Jeon, H. S., Cha, S. I., Kim, C. H., Park, J. H., Kam, S., Park, R.
W., Kim, I. S., and Jung, T. H. (2002) Cancer Epidemiol. Biomarkers
Prev., 11, 993-997.
64.Wu, X., Zhao, H., Wei, Q., Amos, C. I., Zhang,
K., Guo, Z., Qiao, Y., Hong, W. K., and Spitz, M. R. (2003)
Carcinogenesis, 24, 505-509.
65.Butkiewicz, D., Popanda, O., Risch, A., Edler,
L., Dienemann, H., Schulz, V., Kayser, K., Drings, P., Bartsch, H., and
Schmezer, P. (2004) Cancer Epidemiol. Biomarkers Prev.,
13, 2242-2246.
66.Neer, E. J., Schmidt, C. J., Nambudripad, R., and
Smith, T. F. (1994) Nature, 371, 297-300.
67.Christiansen, M., Stevnsner, T., Modin, C.,
Martensen, P. M., Brosh, R. M., Jr., and Bohr, V. A. (2003) Nucleic
Acids Res., 31, 963-973.
68.Selby, C. P., and Sancar, A. (1997) Proc.
Natl. Acad. Sci. USA, 94, 11205-11209.
69.Tantin, D., Kansal, A., and Carey, M. (1997)
Mol. Cell Biol., 17, 6803-6814.
70.Van Gool, A. J., Citterio, E., Rademakers, S.,
van Os, R., Vermeulen, W., Constantinou, A., Egly, J. M., Bootsma, D.,
and Hoeijmakers, J. H. (1997) Embo J., 16, 5955-5965.
71.Bradsher, J., Auriol, J., Proietti de Santis, L.,
Iben, S., Vonesch, J. L., Grummt, I., and Egly, J. M. (2002) Mol.
Cell, 10, 819-829.
72.Yuan, X., Feng, W., Imhof, A., Grummt, I., and
Zhou, Y. (2007) Mol. Cell, 27, 585-595.
73.Balajee, A. S., May, A., Dianov, G. L.,
Friedberg, E. C., and Bohr, V. A. (1997) Proc. Natl. Acad. Sci.
USA, 94, 4306-4311.
74.Proietti-De-Santis, L., Drane, P., and Egly, J.
M. (2006) EMBO J., 25, 1915-1923.
75.Filippi, S., Latini, P., Frontini, M., Palitti,
F., Egly, J. M., and Proietti-De-Santis, L. (2008) EMBO J.,
27, 2545-2556.
76.Stevnsner, T., Muftuoglu, M., Aamann, M. D., and
Bohr, V. A. (2008) Mech. Ageing Dev., 129, 441-448.
77.Groisman, R., Polanowska, J., Kuraoka, I.,
Sawada, J., Saijo, M., Drapkin, R., Kisselev, A. F., Tanaka, K., and
Nakatani, Y. (2003) Cell, 113, 357-367.
78.Fousteri, M., Vermeulen, W., van Zeeland, A. A.,
and Mullenders, L. H. (2006) Mol. Cell, 23, 471-482.
79.Anindya, R., Mari, P. O., Kristensen, U., Kool,
H., Giglia-Mari, G., Mullenders, L. H., Fousteri, M., Vermeulen, W.,
Egly, J. M., and Svejstrup, J. Q. (1010) Mol. Cell, 38,
637-648.
80.Tuo, J., Muftuoglu, M., Chen, C., Jaruga, P.,
Selzer, R. R., Brosh, R. M., Jr., Rodriguez, H., Dizdaroglu, M., and
Bohr, V. A. (2001) J. Biol. Chem., 276, 45772-45779.
81.Tuo, J., Jaruga, P., Rodriguez, H., Dizdaroglu,
M., and Bohr, V. A. (2002) J. Biol. Chem., 277,
30832-30837.
82.Muftuoglu, M., de Souza-Pinto, N. C., Dogan, A.,
Aamann, M., Stevnsner, T., Rybanska, I., Kirkali, G., Dizdaroglu, M.,
and Bohr, V. A. (2009) J. Biol. Chem., 284,
9270-9279.
83.Thorslund, T., von Kobbe, C., Harrigan, J. A.,
Indig, F. E., Christiansen, M., Stevnsner, T., and Bohr, V. A. (2005)
Mol. Cell Biol., 25, 7625-7636.
