Primary Electron Transfer in Reaction Centers of YM210L and YM210L/HL168L Mutants of Rhodobacter sphaeroides
A. G. Yakovlev1*, L. G. Vasilieva2, T. I. Khmelnitskaya2, V. A. Shkuropatova2, A. Ya. Shkuropatov2, and V. A. Shuvalov1,2
1Department of Photobiophysics, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: yakov@genebee.msu.su2Institute of Basic Biological Problems, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: (496) 773-0532; E-mail: shuvalov@issp.serpukhov.su
* To whom correspondence should be addressed.
Received February 8, 2010
The role of tyrosine M210 in charge separation and stabilization of separated charges was studied by analyzing of the femtosecond oscillations in the kinetics of decay of stimulated emission from P* and of a population of the primary charge separated state P+BA– in YM210L and YM210L/HL168L mutant reaction centers (RCs) of Rhodobacter sphaeroides in comparison with those in native Rba. sphaeroides RCs. In the mutant RCs, TyrM210 was replaced by Leu. The HL168L mutation placed the redox potential of the P+/P pair 123 mV below that of native RCs, thus creating a theoretical possibility of P+BA– stabilization. Kinetics of P* decay at 940 nm of both mutants show a significant slowing of the primary charge separation reaction in comparison with native RCs. Distinct damped oscillations in these kinetics with main frequency bands in the range of 90-150 cm–1 reflect mostly nuclear motions inside the dimer P. Formation of a very small absorption band of BA– at 1020 nm is registered in RCs of both mutants. The formation of the BA– band is accompanied by damped oscillations with main frequencies from ~10 to ~150 cm–1. Only a partial stabilization of the P+BA– state is seen in the YM210L/HL168L mutant in the form of a small non-oscillating background of the 1020-nm kinetics. A similar charge stabilization is absent in the YM210L mutant. A model of oscillatory reorientation of the OH-group of TyrM210 in the electric fields of P+ and BA– is proposed to explain rapid stabilization of the P+BA– state in native RCs. Small oscillatory components at ~330-380 cm–1 in the 1020-nm kinetics of native RCs are assumed to reflect this reorientation. We conclude that the absence of TyrM210 probably cannot be compensated by lowering of the P+BA– free energy that is expected for the double YM210L/HL168L mutant. An oscillatory motion of the HOH55 water molecule under the influence of P+ and BA– is assumed to be another potential contributor to the mechanism of P+BA– stabilization.
KEY WORDS: photosynthesis, charge separation, reaction center, wave packet, electron transferDOI: 10.1134/S0006297910070047
Abbreviations: ΔA, absorbance changes (light-minus-dark); BChl, bacteriochlorophyll; BA and BB, monomeric BChl in A- and B-branch, respectively; BPheo, bacteriopheophytin; FT, Fourier transform; HA and HB, BPheo in A- and B-branch, respectively; P, primary electron donor dimer BChl; PA and PB, BChl molecules constituting P; QA and QB, primary and secondary electron acceptor quinone, respectively; RC, reaction center.
The reaction center (RC) of photosynthetic bacteria and green plants is
a pigment–protein complex in which light energy is converted into
the free energy of charge-separated states. In RCs of purple bacteria
two strongly interacting bacteriochlorophylls (BChls), PA
and PB, constitute the special pair P that serves as the
primary electron donor and is localized near the periplasmic side of
the membrane [1, 2]. Two BChl
monomers (BA and BB) and two bacteriopheophytins
(BPheos; HA and HB) are located in the
hydrophobic central part of the membrane in these RCs. The quinones
(QA and QB) terminate two branches (A and B) of
the cofactors. The Fe atom is located between QA and
QB near the cytoplasmic side of the membrane. Light-induced
charge separation takes place in the A branch [3].
On the basis of time-resolved spectroscopic measurements [4-6], it has been suggested that BA is the primary electron acceptor and the state P+BA– can be the first charge-separated state. A possibility of direct electron transfer from P* to HA by the superexchange mechanism involving the vacant orbital of BA is also discussed [7, 8]. The formation of the long-lived (~1 nsec) state P+BA– has been observed in Pheo-modified RCs of Rhodobacter sphaeroides R-26 [5]. The 1020-nm absorption band registered in these RCs was found to be characteristic for the radical anion of BChl. The mutual arrangement of PA, PB, and BA determined by X-ray analysis [1, 2] suggests several possibilities for the electron transfer pathway from P* to BA. To study the electron transfer from P* to BA, femtosecond coherent spectroscopy and frequency analysis of vibration and rotation of the molecular groups involved in charge separation have been applied [9, 10]. This approach in combination with chemical chromophore exchange and site-directed mutagenesis of the RC protein proved to be very useful.
Excitation of P by extremely short (<30 fsec) pulses creates a superposition of several vibrational wavefunctions, or a wavepacket. This nuclear wavepacket moves on the potential energy surface of P* with frequency corresponding to the energy difference between the vibrational levels involved. Such motions were first visualized by measurements of the coherent oscillations in the kinetics of stimulated emission from P* [9]. It has been shown that the motion of the wavepacket to the long-wavelength emission region on the P* potential energy surface is accompanied by reversible electron transfer from P* to BA with a formation of the 1020-nm absorption band of BA– [10]. From measurements on dry films of Rba. sphaeroides R-26 RCs and on deuterated RCs of Rba. sphaeroides R-26, it has been suggested that the 32-cm–1 fundamental mode and its overtones, observed in the kinetics of the 1020-nm band of BA– and of the 935-nm stimulated emission from P*, reflect the rotation of a water molecule [10]. It has been proposed that one of the molecular pathways for the electron transfer from P* to BA in Rba. sphaeroides RCs might be along the chain of polar groups according to Protein Data Bank (file 1AIJ): Mg(PB)–N–C–N(HisM202)–HOH(55)–O=(BA).
