Novel Mitochondria-Targeted Antioxidants, “Skulachev-Ion” Derivatives, Accelerate Dermal Wound Healing in Animals
I. A. Demianenko1,3, T. V. Vasilieva1,3, L. V. Domnina2,3, V. B. Dugina2,3, M. V. Egorov3, O. Y. Ivanova2,3, O. P. Ilinskaya1,3, O. Y. Pletjushkina2,3, E. N. Popova2,3, I. Y. Sakharov4, A. V. Fedorov1,3, and B. V. Chernyak2,3*
1Biological Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: bchernyak@yahoo.com
3Institute of Mitoengineering, Lomonosov Moscow State University, 119991 Moscow, Russia
4Chemical Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received November 30, 2009
It is shown that the novel mitochondria-targeted antioxidant SkQ1, (10-(6′-plastoquinonyl) decyltriphenylphosphonium) stimulates healing of full-thickness dermal wounds in mice and rats. Treatment with nanomolar doses of SkQ1 in various formulations accelerated wound cleaning and suppressed neutrophil infiltration at the early (7 h) steps of inflammatory phase. SkQ1 stimulated formation of granulation tissue and increased the content of myofibroblasts in the beginning of regenerative phase of wound healing. Later this effect caused accumulation of collagen fibers. Local treatment with SkQ1 stimulated re-epithelization of the wound. Lifelong treatment of mice with SkQ1 supplemented with drinking water strongly stimulated skin wounds healing in old (28 months) animals. In an in vitro model of wound in human cell cultures, SkQ1 stimulated movement of epitheliocytes and fibroblasts into the “wound”. Myofibroblast differentiation of subcutaneous fibroblasts was stimulated by SkQ1. It is suggested that SkQ1 stimulates wound healing by suppression of the negative effects of oxidative stress in the wound and also by induction of differentiation. Restoration of regenerative processes in old animals is consistent with the “rejuvenation” effects of SkQ1, which prevents some gerontological diseases.
KEY WORDS: mitochondria-targeted antioxidants, SkQ1, myofibroblasts, inflammation, skin woundsDOI: 10.1134/S000629791003003X
Abbreviations: DMEM, Dulbecco’s modified Eagle’s medium; ICAM1, intercellular adhesion molecule-1; MitoQ, 10-(6′-ubiquinonyl) decyltriphenylphosphonium; ROS, reactive oxygen species; SkQ1, 10-(6′-plastoquinonyl) decyltriphenylphosphonium; TGFβ1, transforming growth factor β1; TNFα, tumor necrosis factor α; VCAM1, vascular cell adhesion molecule-1.
Wound healing is a complex reaction of the organism to injury. Various
cell types including leucocytes, epitheliocytes, fibroblasts, and
macrophages and humoral factors (cytokines) are involved in this
process [1]. Three major phases of wound healing
are distinguished: inflammation, regeneration (formation of granulation
tissue), and tissue remodeling (epithelization and scar formation).
Neutrophils migrate to the wound during the first hours, while
monocytes and tissue macrophages migrate later. Formation of
granulation tissue begins after cessation of inflammation and cleaning
of the wound. This tissue is formed mostly by fibroblasts that
differentiate to myofibroblasts in the wound [2, 3]. This phase is followed by vascularization of
granulation tissue and epithelization of the wound. The latter process
depends on migration, proliferation, and differentiation of epithelial
cells.
Production of reactive oxygen species (ROS) in the wound plays an important bactericidal role as well as a role in cell signaling [4-7]. The concentration of hydrogen peroxide reaches hundreds of micromoles per liter and then slowly declines. In the regeneration phase formation of granulation tissue and vascularization are stimulated by ROS at low concentration. Decline of ROS is not effective in chronic inflammation, and this is one of the major reasons for unhealed wound formation in diabetes, atherosclerosis, and in elderly patients. Unhealed and chronic wounds accompanying various pathologies remain a serious medical problem. In some cases (in diabetes, in particular [8]) antioxidants stimulate wound healing. Development of a new wound cover with antioxidant activity is an important direction in wound therapy. This cover causes cessation of inflammation at an early phase and stimulates regenerative processes at the later phase of wound healing.
