REVIEW: Principles of Control over Formation of Structures Responsible for Respiratory Functions of Mitochondria
V. N. Luzikov*
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: bogdanov@genebee.msu.su## The Editorial Board is deeply grateful to Academician A. A. Bogdanov for editing this review.
* Deceased.
Received December 12, 2008
Topogenesis of mitochondrial proteins includes their synthesis in cytosol and mitochondria, their translocation across the outer and inner membranes, sorting to various mitochondrial compartments, and assembly of different protein complexes. These complexes are involved in transport functions, electron transfer through the respiratory chain, generation of transmembrane electrochemical potential, oxidative phosphorylation of ADP into ATP, etc. To perform these functions, a special stringent control is required over formation of submitochondrial structures and the mitochondrion as a whole. Such control is expected to rigorously eliminate not only misfolded proteins but also incorrectly incorporated subunits and is realized in mitochondria by means of numerous proteases with different functions and localizations. In the case of more complicated protein formations, e.g. supercomplexes, the protein quality is assessed by their ability to realize the integral function of the respiratory chain and, thus, ensure the stability of the whole system. Considering supercomplexes of the mitochondrial respiratory chain, the present review clearly demonstrates that this control is realized by means of various (mainly vacuolar) proteases with different functions and localizations. The contemporary experimental data also confirm the author’s original idea that the general mechanism of assembly of subcellular structures is based on the “selection by performance criterion” and “stabilization by functioning”.
KEY WORDS: biogenesis of mitochondria, protein quality control, respiratory complexes, mitochondrial proteasesDOI: 10.1134/S0006297909130021
Topogenesis of mitochondrial proteins is a part of the biogenesis of mitochondria, the cellular organelles that provide for energy needs and perform some metabolic functions of the cell [1]. Also, mitochondria are now thought to play an important role in programmed cell death (apoptosis) [2].
Topogenesis of mitochondrial proteins includes their synthesis in cytosol and mitochondria, their translocation across the outer and inner membranes, sorting to various mitochondrial compartments, and assembly of different protein complexes. These complexes are involved in transport functions, electron transfer through the respiratory chain, generation of transmembrane electrochemical potential, oxidative phosphorylation of ADP into ATP, etc. [3].
Two main ideas are emphasized in the present review. First, compartmentalization of mitochondria is the only possible structure allowing these organelles to perform their main functions. Second, these functions require especially stringent control over formation of submitochondrial structures and the mitochondrion as a whole. Due to these two features, mitochondria are capable of maintaining transmembrane potential on the inner membrane. In the cell, such unique properties are inherent only in mitochondria.
The bulk of mitochondrial proteins are soluble matrix proteins and some proteins of the mitochondrial inner membrane synthesized in the cytosol. As a rule, translocation of these proteins across the mitochondrial membrane requires a positively charged N-terminal amino acid sequences with features of amphiphilic helices. They act as unique addressing signals. These sequences are detached under the influence of specific processing peptidases.
Mitochondrial proteins must be transferred from the cytosol into the mitochondrial matrix driven by the membrane potential. This is associated with a Δψ-dependent import from the cytosol of a large group of the inner membrane proteins with uncleavable sequences that is ensured by a specific arrangement of positively charged sequences of amino acid residues.
Translocation of mitochondrial matrix proteins is served by a system of pores (protein translocons) in the mitochondrial outer and inner membranes and also by a set of accessory transport proteins.
It has been mentioned above that the generation of transmembrane electrochemical potential on the mitochondrial inner membrane and oxidative phosphorylation of ADP into ATP are provided for by the respiratory chain components including enzyme complexes I-IV and by the presence of H+-ATPase. According to contemporary data, these complexes form supercomplexes. Respiratory complexes can include up to 50 subunits, and the whole respiratory chain of higher eucaryotes contains about 90 subunits [4]. Just the existence of such a great number of subunits dictates the necessity for strict rejection not only of misfolded proteins but also of incorrectly incorporated subunits — this has been called “protein quality control” in the literature. This control is realized in mitochondria by means of numerous proteases with different functions and localizations.
The “protein quality control” concerns not only individual proteins but also more complicated protein formations, e.g. supercomplexes, each of which has its own enzymatic activity. In this case, protein quality can be controlled by their ability to realize the integral function of the respiratory chain that is associated with maintaining stability of the system as a whole.
If the respiratory apparatus of a mitochondrion does not function or functions incorrectly, the mitochondrion is destroyed by means of different mechanisms, such as digestion of individual complexes or micro- and macroautophagy under the influence of vacuolar proteases.