The stabilization of the state P+BA– is completed within ~1.5 psec in Pheo-modified and native RCs of Rba. sphaeroides R-26 at 90 K [10]. The mechanism of the stabilization is not yet completely clear. One possibility is related to a reorientation of the polar groups of the amino acid residues located near P and BA. For example, it might be the OH group of tyrosine located at position M210 in the vicinity of P and BA in Rba. sphaeroides RCs [1, 2].
In the present work, we study a role of tyrosine M210 in charge separation and stabilization of separated charges by measuring the oscillations in kinetics of decay of stimulated emission from P* and of a population of the primary charge-separated state P+BA– in YM210L and YM210L/HL168L mutant RCs of Rba. sphaeroides. The YM210L mutation replaces tyrosine M210 by apolar leucine. In the YM210L mutant of Rba. sphaeroides, the redox potential of P+/P lies approximately 30 mV above that of native RCs [11]. The lifetime of P* in this mutant is increased to 190 psec at 10 K in comparison with 1-2 psec for native RCs of Rba. sphaeroides [12]. The HL168L mutation replaces histidine L168 by leucine. In the HL168L(F) mutant of Rba. sphaeroides, the redox potential of P+/P lies 123(115) mV below that of native RCs [12], which might cause some acceleration of the P* decay. Indeed, in the HL168F mutant of Rba. sphaeroides the lifetime of P* is ~0.8 psec [13]. In the HL168F mutant of Blastochloris viridis the lifetime of P* is as short as 0.25 psec at 30 K in comparison with 1.1 psec for native RCs of Blastochloris viridis [14].
We found in the present work that the kinetics of P* decay at 940 nm of the YM210L and YM210L/HL168L mutant RCs of Rba. sphaeroides showed a significant slowing of the primary charge separation reaction in comparison with native RCs. Formation of a very small absorption band of BA– at 1020 nm is registered in RCs of both mutants. The P* decay and the formation of the BA– band is accompanied by damped oscillations. The Fourier transform (FT) spectrum of P* oscillations for the YM210L/HL168L mutant is closer to the analogous spectrum for native Rba. sphaeroides than to the FT spectrum for the YM210L mutant. The kinetics of the BA– absorption band at 1020 nm in the mutant YM210L/HL168L RCs show a weak stabilization of the P+BA– state accompanied by distinct oscillations that are not damped even at delays more than 2 psec. On the contrary, the analogous kinetics for the YM210L mutant show the absence of P+BA– state stabilization and complete damping of the oscillations within 1.5 psec after excitation. The amplitude of the oscillations at 1020 nm in the RCs of the YM210L/HL168L mutant is somewhat larger than for the YM210L mutant but much smaller than for the native RCs of Rba. sphaeroides. The FT spectrum of the 1020 nm oscillations for the YM210L/HL168L mutant is very rich in the 16-150 cm–1 range.
These data are discussed in terms of dynamic stabilization of separated charges in the state P+BA– by reorientation of the OH group of tyrosine M210 and of the HOH55 molecule in Rba. sphaeroides RCs.
MATERIALS AND METHODS
The HL168L and YM210L mutations were introduced in the pufL gene encoding the L protein subunit and the pufM gene encoding the M protein subunit of the reaction center of Rba. sphaeroides using bacterial strains and plasmids as described in [15]. Mutant and native RCs of Rba. sphaeroides were isolated by treatment of membranes with LDAO followed by DEAE-cellulose chromatography [16]. The RCs were suspended in 10 mM Tris-HCl, pH 8.0, 0.1% Triton X-100 buffer. Low temperature (90 K) measurements were performed on samples containing 65% glycerol (v/v). The optical density of the samples was 0.5 at 860 nm at room temperature. Sodium dithionite (5 mM) was added to keep the RCs in the state PBAHAQA–.
Femtosecond transient absorption was measured with a Tsunami Ti:sapphire mode-locked laser (Spectra Physics, USA). The femtosecond pulses were amplified by a home-built Ti:sapphire amplifier. The output of the amplifier was used to generate a white-light continuum. Then the continuum was divided into the major and minor parts. The major part was filtered and used as a pump beam in the standard pump–probe scheme. The minor part was used as a probe beam. After passing the sample, the probe beam was directed into a spectrograph coupled to a CCD matrix camera. The operating frequency of the femtosecond spectrometer was 15 Hz. The duration of pump and probe pulses was 16-18 fsec. Spectrally broadband pump pulses were centered at 870 nm. The delay between pump and probe pulses was changed in the range of 0-4 psec with an accuracy of ~1 fsec. The temporal dispersion in the 940-1060 nm range was compressed to less than 30 fsec.