NADPH oxidase of phagocytes is the major source of extracellular ROS during inflammation [9, 10]. Oxidative stress induced by exogenous oxidants results in endogenous ROS formation predominantly in mitochondria [11]. The new efficient mitochondria-targeted antioxidants (SkQ family) have been developed recently by Professor V. P. Skulachev and coworkers [12-15]. These compounds consist of an antioxidant part (plastoquinone) and a penetrating cation (“Skulachev ion”). The cation drives selective accumulation of SkQ in mitochondria and imparts high efficiency to these antioxidants at extremely low concentrations. High efficiency of SkQ was confirmed in experiments with cell cultures [13, 16] and with the animals [17-19]. The new antioxidants demonstrated significant therapeutic action in model pathologies of heart, kidney, brain, and eye. Clear geroprotective effects of SkQ were shown in experiments with invertebrates (Ceriodaphnia and Drosophila) and with mice [19].
In the present work the effect of mitochondria-targeted antioxidant SkQ1 (10-(6′-plastoquinonyl) decyltriphenylphosphonium) on healing of full-thickness dermal wounds in animals was studied.
MATERIALS AND METHODS
Wound healing was studied in three series with different SkQ1 applications in mice and rats. Full-thickness dermal wounds were made under ether anesthesia or after intraperitoneal injection of ketamine (80 mg/kg) and xylazine (10 mg/kg). Animals were kept in plastic cages under standard temperature, light, and feeding regimes.
In the first series of experiments, outbred white rats (170-200 g) were daily injected subcutaneously around the wound with 200 nM SkQ1 solution in 50 µl of 0.9% NaCl. In the control group animals were injected with 0.9% NaCl. Samples for histological analysis were collected after 7 h, 1-3, 5, and 14 days.
In the second series of experiments in Sprague–Dawley rats (450-520 g) wounds were treated with film cover Bioplen [20] containing SkQ1 (0.019 µg per g film). The cover was changed every three days during 14 days. In control group wounds were treated with the same cover but without SkQ1. The samples for histological analysis were collected after 14 days.
In the third series of experiments old (28 months) BALB/c mice were administered SkQ1 in drinking water (200 nM, equivalent to daily dose 30 nmol/kg) starting from three months. The control groups contained young (three months) and old (28 months) mice not treated with SkQ1. Samples for histological analysis were collected after nine days.
Wound surface area was measured in photographs using ImageJ (National Institutes of Health (NIH); http:/rsb.info.nih.gov/ij/).
For histological analysis tissue samples were fixed with Bouin's fixative, dehydrated, and embedded in paraffin. Cross-sections (4 µm width) were stained with hematoxylin and eosin (Mallory’s connective tissue stain). Neutrophils were counted in 100 fields of vision. For immunostaining, slices were treated with 3% H2O2 for 10 min to suppress endogenous peroxidase, treated with monoclonal antibodies against smooth muscle α-actin (DAKO, USA) and biotinylated horse antibodies against mice IgG (Vector, USA), and stained with avidin–peroxidase conjugate (Vector) diaminobenzidine solution (Vector). Images were analyzed with a Leica DM 5000 B (Germany) microscope equipped with Leica DFC 320 (Germany) digital camera.
Model of aseptic inflammation. CD1 mice (35-40 g) were supplied with SkQ1 (200 nM, equivalent to daily dose 30 nmol/kg) for three weeks before operation and then for up to three days of experiment. Inflammation was induced by intrusion of round cover glass (12 mm diameter; Fisher, USA) in the interscapular region. Glasses were removed after 12 h and 2 and 3 days.
Cell culture of rat IAR2 epitheliocytes (a kind gift from Dr. P. Montesano [21]) and human subcutaneous fibroblasts (Russian Cell Culture Collection, Institute of Medical Genetics, Russian Academy of Sciences) were grown in DMEM (Dulbecco’s modified Eagle’s medium) (Gibco, USA) supplemented with 10% fetal calf serum (Hyclone, USA), penicillin (100 U/ml), and streptomycin (100 U/ml) at 37°C in a humidified atmosphere containing 5% CO2. Cells were incubated with 20 nM SkQ1 for seven days and transferred to Petri dishes with cover glasses in the same medium without SkQ1. Conditioned medium was collected three days after the transfer. Cells on the cover glasses were grown to form monolayers. Then half of the monolayer was removed, cells were washed with PBS and placed in the growth medium for 24 or 48 h. The conditioned medium was diluted three times with the growth medium and added to the cells after preparation of the “wound”. The samples were fixed with 2% paraformaldehyde in DMEM (pH 7.2) and then with methanol (–20°C). For immunostaining monoclonal antibodies against smooth muscle α-actin (DAKO) and rabbit antibodies against actin (Sigma, USA) were applied. Secondary goat antibodies against mouse IgG were conjugated with rhodamine or with fluorescein (Sigma). Images were analyzed with an Axiovert microscope (Carl Zeiss, Germany) equipped with Axiocam camera (Carl Zeiss) using ImageJ (National Institutes of Health (NIH); http:/rsb.info.nih.gov/ij/).