The main purpose of the present paper is to show the realization of the “selection by performance criterion” and “stabilization by functioning” principles, which were formulated by the author and colleagues more than 25 years ago [5]. Many data of recent years have confirmed these principles, and they are discussed here to design a possible line of further studies.
FORMATION OF PROTEIN COMPLEXES AND SUPERCOMPLEXES OF THE
MITOCHONDRIAL INNER MEMBRANE
Most of the mitochondrial inner membrane proteins are components of the energy transformation system, which involves the respiratory chain and H+-ATPase coupled with the carriers of adenine nucleotides (ANC) and inorganic phosphate (PIC). As stated earlier, the respiratory chain consists of four protein complexes catalyzing partial reactions of electron transfer (complexes I, II, III, IV). In cooperation with H+-ATPase (complex V), they occupy up to 40% of the mitochondrial inner membrane surface in rat liver [6]. The energy transformation system is characterized by the unique involvement of proteins synthesized on both cytosolic and mitochondrial ribosomes. In mammals, complexes I-V include up to 76 polypeptides. Complex I contains seven and 38 products of mitochondrial and nuclear genes, respectively; complex II includes only four products of nuclear genes; complex III is performed by a single product encoded in the mitochondrial genome and by 10 products encoded in the nuclear genome; three proteins of complex IV are encoded in mitochondria and 10 products are encoded in the nucleus; and, finally, complex V contains two products of the mitochondrial genome and 14 products of the nuclear genome [7]. In the yeast Saccharomyces cerevisiae the respiratory chain and H+-ATPase contain 34-36 polypeptides (proteins) because they lack complex I and have fewer proteins synthesized in the cytosol: complex III contains eight proteins instead of 10, complex IV has six to eight proteins instead of 10, and complex V consists of eight proteins instead of 11 [8]. Some aspects of formation of protein complexes of the mitochondrial inner membrane will be considered below. Special attention will be paid to complex IV (cytochrome c oxidase) because this complex plays a crucial role in the general regulation of energy production in the cells of eucaryotes [9].
Protein Complexes of the Mitochondrial Inner Membrane
Composition, structure, and principles of cytochrome c oxidase formation. Mitochondrial cytochrome c oxidase contains four active metal-containing sites (heme a, heme a3, CuA, and CuB) submerged into protein matrix of multisubunits. In S. cerevisiae this matrix includes three large subunits I, II, and III synthesized in mitochondria and six small subunits (subunits IV, Va or Vb, VI, VII, VIIa, and VIII) encoded by the nuclear genes COX4, COX5a or COX5b, COX6, COX7, COX8, and COX9, respectively. Subunit I (COXI) binds heme a and heme a3 and also the reactive site CuB, whereas subunit II includes the CuA-site and is involved in the binding of cytochrome c. Subunit III is involved in proton conductivity of cytochrome c oxidase. Thus, three mitochondrial subunits of the enzyme carry all the catalytic sites of the enzyme, which is confirmed by X-ray crystallographic analysis data obtained at the resolution of 2.8 Å for bovine cytochrome c oxidase. In particular, the same data also show that the enzyme contains single copies of all 13 subunits [10]. Cytochrome c oxidase from Paracoccus denitrificans was earlier shown to contain only two subunits, which are homologous to subunits I and II of the enzyme from yeast and include two heme residues and two Cu-sites [11].
Nevertheless, deletions in COX4, COX5a and COX6, COX7 and COX9 result in complete loss of cellular respiration, cytochromes a and a3, and activity of cytochrome c oxidase [8]. At present, the nuclear-coded corresponding subunits are thought to be responsible for stabilization of specific mitochondrial subunits in the holoenzyme or for promoting formation of the holoenzyme catalytic site. Among the cytochrome c oxidase subunits encoded in the nucleus only subunit VIII is not indispensable — the Null mutant of this subunit retains up to 80% of the activity of the wild type enzyme. Subunit VIII is believed to play a regulatory role in the enzyme functioning by influencing electron transfer between it and cytochrome c [8].