Transient absorption difference (light-minus-dark) spectra were obtained by averaging 5000-10,000 measurements at each time delay. The accuracy of absorbance measurements was (1-2)·10–5 optical density units. The amplitude of the spectral band at 1020 nm was taken at its maximum after subtraction of the superimposed broad background (Fig. 1). The kinetics of absorbance changes (ΔA) at fixed wavelength were plotted using the measured difference spectra. Polynomial non-oscillating fits were subtracted from the kinetics. The residual oscillatory parts of the kinetics were Fourier transformed to obtain the frequency spectra of oscillations. The fits corresponded to minimal amplitude of the oscillations and to minimal noise in FT spectra.
RESULTS
Figure 1 shows the spectra of absorbance changes (ΔA) measured in RCs of the Rba. sphaeroides mutants YM210L/HL168L (a) and YM210L (b) at different femtosecond delays at 90 K in the range of 980-1060 nm. These difference (light-minus-dark) spectra are similar in shape and amplitude. The formation of the BA– absorption band centered at 1020 nm is observed for RCs of both mutants. This absorption band is, however, much weaker than that found for native Rba. sphaeroides RCs [10]. The amplitude of the 1020 nm band of both mutants varies with time in an oscillatory manner (Fig. 1; see Fig. 4 for more details).
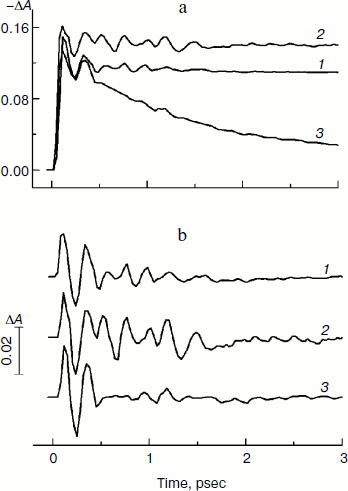
Figure 2 presents the kinetics of P* stimulated emission at 940 nm measured for the mutant YM210L/HL168L and YM210L RCs as well as for RCs of native Rba. sphaeroides at 90 K. The kinetics for the mutants are similar to each other and show very slow P* decay, whereas the P* decay in native RCs is much faster. Remarkable damped oscillations are seen in the kinetics of the mutant and native RCs at 940 nm (Fig. 2b). The characteristic time of damping is ~1 psec for the YM210L/HL168L mutant, ~1.5 psec for the YM210L mutant, and ~0.5 psec for the native Rba. sphaeroides. One can see seven distinct peaks of the oscillations in the YM210L/HL168L mutant kinetics, two intense and four or five weaker peaks of the oscillations in the YM210L mutant kinetics, and only two intense peaks of the oscillations in the native Rba. sphaeroides kinetics.Fig. 1. Difference (light-minus-dark) absorption spectra in the range of 980-1060 nm acquired at various femtosecond delays at 90 K in mutant YM210L/HL168L (a) and YM210L (b) RCs of Rba. sphaeroides. Here and in Figs. 2-5, the RCs were excited by 17-fsec pulses at 870 nm. Numbers indicate delays in femtoseconds.
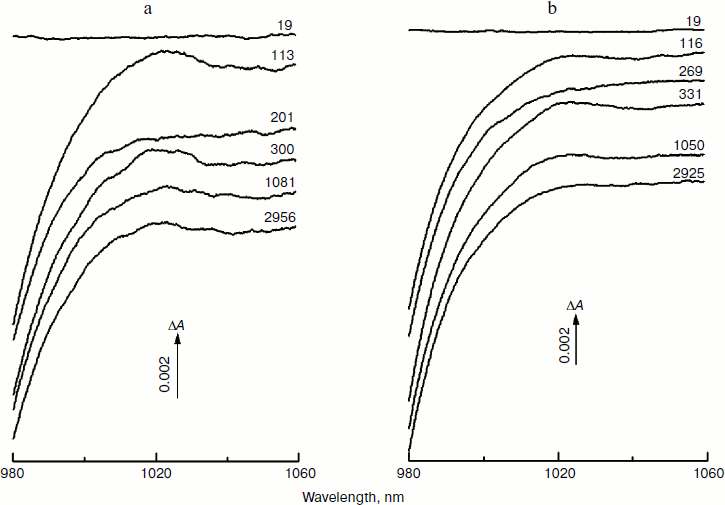
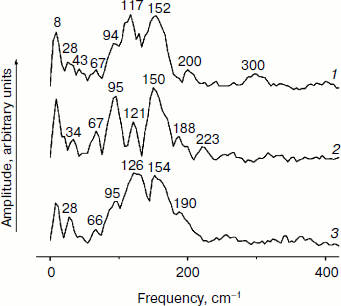
Figure 3 shows the Fourier transform spectra of oscillations from Fig. 2. These FT spectra are similar for mutant and native RCs with slight differences in the spectral position and amplitude of the peaks. The maxima of the three dominant peaks in the FT spectra have the frequencies around 94-95, 117-126, and 150-154 cm–1. The smaller peaks have maxima at 28, 34-43, 66-67, 188-200, 223, and 300 cm–1. The peak at 8 cm–1 seems to have an artificial nature and reflects an overall non-symmetry of oscillation curves. The shape of the FT spectrum for the YM210L/HL168L mutant is closer to the shape of the spectrum for native RCs. The main peaks in these two spectra are not separated as completely as in the FT spectrum of the YM210L mutant.Fig. 2. Femtosecond kinetics of ΔA (a) and its oscillatory part (b) for the 940-nm band of P* stimulated emission in mutant YM210L/HL168L (1), YM210L (2), and native (3) RCs of Rba. sphaeroides at 90 K.