Statistical analysis was done with STATISTICA 7.0 software. The data were expressed as mean ± standard deviation. Student’s unpaired t-test and Mann–Whitney U-test were conducted for comparisons, and significance was set at level p < 0.05 (*) or p < 0.001 (**).
RESULTS
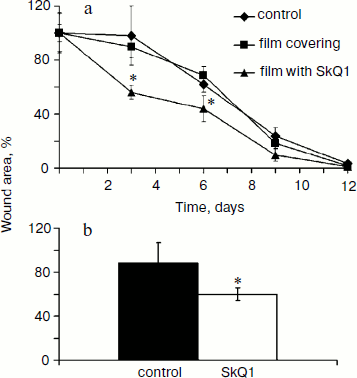
SkQ1 accelerates healing of full-thickness dermal wounds. The effect of SkQ1 on wound healing was studied in two models with different methods of drug delivery (see “Materials and Methods”). SkQ1 incorporated in film wound covering Bioplen significantly accelerated wound healing at every phase of the process. Significant acceleration of wound closure was observed at the fourth and seventh day of the experiment (Fig. 1a). Injections of SkQ1 (200 nM) also accelerated wound closure observed at the fifth day (Fig. 1b).
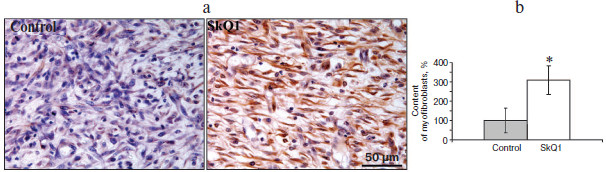
Myofibroblasts play an important role in the formation of granulation tissue; they express smooth muscle α-actin and produce an excess of extracellular matrix proteins [2, 3]. Independently from the method of delivery, SkQ1 caused 3-fold increase (p < 0.05) in myofibroblast content in granulation tissue (Fig. 2; see color insert). Histological analysis of protracted wounds on the 14th day of the experiment demonstrated that SkQ1 incorporated in the film wound covering increased the size of collagen fibers (not shown). This could be the result of myofibroblast activity.Fig. 1. SkQ1 stimulated healing of full-thickness dermal wounds. Rats were treated with SkQ1 incorporated in film covering (0.019 µg per g of film) (a) or injected (200 nM) in the wound area (b). The size of wounds was determined from photographs. * p < 0.05.
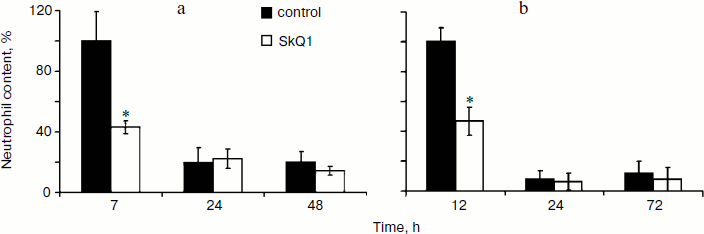
Effect of SkQ1 on inflammatory phase of wound healing. Visual observations of the wounds of animals revealed a pronounced weakening of the signs of inflammation in both models of SkQ1 delivery. Accelerated wound cleaning and decrease of edema were observed at the early phase of wound healing (not shown). Analysis of neutrophil infiltration revealed more than 2-fold increase (p < 0.05) in neutrophil content at 7 h after operation in SkQ1 treated animals (Fig. 3a). The neutrophil content dropped to the initial level 24 h after the operation.Fig. 2. SkQ1 stimulated accumulation of myofibroblasts in the wound. Rats were treated with injections of SkQ1. Staining of transverse sections of wound samples on the fifth day after operation with antibodies against smooth muscle α-actin (brown stain). The slices were co-stained with hematoxylin (a); b) content of myofibroblasts in wound. * p < 0.05.
The effect of SkQ1 on neutrophil infiltration was studied in more detail in the model of aseptic inflammation where a sterile cover glass was introduced subcutaneously. Treatment with SkQ1 (30 nmol/kg per day with drinking water) decreased 2-fold (p < 0.05) the neutrophil content in the inflammation area 14 h after operation (Fig. 3b). Thus in both models SkQ1 suppressed neutrophil infiltration at the early stages of inflammation.Fig. 3. SkQ1 affected neutrophil infiltration in the inflammatory focus: a) dermal wound; b) aseptic inflammation. The content of neutrophils was compared to the maximal value in both models. * p < 0.05.