Cytochrome c oxidase from bovine heart includes 13 subunits, and three large ones (I, II, III) with molecular weights of 51.4, 22.7, and 26.7 kDa, respectively, are synthesized in mitochondria. These subunits form a compact “nucleus” consisting of 12 transmembrane helices of subunit I, two transmembrane helices of subunit II, and seven transmembrane helices of subunit III. Subunits IV-VI with molecular weights from 0.6 to 14.7 kDa synthesized in the cytosol are disposed on the periphery of this “nucleus” leaving a vacant zone for contact with a second molecule of the enzyme and resulting dimerization. Only three subunits of cytochrome c oxidase encoded in the nucleus lack transmembrane domains and are exposed either to the matrix or to the intermembrane space. The general arrangement of the enzyme subunits synthesized in the cytosol allows them to group catalytic subunits I, II, and III in the inner membrane and stabilize the whole construction [11].
Cytochrome c oxidase is assembled in the mitochondrial inner membrane with involvement of two groups of accessory proteins. One group ensures synthesis of heme a (proteins encoded by genes COX10, COX15, and YAH1) and also inclusion of CuA,B into the enzyme molecule (proteins encoded by genes SCO1, COX11, COX17, and COX23). The second group is necessary for assembly of the holoenzyme from subunits (encoded by genes PET100, PET117, PET191, COX14, COX16, etc.) [12]. Proteins of the second group imported into mitochondria from the cytosol are not included into the cytochrome c oxidase holoenzyme, but mutations in any of these proteins prevent formation of the holoenzyme. Concrete functions of these proteins are not always known. Interesting data were recently obtained for Pet100 [12]. This 13-kDa protein is an integral protein of the inner membrane of yeast mitochondria. Mitochondria were solubilized in Triton X-100, centrifuged in a density gradient, and subsequent immunoprecipitation of protein fractions with antiserum against Pet100 indicated that this protein selectively bound subunits VIIa, VII, and VIII of cytochrome c oxidase with production of a common complex (complex A). This was confirmed by cross-immunoprecipitation. At the same time, a fraction containing cytochrome c oxidase activity and the complete set of holoenzyme subunits was not associated with Pet100.
It seems that in the presence of Pet100 complex A consisting of subunits VII, VIIa, and VIII acts as a “trigger” for further stages of the holoenzyme assembly in the membrane. The Pet100 null mutant does not contain the cytochrome c oxidase holoenzyme although the whole set of its subunits (I, II, III, IV, V, Va, VI, VII, VIIa, and VIII) was detected in it in slightly reduced amounts. In this case, the sucrose gradient of the Triton X-100 lysate of mitochondria revealed two tightly bound complexes, which included subunits VII, VIIa, and VIII (complex A1 without Pet100) and subunits V and VI (complex B).
The lack of the cytochrome c oxidase holoenzyme formation in the absence of Pet100 suggests that it is required for maintaining a certain order in interactions between the subunits (or their groups), and disorders in this order can cause disorders in the assembly including stages of attachment of the heme residue and formation of the CuA,B sites. Thus, although Pet100 is not involved in the early stages of the holoenzyme formation, including transcription, translation, addressing into mitochondria, and sorting to mitochondria, it acts as a chaperone responsible for correct assembly of certain regions (or stages) of cytochrome c oxidase. This accessory protein is necessary for formation of the active site of the enzyme [12].
Formation of bc1 complex (complex III of the mitochondrial respiratory chain). Complex III includes three catalytic subunits: cytochrome b (encoded in mitochondria), cytochrome c1, and FeS-Riske protein (the latter two proteins are encoded in the nucleus, imported into mitochondria, and incorporated into the mitochondrial inner membrane). Complex III also contains several subunits without catalytic but with structural functions. In S. cerevisiae, such subunits include Core1, Core2, Qcr6p, Qcr7p, Qcr8p, Qcr9p, and Qcr10p [13]. Complex III is assembled through some intermediate states. The current model suggests that initially a complex of cytochrome b with Qcr7p and Qcr8p is produced that later binds with Core1 and Core2. Another subcomplex consists of cytochrome c1, Qcr6p, and Qcr9p. Formation of these subcomplexes impart to them increased resistance to proteolysis as compared to the individual subunits. Then subcomplexes of cytochrome b and cytochrome c1 combine into a precomplex bc1 with the FeS-Riske protein and, possibly, with the subunit Qcr10p. For assembly of the complete complex bc1 a specific protein Bcs1p is required, which is a 45.6-kDa integral protein of the mitochondrial inner membrane but not a direct component of the cytochrome bc1 complex [14]. Nevertheless, deletion of the Bcs1 gene leads to formation of a deficient nonfunctional complex bc1 (complex III). From this viewpoint, Bcs1p may be considered as an “assembling chaperone”, and its action is characterized by a coupled hydrolysis of ATP. These criteria allow us to refer Bcs1p as a member of the AAA family of proteins involved in various processes of protein association and dissociation.