In Fig. 4, the kinetics of ΔA at 1020 nm and the oscillatory parts of the kinetics are presented for the YM210L/HL168L and YM210L mutants and for the native Rba. sphaeroides RCs. The kinetics reflect the formation of the BA– absorption band that is accompanied by strong oscillations. The first, most intense peak in the 1020-nm kinetics is observed at 100-120 fsec delay for mutant and native RCs. The amplitude of this peak is as low as ~0.0005 optical density units in YM210L/HL168L and ~0.0003 optical density units in the YM210L mutants. These values are several times less than those for native RCs (Fig. 4, curve 3). Note that the 1020-nm absorption band is very weak in comparison with the P* stimulated emission band (Fig. 2) and with the analogous 1020-nm band of Pheo-modified RCs of Rba. sphaeroides (ΔA ~ 0.02 at 4-psec delay) [10]. A non-oscillating background of the coherent oscillations is clearly seen in the 1020-nm kinetics of the YM210L/HL168L mutant (Fig. 4, curve 1). This background reflects a quasi-exponential stabilization of the P+BA– state. This stabilization is almost absent in the YM210L mutant (Fig. 4, curve 2), in which the value of ΔA is as low as ~0.0001 at 3 psec delay. The non-oscillating background in the kinetics of native RCs reflects two very fast reactions: P* → P+BA– with a time constant of ~1.2 psec and P+BA– → P+HA– with a time constant of ~0.2-0.3 psec in accordance with earlier results [10]. The initial oscillations in the 1020-nm band with a period of ~200-220 fsec are in phase with the oscillations in the 940 nm band of P* stimulated emission (Fig. 2). The approximate damping time of the initial oscillations in the 1020-nm band is ~1 psec for mutant RCs and ~0.5 psec for native RCs. The low frequency oscillations are damped much slower: in the YM210L/HL168L mutant non-regular oscillations are observed even at more than 2 psec delay times, and in the YM210L mutant and native RCs the low-frequency peaks are seen at delays between 1 and 2 psec. The amplitude of the low-frequency component of the oscillations in the 1020-nm band does not exceed ~0.00015 in mutant RCs and 0.0005 in native RCs.Fig. 3. Spectra of Fourier transform of the oscillatory parts from Fig. 2b for the 940-nm band of P* stimulated emission in mutant YM210L/HL168L (1), YM210L (2), and native (3) RCs of Rba. sphaeroides at 90 K.
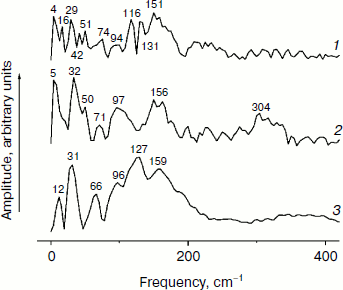
In Fig. 5, the Fourier transformed spectra of the oscillatory parts of the kinetics at 1020 nm from Fig. 4 are shown. The main difference from the analogous FT spectra of the oscillations at 940 nm (Fig. 3) is the increased amplitudes of the low-frequency modes. The FT spectrum of the YM210L/HL168L mutant is very rich in the range of 16-151 cm–1 (curve 1). The main peaks are observed at 29, 116, 131, and 151 cm–1, and smaller peaks are at 16, 42, 51, 74, and 94 cm–1. Some of these frequency modes are also observed in the FT spectrum of the oscillations at 940 nm (Fig. 3, curve 1). In the YM210L mutant, the FT spectrum of the oscillations at 1020 nm shows the main peaks at 32, 97, and 156 cm–1 and smaller peaks at 50, 71, 97, and 304 cm–1 (Fig. 5, curve 2). The 304-cm–1 mode seems to be close to the double frequency mode of 156 cm–1. The 32-cm–1 mode is not observed in the FT spectrum of the YM210L/HL168L mutant, but a number of other low frequency modes below 100 cm–1 are seen in the FT spectra of both mutants at slightly different frequencies. In native RCs, the FT spectrum of oscillations at 1020 nm shows peaks at 31-32, 66, 96, 127, 159, and 190 cm–1 in accordance with earlier results [10] (Fig. 5, curve 3). These frequencies are close to progression of overtones of the fundamental frequency at 32 cm–1. Note that this progression is not observed in the analogous spectra of the YM210L/HL168L and YM210L mutants.Fig. 4. Femtosecond kinetics of ΔA (a) and its oscillatory part (b) for the 1020-nm band of BA– absorption in mutant YM210L/HL168L (1), YM210L (2), and native (3) RCs of Rba. sphaeroides at 90 K.
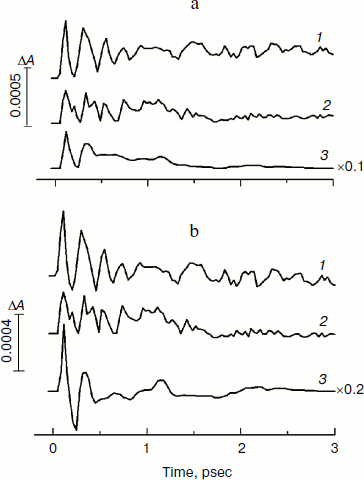
Fig. 5. Spectra of Fourier transform of the oscillatory parts from Fig. 4b for the 1020-nm band of BA– absorption in mutant YM210L/HL168L (1), YM210L (2), and native (3) RCs of Rba. sphaeroides at 90 K.