84.Wong, H. K., Muftuoglu, M., Beck, G., Imam, S.
Z., Bohr, V. A., and Wilson, D. M., 3rd. (2007) Nucleic Acids
Res., 35, 4103-4113.
85.Kamenisch, Y., Fousteri, M., Knoch, J., von
Thaler, A. K., Fehrenbacher, B., Kato, H., Becker, T., Dolle, M. E.,
Kuiper, R., Majora, M., Schaller, M., van der Horst, G. T., van Steeg,
H., Rocken, M., Rapaport, D., Krutmann, J., Mullenders, L. H., and
Berneburg, M. (2010) J. Exp. Med., 207, 379-390.
86.Imam, S. Z., Indig, F. E., Cheng, W. H., Saxena,
S. P., Stevnsner, T., Kufe, D., and Bohr, V. A. (2007) Nucleic Acids
Res., 35, 4941-4951.
87.Licht, C. L., Stevnsner, T., and Bohr, V. A.
(2003) Am. J. Hum. Genet., 73, 1217-1239.
88.Nardo, T., Oneda, R., Spivak, G., Vaz, B.,
Mortier, L., Thomas, P., Orioli, D., Laugel, V., Stary, A., Hanawalt,
P. C., Sarasin, A., and Stefanini, M. (2009) Proc. Natl. Acad. Sci.
USA, 106, 6209-6214.
89.De Waard, H., de Wit, J., Andressoo, J. O., van
Oostrom, C. T., Riis, B., Weimann, A., Poulsen, H. E., van Steeg, H.,
Hoeijmakers, J. H., and van der Horst, G. T. (2004) Mol. Cell
Biol., 24, 7941-7948.
90.Leadon, S. A., and Cooper, P. K. (1993) Proc.
Natl. Acad. Sci. USA, 90, 10499-10503.
91.Ropolo, M., Degan, P., Foresta, M.,
D’Errico, M., Lasiglie, D., Dogliotti, E., Casartelli, G., Zupo,
S., Poggi, A., and Frosina, G. (2007) Free Radic. Biol. Med.,
42, 1807-1817.
92.D’Errico, M., Parlanti, E., Teson, M.,
Degan, P., Lemma, T., Calcagnile, A., Iavarone, I., Jaruga, P., Ropolo,
M., Pedrini, A. M., Orioli, D., Frosina, G., Zambruno, G., Dizdaroglu,
M., Stefanini, M., and Dogliotti, E. (2007) Oncogene, 26,
4336-4343.
93.Spivak, G., and Hanawalt, P. C. (2006) DNA
Repair (Amst.), 5, 13-22.
94.Osterod, M., Larsen, E., Le Page, F., Hengstler,
J. G., van der Horst, G. T., Boiteux, S., Klungland, A., and Epe, B.
(2002) Oncogene, 21, 8232-8239.
95.Citterio, E., van den Boom, V., Schnitzler, G.,
Kanaar, R., Bonte, E., Kingston, R. E., Hoeijmakers, J. H., and
Vermeulen, W. (2000) Mol. Cell Biol., 20, 7643-7653.
96.Newman, J. C., Bailey, A. D., and Weiner, A. M.
(2006) Proc. Natl. Acad. Sci. USA, 103, 9613-9618.
97.Mosesso, P., Penna, S., Pepe, G., Lorenti-Garcia,
C., and Palitti, F. (2004) Cytogenet. Genome Res., 104,
178-181.
98.Kyng, K. J., May, A., Brosh, R. M., Jr., Cheng,
W. H., Chen, C., Becker, K. G., and Bohr, V. A. (2003) Oncogene,
22, 1135-1149.
99.Dianov, G., Bischoff, C., Sunesen, M., and Bohr,
V. A. (1999) Nucleic Acids Res., 27, 1365-1368.
100.Van der Horst, G. T., van Steeg, H., Berg, R.
J., van Gool, A. J., de Wit, J., Weeda, G., Morreau, H., Beems, R. B.,
van Kreijl, C. F., de Gruijl, F. R., Bootsma, D., and Hoeijmakers, J.
H. (1997) Cell, 89, 425-435.
101.Van der Horst, G. T., Meira, L., Gorgels, T.
G., de Wit, J., Velasco-Miguel, S., Richardson, J. A., Kamp, Y.,
Vreeswijk, M. P., Smit, B., Bootsma, D., Hoeijmakers, J. H., and
Friedberg, E. C. (2002) DNA Repair (Amst.), 1,
143-157.