Formation of NADH:coenzyme Q oxidoreductase (complex I of the mitochondrial respiratory chain). Complex I of bovine heart mitochondria contains 45 subunits, the majority of which is encoded by the nucleus and the rest (seven) by the mitochondrial genome. In the case of single copies of every subunit, the molecular weight of complex I should be 980 kDa (900 kDa by gel filtration data). Complex I consists of domains and is easily dissociated under the influence of nonionic detergents. With a Mono-Q chromatographic column three domains are distinctly separated, one of which is peripheral and directed to the mitochondrial matrix, whereas two other domains, a large and a small one, are localized inside the inner membrane. The peripheral domain contains about 22 subunits, and the membrane domains include eight and 13 subunits. Electron microscopy shows that complex I is L-shaped, with the right angle formed by the peripheral and the two membrane domains and the peripheral domain extending into the matrix by about 150 Å [15, 16]. Note that proteins synthesized in mitochondria (ND1-ND6 and ND4L) are highly hydrophobic and are constituents only of the small and large membrane domains.
According to the domain structure of complex I, it is formed from the abovementioned blocks, each capable of existing independently. This can be illustrated by disappearance of individual blocks as a result of selective mutations in their proteins with the retention of other blocks [17]. Some data suggest that the interaction between proteins can be controlled by special protein chaperones. In particular, the membrane domain is formed by two intermediates: the 200-kDa small and the 350-kDa large ones. The latter intermediate is associated with 30- and 84-kDa accessory proteins which are not components of the mature complex I. In mutants with destroyed genes of one or of two of these proteins, the peripheral and the small membrane blocks are accumulated, but the large intermediate is not produced. Thus, the 30- and 84-kDa accessory proteins are recycled providing for formation of the complete membrane block and functioning as associating chaperones.
Formation of mitochondrial H+-ATPase. Mitochondrial H+-ATPase (F1Fo-ATPase), which synthesizes the major part of ATP of the eucaryotic cell, includes the membrane-associated catalytic site F1 (subunits α3β3γδε) and a membrane sector F0 (subunits Atp6p, Atp8p, and Atp9p, which are also called a, b, and c, respectively) [18-21]. According to a simplified model, in the first stage of assembly of the complex in the membrane plane a ring is formed which contains 10-12 homooligomeric subunits of Atp9p [22]. A pre-assembled sector F1 is attached to the Atp9p ring and then binds two subunits (Atp8p and Atp4p) and, possibly, other components of the stator sector. Finally, this intermediate complex binds Atp6p. Products ATP10, ATP11, and ATP12 of nuclear genes are not components of F1Fo-ATPase but influence its formation. A mutation (atp10 null) in Atp10p associated with the mitochondrial inner membrane causes a deficiency in F0 as a result of destabilization of the Atp6p subunit. However, mutations in matrix-localized proteins Atp11p (atp11 null) and Atp12p (atp12 null) prevent oligomerization of sector F1 subunits [22, 23]. Note that at present 16 nuclear-coded proteins of F1Fo-ATPase are known; thus, the regulation of this complex (complex V) is likely to be more complicated.
The presented examples demonstrate that assembly of multisubunit complexes of the mitochondrial inner membrane follows certain rules. First, the assembly requires a definite order of subunit interactions and, second, accessory proteins, which are not direct components, must be involved. Note also that accessory proteins are rather numerous and are not universal in their features, as discriminated, in particular, from complex IV formation (see above). Thus, currently only the most general principles of formation of respiratory complexes of the mitochondrial inner membrane are known.
It should be mentioned that the idea of controlled assembly of multisubunit complexes with involvement of some “patterns” exemplified, in particular, by chaperone proteins was supposed even in 1965 by Green and Hechter [24].
Supercomplexes of the Mitochondrial Respiratory Chain and H+-ATPase
There is some evidence that respiratory complexes, H+‑ATPase, and some proteins functionally bound with it in the mitochondrial inner membrane form even more complicated structures, or supercomplexes [4]. In particular, these supercomplexes are supposed to promote faster diffusion of substrates or realization of the tunnel effect in enzymatic reactions.