DISCUSSION
Stabilization of the separated charges in the state P+BA– is clearly observed in native RCs, while only partial stabilization is seen in the YM210L/HL168L mutant RCs and it is suppressed in the YM210L mutant (Figs. 2 and 4). A reason for this can be related to an increase in the free energy of P+BA– in both mutant RCs, whose energy can be slightly higher than that of P* due to replacement of the tyrosine YM210 by an apolar residue (leucine) [17]. In the HL168L mutant of Rba. sphaeroides, the redox potential of P+/P lies 123 mV below that of native RCs [12]. This can lower the free energy of P+BA– in the double mutant YM210L/HL168L and increase the stabilization of the separated charges in comparison with the single YM210L mutant. However, the experimentally observed effect of the addition of the HL168L mutation is much smaller than expected. The reversible femtosecond electron transfer from P* to BA is clearly seen in the 1020-nm kinetics of the YM210L/HL168L and YM210L mutants (Fig. 4). The reversibility of this electron transfer reflects a nuclear wavepacket motion on the P* potential energy surface. The wavepacket energy seems to be enough to approach the intercrossing area between the P* and P+BA– potential energy surfaces but not sufficient to provide a stabilization of P+BA– in the mutants.
Two possible mechanisms of the dynamic stabilization of P+BA– in native RCs can be considered. The first possibility is that an electron can be transferred from P* to the higher vibrational level on the P+BA– potential energy surface with subsequent vibrational relaxation to the lowest level [18]. This possibility requires non-symmetrical arrangement of the potential energy surfaces of P* and P+BA– with free energy difference between the two states ΔG of around –500 cm–1 [19, 20]. According to the well-known Marcus theory [21], the rate constant K for the charge separation reaction in the high-temperature limit can be written as:
where ħ is the Plank constant, V is the electronic coupling matrix element, λ is the reorganization energy, kB is the Boltzmann constant, T is temperature, and Ea = 0.25λ(1 + ΔG/λ)2 is the activation energy of the reaction. In RCs of native and mutant Rba. sphaeroides, it is reasonable to take the values of λ ~ 800 cm–1 and V ~ 30 cm–1 [14]. Then for T = 90 K and ΔG = –λ (activation-less regime) we obtain a maximal speed of electron transfer reaction with K = 1/0.74 psec–1. In native RCs of Rba. sphaeroides, the experimentally determined value of ΔG = –(350-550) cm–1 [19]. Taking the value of ΔG = –550 cm–1, we obtain Ea = 19.5 cm–1 and K = 1/1.1 psec–1, which that is close to the experimental value of K for native RCs at 90 K (Fig. 2). For ΔG = 110 cm–1, Ea = 253 cm–1 and expression (1) gives the value of K = 1/50 psec–1 that is approximately equal to the rate constant for the double mutant YM210L/HL168L (Fig. 2, curve 1). For ΔG = 240 cm–1, the activation energy Ea = 338 cm–1 and the calculated value of K = 1/190 psec–1 is close to the experimental value for YM210L mutant [12]. So, the increase in ΔG to positive values in the mutants YM210L/HL168L and YM210L leads to an increase in the activation energy of the reaction and strong decrease in the reaction rate. Formula (1) was obtained for the case of fast vibrational relaxation. If this relaxation is comparable or slower than the lifetime of the P+BA– state, then the rate of primary reaction is limited by the vibrational relaxation (usually several picoseconds).
The second possibility is that stabilization of P+BA– might be due to a reorientation of surrounding groups when the P+BA– state is reversibly formed. When the nuclear wavepacket approaches the intercrossing area of the P* and P+BA– surfaces, both states, P* and P+BA–, are registered. The wavepacket moves in an oscillatory manner on the P* surface if there are no additional non-coherent changes in the nuclear position. This seems to occur in mutants YM210L/HL168L and YM210L having several peaks of femtosecond oscillations in the formation of the P+BA– state (Fig. 4). In native RCs, the nuclear position can be changed non-coherently by reorientation of the surrounding polar groups like Oδ–Hδ+ of TyrM210 (Fig. 6). The motion of Hδ towards BA– could lower the energy of P+BA– with respect to that of P* and stabilize P+BA–. The Hδ+ of OH-TyrM210 can occupy two positions with respect to PA and BA having mostly positive and negative charges, respectively, in the state P+BA– [22]. In the first (“neutral”) position, dipole Oδ–Hδ+ of TyrM210 is placed perpendicular to a line running between C–N(IV) of PA and N(II) of BA. These atoms are the closest neighbors to TyrM210 and are mostly populated by positive and negative charges, respectively, in the state P+BA– [22]. The first position can be realized in the neutral states PBA or P*BA with close to zero Coulomb interaction. In the second position, Hδ+ of OH-TyrM210 is on a line connecting Oδ– of OH-TyrM210 and N(II) of BA. This position can be realized when P+BA– is stabilized due to the motion of Hδ+ towards BA attracted and repulsed by BA– and PA+, respectively.