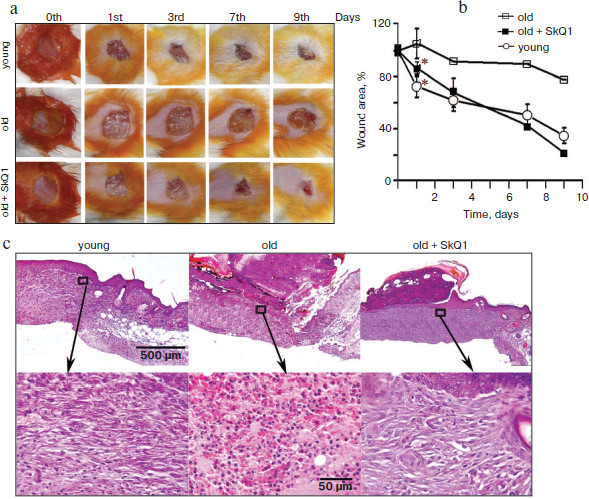
Lifelong treatment with SkQ1 accelerates wound healing in aged mice. The aged are characterized by a violation of the repair processes. Wound healing is accompanied with prolonged inflammation and takes much more time than in young counterparts. It was shown earlier that lifelong treatment with SkQ1 decelerated development of various features of aging and increased lifespan of mice [19]. We have studied the effect of lifelong treatment with SkQ1 (30 nmol/kg per day with drinking water) on wound healing in old (28 months) mice. It was shown that closure of the wound after 24 h was significantly (p < 0.05) more pronounced in SkQ1 treated mice than in control mice of the same age (Fig. 4, a and b; see color insert). Unfortunately, poor survival of the aged mice did not allow statistical analysis at the later stages, but significant acceleration of wound healing in SkQ1 treated mice was clearly visible. The maximal effect was observed at the ninth day after operation. Histological analysis of the wound in old mice without SkQ1 treatment revealed the absence of granulation, neutrophil infiltration, and poor epithelization on the ninth day after operation, while in SkQ1 treated mice complete epithelization and formation of connective tissue was observed. Collagen fibers were pronounced but less mature than in the healed wounds of young mice (Fig. 4c; see color insert).
SkQ1 stimulates healing of “wound” in in vitro models. To study the molecular mechanisms of SkQ1 action, we have studied the effects of SkQ1 in the model of wound in cultures of fibroblasts and epithelial cells.Fig. 4. Lifelong treatment with SkQ1 (30 nmol/kg per day) accelerated full-thickness dermal wound healing in old (28 months) mice. Young (three months) or old (28 months) mice untreated with SkQ1 were used as controls. a) Wound closure (photo); b) plots of wound area decrease, * p < 0.05. c) Histological sections on the ninth day after operation: general view and magnified fragments of granulation tissue. Staining with hematoxylin and eosin.
It was shown that SkQ1 (20 nM) significantly (2-fold) stimulates movement of human subcutaneous fibroblasts into the “wound” made in a cellular monolayer. In the presence of SkQ1 the content of myofibroblasts (cells expressing smooth muscle α-actin) was increased, especially near the “wound” (Fig. 5; see color insert). These data are in complete agreement with increased accumulation of myofibroblasts in wounds of SkQ1 treated animals (Fig. 2).
Significant acceleration of cell movement into the “wound” was shown in monolayer of immortalized rat IAR2 epithelial cells treated with SkQ1 (Fig. 6, a and b). In this model, “wound” closure depended on proliferation as well as on migration of the cells. It was shown that SkQ1 did not increase the content of cells in metaphase of mitosis (2.9 vs. 2.5% in control vs. SkQ1 treated cells). These data indicate that stimulation of “wound” closure in monolayer of epithelial cells by SkQ1 was mediated by accelerated movement of the cells.Fig. 5. SkQ1 stimulated closure of “wound” in monolayer of human subcutaneous fibroblasts. Analysis on the second day after operation. Bar, 20 µm. a) Immunostaining of smooth muscle α-actin (green) and total actin (red); arrow indicates the direction of migration. The image is composed from fragments of the “wound” area. b) Concentration-dependence of the effect of SkQ1 on migration of fibroblasts into the “wound”. ** p < 0.001.