102.Berg, R. J., Rebel, H., van der Horst, G. T.,
van Kranen, H. J., Mullenders, L. H., van Vloten, W. A., and de Gruijl,
F. R. (2000) Cancer Res., 60, 2858-2863.
103.Gorgels, T. G., van der Pluijm, I., Brandt, R.
M., Garinis, G. A., van Steeg, H., van den Aardweg, G., Jansen, G. H.,
Ruijter, J. M., Bergen, A. A., van Norren, D., Hoeijmakers, J. H., and
van der Horst, G. T. (2007) Mol. Cell Biol., 27,
1433-1441.
104.Wong, A. W., McCallum, G. P., Jeng, W., and
Wells, P. G. (2008) J. Neurosci., 28, 9047-9054.
105.McCallum, G. P., Wong, A. W., and Wells, P. G.
(2010) Antioxid. Redox Signal., Epub ahead of print.
106.Trapp, C., Schwarz, M., and Epe, B. (2007)
Cancer Res., 67, 5156-5161.
107.Osenbroch, P. O., Auk-Emblem, P., Halsne, R.,
Strand, J., Forstrom, R. J., van der Pluijm, I., and Eide, L. (2009)
FEBS J., 276, 2811-2821.
108.Aamann, M. D., Sorensen, M. M., Hvitby, C.,
Berquist, B. R., Muftuoglu, M., Tian, J., de Souza-Pinto, N. C.,
Scheibye-Knudsen, M., Wilson, D. M., 3rd, Stevnsner, T., and Bohr, V.
A. (2010) FASEB J., 24, 2334-2346.
109.Itoh, T., Ono, T., and Yamaizumi, M. (1994)
Mutat. Res., 314, 233-248.
110.Hayashi, M., Itoh, M., Araki, S., Kumada, S.,
Shioda, K., Tamagawa, K., Mizutani, T., Morimatsu, Y., Minagawa, M.,
and Oda, M. (2001) J. Neuropathol. Exp. Neurol., 60,
350-356.
111.Stevnsner, T., Nyaga, S., de Souza-Pinto, N.
C., van der Horst, G. T., Gorgels, T. G., Hogue, B. A., Thorslund, T.,
and Bohr, V. A. (2002) Oncogene, 21, 8675-8682.
112.Laugel, V., Dalloz, C., Durand, M., Sauvanaud,
F., Kristensen, U., Vincent, M. C., Pasquier, L., Odent, S.,
Cormier-Daire, V., Gener, B., Tobias, E. S., Tolmie, J. L.,
Martin-Coignard, D., Drouin-Garraud, V., Heron, D., Journel, H., Raffo,
E., Vigneron, J., Lyonnet, S., Murday, V., Gubser-Mercati, D., Funalot,
B., Brueton, L., Sanchez Del Pozo, J., Munoz, E., Gennery, A. R.,
Salih, M., Noruzinia, M., Prescott, K., Ramos, L., Stark, Z., Fieggen,
K., Chabrol, B., Sarda, P., Edery, P., Bloch-Zupan, A., Fawcett, H.,
Pham, D., Egly, J. M., Lehmann, A. R., Sarasin, A., and Dollfus, H.
(2010) Hum. Mutat., 31, 113-126.
113.Ma, H., Hu, Z., Wang, H., Jin, G., Wang, Y.,
Sun, W., Chen, D., Tian, T., Jin, L., Wei, Q., Lu, D., Huang, W., and
Shen, H. (2009) Cancer Lett., 273, 172-176.
114.Cleaver, J. E., Hefner, E., Laposa, R. R.,
Karentz, D., and Marti, T. (2007) Neuroscience, 145,
1300-1308.
115.Van der Pluijm, I., Garinis, G. A., Brandt, R.
M., Gorgels, T. G., Wijnhoven, S. W., Diderich, K. E., de Wit, J.,
Mitchell, J. R., van Oostrom, C., Beems, R., Niedernhofer, L. J.,
Velasco, S., Friedberg, E. C., Tanaka, K., van Steeg, H., Hoeijmakers,
J. H., and van der Horst, G. T. (2007) PLoS Biol., 6,
e304.
116.Le May, N., Mota-Fernandes, D., Velez-Cruz, R.,
Iltis, I., Biard, D., and Egly, J. M. (2010) Mol. Cell,
38, 54-66.