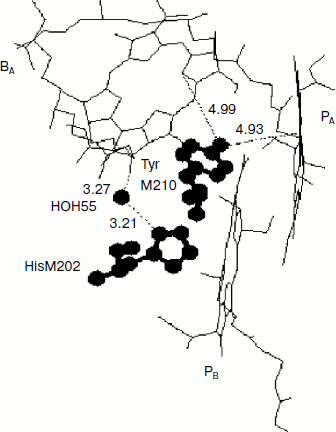
Rotation of the OH-group of TyrM210 around the C–O bond at T = 0 can be approximately described by the following equation [23]:Fig. 6. Fragment of the structure (Protein Data Bank, file 1AIJ) of special pair bacteriochlorophylls PA and PB, monomeric bacteriochlorophyll BA, and TyrM210 in Rba. sphaeroides RCs. Numbers are distances in Å. The oxygen of the OH-group of TyrM210 is located symmetrically between PA and BA and separated from C–N(IV) of PA (positively charged) and from N(II) of BA (negatively charged) by ~5 Å. The oxygen of HOH55 is separated from the oxygen of the keto carbonyl group of ring V of BA by 3.27 Å and from the nitrogen of HisM202 by 3.21 Å.
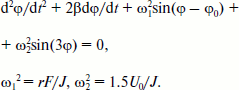 (2)
(2)
Here, φ is the angle of rotation, φ(t = 0) = 0, β is the frictional coefficient, ω12 = rF/J, r is the shortest distance from Hδ+ of the OH-group to the axe of rotation, F is the sum value of Coulomb forces acting between Hδ+ and P+ and BA–, J is the moment of inertia of the OH-group, ω22 = 1.5U0/J, U0 is the potential barrier of torsional rotation of the OH-group, φ0 is the final angle at t = in the case U0 = 0. For the OH-group of Tyr the value of U0 can be estimated as ~1000 cm–1 [24]. Note that the potential of elasticity of torsional rotation of the mentioned C–O bond can be approximately written as U = 0.5(1 – cos(3φ)). To achieve the values of φ > 60°, the system must get over the potential barrier of U0. At φ = 120, 240, 360° the value of U = 0. Equation (2) describes damping oscillations around the angle of equilibrium φeq, which can be obtained from the following formula:
In the case of small φ the frequency ω of the oscillations is given by the formula (4):
Numerical estimations give the value of ω/2π ~ 400 cm–1. The oscillations of the orientation of the OH-group can influence the free energy of the P+BA– state. These oscillations can be translated to the kinetics of the primary reaction via the dependence of the rate constant K on ΔG according to formula (1).
The Coulomb energy interaction between the charges for the mentioned above second position of Hδ+ was estimated to be ~600-900 cm–1. The experimental energy difference between P* and P+BA– in the stabilized state P+BA– in Pheo-modified RCs was found to be 350-550 cm–1 [19, 20]. So, the estimated energy difference between the two positions of Hδ+ is enough to stabilize the P+BA– state. The estimation of the time of shift of Hδ+ from the first to the second position due to Coulomb attraction and repulsion by charged atoms of BA– and PA+, respectively, gave the value of ~1-2 psec, which is close to the lifetime of P* in native RCs (Fig. 2, curve 3). Note that the attraction and repulsion of Hδ+ of OH-TyrM210 by BA– and PA+, respectively, occur when P+BA– is formed and are absent in the neutral state P*. The increase in the stabilization time with temperature [8] is consistent with the suggested mechanism since the interaction of Hδ+ of OH-TyrM210 with phonons can produce some additional Hδ+ motions leading to increase in the stabilization time.
Molecular dynamics calculations of the reorientation of the OH-group in TyrM210 of Rba. sphaeroides RCs show that the spectrum of autocorrelation function of stochastic rotations of the OH-group consists of several narrow (~4 cm–1) peaks in the range from 270 to 390 cm–1 with two main peaks at 356 and 368 cm–1 [25]. These peaks are absent in the analogous spectrum of mutant RCs without Tyr at position M210. Note that the spectrum of resonance Raman scattering of tyrosine does not contain any peaks in the range from 300 to 450 cm–1 [26]. This proves the idea that the reorientation of the OH-group of TyrM210 appears only when charge separation between P and BA takes place. The Fourier transform spectrum of oscillations in the BA– band of native RCs of Rba. sphaeroides at 1020 nm contains two small bands at 329 and 379 cm–1 with a width of ~20 cm–1 (damping time of ~1.5 psec) (Fig. 5, curve 3). Two small bands at 341 and 365 cm–1 are observed in the FT spectrum of oscillations in the P* stimulated emission band of native RCs of Rba. sphaeroides at 940 nm (Fig. 3, curve 3). These bands are absent in the analogous FT spectra of YM210L/HL168L and YM210L mutant RCs (Figs. 3 and 5) and in the YM210W mutant too [27]. In Pheo-modified RCs of Rba. sphaeroides, the FT spectrum of the oscillations at 940 and 1020 nm excited by 20-fsec pulses contains small bands at 340 and 325 cm–1 [10]. If the observed bands reflect the rotation of the OH-group of TyrM210 then small amplitudes of these bands might signify that the influence of the OH-group reorientation on the rate of charge transfer is small. The same conclusion was made in [25]: the presence of the polar OH-group of TyrM210 lowers the energy level of P+BA– by ~1500 cm–1 due to a static redistribution of charges in the near vicinity. The dynamic effect of reorientation of the OH-group due to charge separation was found to have a modest additive influence on the rate of primary reaction. The reasonable coincidence of estimations by formulas (1)-(4), molecular dynamic calculations, and measurements seems to support the suggestion that the stabilization of P+BA– is provided by non-coherent reorientation of Hδ+ of OH-TyrM210.