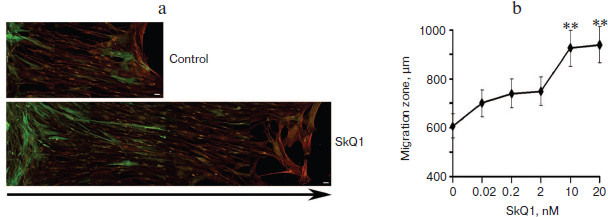
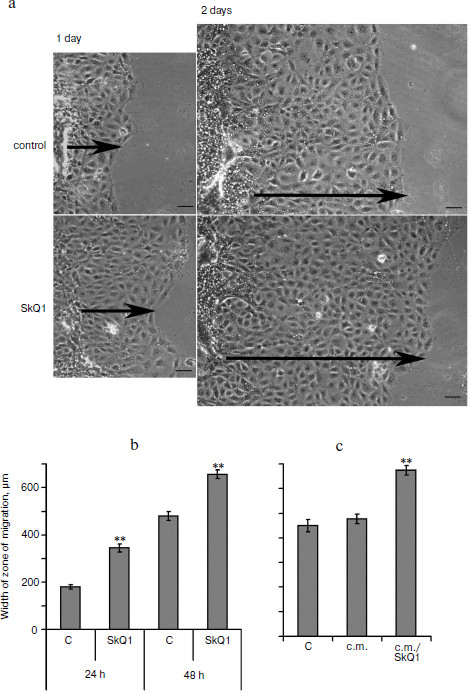
It was found that the conditioned medium of IAR2 cells treated with SkQ1 also stimulated closure of the “wound” (Fig. 6c) indicating that the effect of SkQ1 was mediated by increased production or activity of some cytokines in the growth medium.Fig. 6. SkQ1 (20 nM) stimulated migration of IAR2 rat epithelial cells into “wound” made in the cellular monolayer. a) Migration of cells 24 and 48 h after “wounding” (phase contrast). Arrows indicate the directions of migration. Bar, 100 µm. b) Morphometric analysis of zone of migration. c) Effect of conditioned medium (c.m.) from cells treated with SkQ1 on migration of naïve cells into the “wound”. ** p < 0.001.
DISCUSSION
The data presented above indicated that local treatment with low (nanomolar) doses of mitochondria-targeted antioxidant SkQ1 stimulated wound healing. Significant effects of SkQ1 were observed at the early inflammatory phase and the phase of regeneration including formation of granulation tissue and epithelization. In the models of full-thickness dermal wound and in the model of aseptic inflammation, SkQ1 significantly decreased the content of neutrophils in the area of inflammation at the early stages (7-12 h) of the process (Fig. 3). This effect could be mediated by suppression of inflammation or with acceleration of the inflammatory phase. It is known that inflammation is accompanied with increase in ROS content in the wound [4-7]. The main source of ROS during inflammation is NADPH-oxidase in plasma membrane of neutrophils and macrophages [9, 10]. ROS produced by these cells exert bactericidal action and support the process of inflammation by transmitting intercellular signals [6, 7, 22-30]. Traditional antioxidants suppress neutrophil infiltration by scavenging of extracellular ROS in the wound. The possible role of mitochondrial ROS production in inflammation remains unexplored. Recently in two groups it was found that mitochondrial ROS were involved in inflammatory signaling in cells of vascular endothelium [31, 32]. It was shown that mitochondria-targeted antioxidant MitoQ (10-(6′-ubiquinonyl) decyltriphenylphosphonium) (similar in action to SkQ1) inhibited accumulation of ROS and secretion of inflammatory cytokines (IL-1β, IL-6, IL-8) in animal model of sepsis [31]. Expression of ICAM1 (intercellular adhesion molecule-1) in endothelial cells treated with inflammatory cytokine TNFα (tumor necrosis factor α) was suppressed by 3-10 nM MitoQ [32]. Higher doses of MitoQ (30-100 nM) enhanced the effect of TNFα, probably, due to the prooxidant action of this compound. It was shown earlier that traditional antioxidants suppressed expression of adhesion molecules (ICAM1, VCAM1 (vascular cell adhesion molecule-1), E-selectin) and chemokines (IL-8) during inflammation [33-37]. These effects were mediated by inhibition of redox-sensitive signaling pathways, which resulted in activation of stress-kinases (p-38, JNK) and transcription factor NF-κB [33-37]. It can be suggested that the effect of SkQ1 on inflammation was mediated by the same signaling during wound healing.