The double mutant YM210L/HL168L of Rba. sphaeroides gives an opportunity to see a key role of TyrM210 in the stabilization of the P+BA– state. The HL168L mutation changed the redox potential of P+/P to 123 mV below that of native RCs [12]. This is much more than the 30 mV increase in this potential caused by the YM210L mutation [11]. The resulting ~90 mV decrease of P+/P redox potential in the YM210L/HL168L mutant would provide a significant lowering of the P+BA– free energy level that would speed up the primary reaction P* → P+BA– and improve the stabilization of P+BA– state in comparison with the single YM210L mutant. But this is not a case. The P* lifetime in the YM210L/HL168L mutant is much longer than in native RCs (Fig. 2). The 1020-nm kinetics of the YM210L/HL168L mutant show a slight accumulation of BA– during 3 psec after excitation and approximately double increase in the amplitude of the initial oscillations in comparison with the YM210L mutant (Fig. 4). These facts mean that the absence of TyrM210 cannot be compensated by lowering of the P+BA– free energy. On the other hand, no information is available about the influence of the mentioned mutations on the BA/BA– redox potential. It is possible that the real change in the P+BA– free energy level in the YM210L/HL168L mutant in comparison with the YM210L mutant is not significant.
Another possibility of dynamic stabilization of the P+BA– state might be associated with crystallographically defined water molecule HOH55 located in the structure of purple bacteria RCs between PB and BA [28]. Water HOH55 is within hydrogen-bonding distance of both the oxygen of the 131-keto carbonyl group of BA and residue HisM202 that provides the axial ligand to the magnesium of the PB bacteriochlorophyll [28]. A hydrogen bond interaction with the 131-keto carbonyl group of BA would stabilize BA– in native RCs, particularly when P is oxidized during charge separation [29]. Resonance Raman spectroscopy has shown the presence of a relatively strong hydrogen bond between this group and HOH55 in the ferricyanide-oxidized wild-type RCs (4.1 kcal/mol or 1435 cm–1) [28]. It is important that in neutral RCs this bond is not detected.
Stochastically oriented water dipole can changes its orientation in the electric fields of P+ and BA– according to Eq. (2) with ω2 = 0. In the final orientation, the Hδ+ atom is closer to the 131-keto carbonyl group of BA that makes easier the possible formation of an H-bond. The time constant of H-bond formation can be estimated from molecular dynamics calculations as 0.1 psec [30]. The estimated energy that is necessary for changing the orientation of a water dipole by the angle of π is ~500-600 cm–1. The calculated frequencies of oscillations of water dipole orientation are in the range of ~100-130 cm–1 and depend on the initial orientation. In the case of absence of negative charge on BA these frequencies are ~50-65 cm–1. It is very difficult to detect these frequencies in the kinetics of BA– absorption because the frequency range of interest contains many different bands (Fig. 5). Nevertheless, it might be the 127-cm–1 band in the FT spectrum of native RCs (Fig. 5, curve 3) and 51 cm–1 band in the FT spectrum of YM210L mutant (Fig. 5, curve 2). The absence of the 127-cm–1 band and the presence of the 51-cm–1 band in YM210L spectrum might reflect the absence of negative charge on BA during the first picoseconds. In the GM203L mutant of Rba. sphaeroides water HOH55 is excluded from the RC structure, which leads to ~4-fold slowing of the electron transfer from P* to BA at 90 K [31].
Based on these facts, one can speculate that the interaction of Hδ+ from HOH55 with BA– appears as a response on the accumulating positive charge of P+. This interaction becomes stronger during the primary charge separation between P* and BA, providing a positive feedback of the stabilization process. It was found by femtosecond absorption spectroscopy that in native RCs of Rba. sphaeroides the primary reaction P* → P+BA– is accompanied by a rotation-like motion of water HOH55 with a frequency of ~32 cm–1 [10]. Similar frequency modes are observed in the kinetics of the BA– band of the YM210L/HL168L and YM210L mutants (Fig. 5). This type of motion might play a similar role in the stabilization process as a motion of the OH-group of TyrM210.
Support by the Russian Basic Research Foundation grant No. 08-04-00888 and by the program of basic research of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology” No. 10P is gratefully acknowledged.
REFERENCES
1.Michel, H., Epp, O., and Deisenhofer, J. (1986)
The EMBO J., 5, 2445-2451.
2.Komiya, H., Yeates, T. O., Rees, D. C., Allen, J.
P., and Feher, G. (1988) Proc. Natl. Acad. Sci. USA, 85,
9012-9016.
3.Maroti, P., Kirmaier, C., Wraight, C., Holten, D.,
and Pearlstein, R. M. (1985) Biochim. Biophys. Acta, 810,
132-141.
4.Shuvalov, V. A., Klevanik, A. V., Sharkov, A. V.,
Matveetz, Yu. A., and Krukov, P. G. (1978) FEBS Lett.,
91, 135-139.
5.Kennis, J. T. M., Shkuropatov, A. Ya., van Stokkum,
I. H. M., Gast, P., Hoff, A. J., Shuvalov, V. A., and Aartsma, T. J.
(1997) Biochemistry, 36, 16231-16238.