Treatment with SkQ1 significantly decreased the size of the wound at the stage of formation and maturation of granulation tissue (fifth day) (Fig. 1). Histological analysis of the wound tissues revealed accelerated myofibroblast differentiation of subcutaneous fibroblasts and migration of epithelial cells into the wound (epithelization) induced by SkQ1. Similar effects of SkQ1 were observed in the “wound” model in vitro in cell cultures. It is known that myofibroblasts differentiate from the tissue fibroblasts and from the progenitors that migrate into the wound. These cells perform three main functions during wound healing: contraction of the wound edges due to high contractility of actin stress-fibers and strong attachment to the extracellular matrix, scar formation due to increased production of collagen, and secretion of the growth factors and cytokines [2, 3]. The content of myofibroblasts in the wound on the fifth day after operation was significantly higher in rats treated with SkQ1 than in the control group (Fig. 2). An increase in content and size of the collagen fibers in wounds treated with SkQ1 was observed on the 14th day (not shown) and probably was related to activity of myofibroblasts. One can suggest that early appearance of myofibroblasts in SkQ1 treated wounds depended on shortening of the inflammatory phase, but the experiments with the culture of subcutaneous fibroblasts demonstrated that SkQ1 could directly stimulate myofibroblast differentiation.
It was shown that SkQ1 stimulated the formation of myofibroblasts at the edge of the “wound” in cellular monolayer (Fig. 5). Moreover, SkQ1 stimulated migration of fibroblasts into the “wound” in vitro. It is known that myofibroblasts have large area of attachment and developed focal adhesions that decrease individual motility of these cells [38, 39]. It could be suggested that accumulation of myofibroblasts at the edge of the “wound” resulted in expulsion of the cells from monolayer into the “wound”, accelerating their directed movement. Accumulation of myofibroblasts is a temporary event during normal wound healing. Defects in elimination of myofibroblasts result in excessive accumulation of collagen and formation of hypertrophic scars. Future studies are necessary to clarify the effect of SkQ1 at the later steps of wound healing.
In the culture of IAR2 epithelial cells, SkQ1 also stimulated migration of the cells into the “wound” (Fig. 6) in agreement with accelerated epithelization of wounds in animal models. Accelerated movement in the culture of IAR2 was not accompanied by decomposition of epithelial layer. The experiments with conditioned medium demonstrated that SkQ1 induced production by epithelial cells of some cytokines that stimulated their movement (Fig. 6). We found earlier that SkQ1 stimulated activity of TGFβ1 (transforming growth factor β1) in culture of human subcutaneous fibroblasts that resulted in myofibroblast differentiation (Popova et al., unpublished). However, in experiments with epithelial cells neutralization of TGFβ1 in the growth medium did not block the stimulatory effect of SkQ1 (not shown). No signs of mesenchymal differentiation of epithelial cells treated with SkQ1 (which are inherent to the action of TGFβ1 [40]) were observed. Moreover in SiHa carcinoma cells and in IAR2 cells transformed with oncogene N-Ras, SkQ1 induced restoration of some features of epithelium (cell morphology, E-cadherin containing intercellular contacts) [16]. These data indicated that SkQ1-stimulated migration of epithelium into the wound was mediated by some unknown cytokines rather than by TGFβ1.
Wound healing in aged animals is greatly hampered due to prolonged inflammatory phase and decelerated reparation. Our experiment with 28-month-old mice has confirmed these observations. Lifelong treatment of the old mice with SkQ1 (30 nmol/kg daily) strongly stimulated wound healing (Fig. 4). Histological analysis revealed significant violations of the process of wound healing in old mice. In the old mice treated with SkQ1, in contrast, the complete epithelization and formation of connective tissue without leukocyte infiltration was observed. It was shown earlier that treatment with SkQ1 prevented development of various age-dependent pathologies [17-19]. This could be related to prevention of oxidative damage that accumulated in the organism with age. This explanation seems probable in the case of wound healing. On the other hand, it was found that SkQ1 treatment led to a significant slowdown in the development of signs of “healthy aging” [19]. These observations favored the hypothesis of cancellation of the program of aging by mitochondria-targeted antioxidants suggested by V. P. Skulachev [15]. From this point of view the described effect of SkQ1 could be one of the manifestations of prevention programmed decline in reparation processes during aging.
In conclusion, it was shown that local treatment with SkQ1 incorporated in synthetic coverings or injected in the wound area stimulated wound healing. The novel antioxidant suppressed inflammation and stimulated reparation processes at extremely low doses. The effect was directed on migration of leucocytes into the inflammatory focus, on myofibroblast differentiation, and epithelization of the wound. These data indicated an important role of mitochondrial ROS production in regulation of cell and tissue homeostasis. Future studies in various cellular and animal models are necessary to clarify the precise mechanisms of action of mitochondria-targeted antioxidants on the processes of inflammation and reparation. The data presented above give hope for successful application of these compounds in therapy of poorly healing wounds of various etiology.
The authors express their heartfelt gratitude to V. P. Skulachev for constant interest and support. We give our congratulations on his anniversary and wish many years of good health, happiness, and success in work for the benefit of science.
This work was supported by the Russian Foundation for Basic Research (grant Nos. 07-04-00335, 09-04-00667, 09-04-01454).
REFERENCES
1.Singer, A. J., and Clark, R. A. (1999) N. Engl.
J. Med., 341, 738-746.
2.Hinz, B. (2007) J. Invest. Dermatol.,
127, 526-537.
3.Hinz, B., Phan, S. H., Thannickal, V. J., Galli,
A., Bochaton-Piallat, M. L., and Gabbiani, G. (2007) Am. J.
Pathol., 170, 1807-1816.
4.Roy, S., Khanna, S., Nallu, K., Hunt, T. K., and
Sen, C. K. (2006) Mol. Ther., 13, 211-220.
5.Ojha, N., Roy, S., He, G., Biswas, S., Velayutham,
M., Khanna, S., Kuppusamy, P., Zweier, J. L., and Sen, C. K. (2008)
Free Radic. Biol. Med., 44, 682-691.
6.Sen, C. K., and Roy, S. (2008) Biochim. Biophys.
Acta, 1780, 1348-1361.
7.Roy, S., Khanna, S., and Sen, C. K. (2008) Free
Radic. Biol. Med., 44, 180-192.
8.Galeano, M., Torre, V., Deodato, B., Campo, G. M.,
Colonna, M., Sturiale, A., Squadrito, F., Cavallari, V., Cucinotta, D.,
Buemi, M., and Altavilla, D. (2001) Surgery, 129,
467-477.
9.Babior, B. M. (2000) Am. J. Med.,
109, 33-44.
10.Babior, B. M. (2000) IUBMB Life,
50, 267-269.
11.Chernyak, B. V., Izyumov, D. S., Lyamzaev, K. G.,
Pashkovskaya, A. A., Pletjushkina, O. Y., Antonenko, Y. N., Sakharov,
D. V., Wirtz, K. W., and Skulachev, V. P. (2006) Biochim. Biophys.
Acta, 1757, 525-534.
12.Skulachev, V. P. (2007) Biochemistry
(Moscow), 72, 1385-1396.
13.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L.
E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Y.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Y., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Y., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1273-1287.
14.Antonenko, Y. N., Roginsky, V. A., Pashkovskaya,
A. A., Rokitskaya, T. I., Kotova, E. A., Zaspa, A. A., Chernyak, B. V.,
and Skulachev, V. P. (2008) J. Membr. Biol., 222,
141-149.
15.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) Biochim. Biophys. Acta, 1787, 437-461.
16.Agapova, L. S., Chernyak, B. V., Domnina, L. V.,
Dugina, V. B., Efimenko, A. Y., Fetisova, E. K., Ivanova, O. Y.,
Kalinina, N. I., Khromova, N. V., Kopnin, B. P., Kopnin, P. B.,
Korotetskaya, M. V., Lichinitser, M. R., Lukashev, A. L., Pletjushkina,
O. Y., Popova, E. N., Skulachev, M. V., Shagieva, G. S., Stepanova, E.
V., Titova, E. V., Tkachuk, V. A., Vasiliev, J. M., and Skulachev, V.
P. (2008) Biochemistry (Moscow), 73, 1300-1316.
17.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. B., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashook, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasilyeva, A. K., Victorov, I. V., Zorov, D. B., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1288-1299.
18.Neroev, V. V., Archipova, M. M., Bakeeva, L. E.,
Fursova, A. Z., Grigorian, E. N., Grishanova, A. Y., Iomdina, E. N.,
Ivashchenko, Z., Katargina, L. A., Khoroshilova-Maslova, I. P., Kilina,
O. V., Kolosova, N. G., Kopenkin, E. P., Korshunov, S. S., Kovaleva, N.
A., Novikova, Y. P., Philippov, P. P., Pilipenko, D. I., Robustova, O.
V., Saprunova, V. B., Senin, I. I., Skulachev, M. V., Sotnikova, L. F.,
Stefanova, N. A., Tikhomirova, N. K., Tsapenko, I. V., Shchipanova, A.
I., Zinovkin, R. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1317-1328.
19.Anisimov, V. N., Bakeeva, L. E., Egormin, P. A.,
Filenko, O. F., Isakova, E. F., Manskikh, V. N., Mikhelson, V. M.,
Panteleeva, A. A., Pasyukova, E. G., Pilipenko, D. I., Piskunova, T.