6.Sporlein, S., Zinth, W., Meyer, M., Scheer, H., and
Watchtveitl, J. (2000) Chem. Phys. Lett.,
322, 454-464.
7.Parson, W. W., and Warshel, A. (1987) J. Am.
Chem. Soc., 109, 6152-6163.
8.Fleming, G. R., Martin, J.-L., and Breton, J.
(1988) Nature (London), 333, 190-192.
9.Vos, M. H., Rappaport, F., Lambry, J.-C., Breton,
J., and Martin, J.-L. (1993) Nature, 363,
320-325.
10.Yakovlev, A. G., Shkuropatov, A. Ya., and
Shuvalov, V. A. (2002) Biochemistry, 41, 14019-14027.
11.Beekman, L. M. P., van Stokkum, I. H. M.,
Monshouwer, R., Rijnders, A. J., McGlynn, P., Visschers, R. W., Jones,
M. R., and van Grondelle, R. (1996) J. Phys. Chem., 100,
7256-7268.
12.Vos, M. H., Jones, M. R., Breton, J., Lambry,
J.-C., and Martin, J.-L. (1996) Biochemistry, 35,
2686-2693.
13.Rischel, C., Spiedel, D., Ridge, J. P., Jones, M.
R., Breton, J., Lambry, J.-C., Martin, J.-L., and Vos, M. H. (1998)
Proc. Natl. Acad. Sci. USA, 95, 12306-12311.
14.Huppman, P., Arlt, T., Penzkofer, H., Schmidt,
S., Bibikova, M., Dohse, B., Oesterhelt, D., Wachtveit, J., and Zinth,
W. (2002) Biophys. J., 82, 3186-3197.
15.Vasilieva, L. G., Bolgarina, T. I., Khatypov, R.
A., Shkuropatov, A. Ya., and Shuvalov, V. A. (2001) Dokl. Ros. Akad.
Nauk, 376, 826-829.
16.Shuvalov, V. A., Shkuropatov, A. Y., Kulakova, S.
M., Ismailov, M. A., and Shkuropatova, V. A. (1986) Biochim.
Biophys. Acta, 849, 337-348.
17.Nagarajan, V., Parson, W. W., Davis, D., and
Schenck, C. (1993) Biochemistry, 32, 12324-12336.
18.Bixon, M., Jortner, J., Plato, M., and
Michel-Beyerle, M. E. (1988) in The Photosynthetic Bacterial
Reaction Center: Structure and Dynamics (Breton, J., and Vermeglio,
A., eds.) Plenum Press, NY-London, pp. 399-419.
19.Shuvalov, V. A., and Yakovlev, A. G. (1998)
Membr. Cell Biol., 12, 563-569.
20.Nowak, F. R., Kennis, J. T. M., Franken, E. M.,
Shkuropatov, A. Ya., Yakovlev, A. G., Gast, P., Hoff, A. J., Aartsma,
T. J., and Shuvalov, V. A. (1998) Proc. XI Int. Congr. on
Photosynthesis, Budapest, Hungary, Kluwer, Academic Publishers,
Dordrecht, pp. 783-786.
21.Marcus, S. A., and Sutin, N. (1985) Biochim.
Biophys. Acta, 811, 265-322.
22.Plato, M., Lendzian, F., Lubitz, W., Trankle, E.,
and Mobius, K. (1988) in The Photosynthetic Bacterial Reaction
Center: Structure and Dynamics (Breton, J., and Vermeglio, A.,
eds.) Plenum Press, NY-London, pp. 379-388.
23.Rubin, A. B. (2000) Biophysics [in
Russian], Nauka, Moscow.
24.Volkenstein, M. V. (1981) Biophysics [in
Russian], Nauka, Moscow.
25.Parson, W. W., and Warshel, A. (2009) in The
Purple Phototrophic Bacteria (Hunter, C. N., Daldal, F., Thurnauer,
M. C., and Beatty, J. T., eds.) Springer Science + Business Media
B. V., Amsterdam, pp. 355-377.
26.Nabiev, I. R., Efremov, R. G., and Chumanov, G.
D. (1988) Uspekhi Fiz. Nauk, 154, 459-496.
27.Yakovlev, A. G., Vasilieva, L. G., Shkuropatov,
A. Ya., Bolgarina, T. I., Shkuropatova, V. A., and Shuvalov, V. A.
(2003) J. Phys. Chem. A, 107, 8330-8338.
28.Potter, J. A., Fyfe, P. K., Frolov, D., Wakeham,
M. C., van Grondelle, R., Robert, B., and Jones, M. R. (2005) J.
Biol. Chem., 280, 27155-27164.
29.Fyfe, P. K., Ridge, J. P., McAuley, K. E.,
Cogdell, R. J., Isaacs, N. W., and Jones, M. R. (2000)
Biochemistry, 39, 5953-5960.
30.Voloshin, V. P., Jeligovskaya, E. A., Malenkov,
G. G., Naberuhin, Y. I., and Tuitik, D. L. (2001) Ros. Khim.
Zh., XLV, No. 3, 31-37.
31.Yakovlev, A. G., Jones, M. R., Potter, J. A.,
Fyfe, P. K., Vasilieva, L. G., Shkuropatov, A. Ya., and Shuvalov, V. A.
(2005) Chem. Phys., 319, 297-307.