S., Popovich, I. G., Roshchina, N. V., Rybina, O. Y., Saprunova, V. B.,
Samoylova, T. A., Semenchenko, A. V., Skulachev, M. V., Spivak, I. M.,
Tsybul’ko, E. A., Tyndyk, M. L., Vyssokikh, M. Y., Yurova, M. N.,
Zabezhinsky, M. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1329-1342.
20.Yudanova, T. N., Skokova, I. F., Aleshina, E. Y.,
and Gal’braikh, L. S. (2001) Fiber Chem., 33,
20-24.
21.Montesano, R., Saint Vincent, L., Drevon, C., and
Tomatis, L. (1975) Int. J. Cancer, 16,
550-558.
22.Fraticelli, A., Serrano, C. V., Jr., Bochner, B.
S., Capogrossi, M. C., and Zweier, J. L. (1996) Biochim. Biophys.
Acta, 1310, 251-259.
23.Klyubin, I. V., Kirpichnikova, K. M., and
Gamaley, I. A. (1996) Eur. J. Cell Biol., 70,
347-351.
24.Curzio, M., Esterbauer, H., and Dianzani, M. U.
(1985) Int. J. Tissue React., 7, 137-142.
25.Curzio, M., Di Mauro, C., Esterbauer, H., and
Dianzani, M. U. (1987) Biomed. Pharmacother., 41,
304-314.
26.Curzio, M., Esterbauer, H., Poli, G., Biasi, F.,
Cecchini, G., Di Mauro, C., Cappello, N., and Dianzani, M. U. (1987)
Int. J. Tissue React., 9, 295-306.
27.Curzio, M. (1988) Free Radic. Res.
Commun., 5, 55-66.
28.Rossi, M. A., Di Mauro, C., and Dianzani, M. U.
(2001) Int. J. Tissue React., 23, 45-50.
29.Kumagai, T., Matsukawa, N., Kaneko, Y., Kusumi,
Y., Mitsumata, M., and Uchida, K. (2004) J. Biol. Chem.,
279, 48389-48396.
30.Trevisani, M., Siemens, J., Materazzi, S.,
Bautista, D. M., Nassini, R., Campi, B., Imamachi, N., Andre, E.,
Patacchini, R., Cottrell, G. S., Gatti, R., Basbaum, A. I., Bunnett, N.
W., Julius, D., and Geppetti, P. (2007) Proc. Natl. Acad. Sci.
USA, 104, 13519-13524.
31.Lowes, D. A., Thottakam, B. M., Webster, N. R.,
Murphy, M. P., and Galley, H. F. (2008) Free Radic. Biol.
Med., 45, 1559-1565.
32.Mukherjee, T. K., Mishra, A. K., Mukhopadhyay,
S., and Hoidal, J. R. (2007) J. Immunol., 178,
1835-1844.
33.Lin, S. J., Shyue, S. K., Hung, Y. Y., Chen, Y.
H., Ku, H. H., Chen, J. W., Tam, K. B., and Chen, Y. L. (2005)
Arterioscler. Thromb. Vasc. Biol., 25,
334-340.
34.Radomska-Lesniewska, D. M., Sadowska, A. M., van
Overveld, F. J., Demkow, U., Zielinski, J., and de Backer, W. A. (2006)
J. Physiol. Pharmacol., 57 (Suppl. 4), 325-334.
35.Hashimoto, S., Gon, Y., Matsumoto, K., Takeshita,
I., and Horie, T. (2001) Br. J. Pharmacol., 132,
270-276.
36.Takahashi, M., Suzuki, E., Takeda, R., Oba, S.,
Nishimatsu, H., Kimura, K., Nagano, T., Nagai, R., and Hirata, Y.
(2008) Am. J. Physiol. Heart Circ. Physiol., 294,
H2879-2888.
37.Lu, C., Bambang, I. F., Armstrong, J. S., and
Whiteman, M. (2008) Diabetes Obes. Metab., 10,
347-349.
38.Dugina, V., Fontao, L., Chaponnier, C., Vasiliev,
J., and Gabbiani, G. (2001) J. Cell Sci., 114,
3285-3296.
39.Kondo, M., Cubillo, E., Tobiume, K., Shirakihara,
T., Fukuda, N., Suzuki, H., Shimizu, K., Takehara, K., Cano, A.,
Saitoh, M., and Miyazono, K. (2004) Cell Death Differ.,
11, 1092-1101.
40.Gabbiani, G. (2003) J. Pathol.,
200, 500-503.