Supercomplexes of the respiratory chain. Supercomplexes in the fraction of mitochondrial inner membranes can be identified by their solubilization with detergents and subsequent electrophoresis in polyacrylamide gel under non-denaturing conditions: BN-PAGE, 2D BN-PAGE, and also using a coupled SDS-PAGE [25, 26]. This approach allows researchers to determine both the size and composition of the inner membrane fragments (in the case of a suitable method for identification of individual proteins, such as immunoblotting and N-terminal sequencing).
Studies on the bc1 complex (complex III) and cytochrome c oxidase (complex IV) in digitonin lysates of S. cerevisiae reveal supercomplexes of composition III2IV1 (500 kDa) and III2IV2 (750 kDa). Depending on conditions of the yeast growth, relative contents of these complexes can be up to 30 and 90%, respectively, and the free complex IV content as a dimer can be less than 10%. Therefore, aggregate states of complexes III and IV are supposed to vary and characterize different stages of formation of functionally important structures.
Some data show that subunits of complexes III and IV are co-immunoprecipitated from the digitonin-solubilized mitochondrial membranes, and this suggests a physical interaction of these complexes within the membrane. Similar conclusions are obtained as a result of comparative analysis of effects of digitonin and dodecyl maltoside (DDM) on the partial activities of complexes III and IV (coenzyme Q: cytochrome c reductase and cytochrome c oxidase activities, respectively) in the mitochondrial membrane and their integral coenzyme Q oxidase activity.
In addition to the III2IV1 and III2IV2 complexes (see above), particles with molecular weights of 1500 and 1750 kDa including complexes I, III, and IV were detected by BN-PAGE in digitonin lysates of mitochondrial membranes. Moreover, by two-dimensional BN-PAGE (2D BN-PAGE) and SDS-PAGE, stoichiometry of complexes I, III, and IV was established, and they were shown to directly interact with one another producing supercomplexes of I1III2IV2-4 composition. Based on this and other experiments, it was supposed that respiratory supercomplexes should generate a network in the mitochondrial inner membrane. This network consists of separate “respirasomes” [25]. Electron microscopy data obtained by negative contrasting show the complex I1III2IV1 preparations as triangular particles with a long side of 30-33 nm [27]. These particles seem to include complex I as an L-shaped construction coupled with complexes III and IV (with total molecular weight of 1400 kDa). Generation in the membrane of large supramolecular structures consisting of respirasomes is also likely [28].
A unique feature of the respiratory chain (reconstructed from isolated respiratory complexes or submitochondrial particles) was found about 40 years ago [29]: it can become resistant to trypsin/chymotrypsin and phospholipase A2 under conditions of its functioning, i.e. oxidation of NADH or succinic acid by oxygen. This feature is inherent in the whole respiratory chain but not in its fragments. On the other hand, functioning of individual regions does not protect against the abovementioned destroying agents. Regions at the level of coenzyme Q between cytochromes b and c1 and also at the level of cytochrome c are “weak points” of the respiratory chain with respect to phospholipase A2. These vulnerable spots seem to be shielded during oxidizing with oxygen of corresponding substrates. The above-described model experiments are rather illustrative, but there is no confidence that they are relevant to real physiological conditions.
Supercomplex containing H+-ATPase, adenine nucleotide carrier (ANC), and inorganic phosphate carrier (PIC). Supercomplex with this composition was prepared from a highly purified preparation of rat mitochondrial inner membranes using successive fragmentation to enrich the fragments with components of ATP synthase and subsequent treatment of the fragments with various detergents. Eighty detergents were tested, and only four of them were suitable for the purpose [30]. By SDS-PAGE data, the resulting preparation as vesicles contained only 17 polypeptides and, except regulatory factors IF1 and B, 15 of them belonged to F1Fo-ATPase (ATP synthase or H+-ATPase), one to ANC, and one to PIC. On centrifugation through 25% sucrose, this preparation interacted as a unified protein fraction with antibodies against β-subunit of F1‑ATPase. Thus, this preparation can be considered as a specialized region, a subfraction of the inner membrane and a place of formation of the ATP synthase–ANC–PIC complex called “ATP synthasome” [31].
The ATP synthasome has the following stoichiometry — ATP synthase/ANC/PIC, 1 : 1 : 1. Electron microscopy with resolution of 23 Å of negatively stained preparations of this supercomplex clearly visualized three structural elements: a classic “head” presented the major mass of F0, the central stalk, and a basepiece. This basepiece is ellipsoid-like and includes two domains, the larger of which is connected to the central stalk [31]. Both ANC and PIC are located asymmetrically relative to the basepiece of ATP synthase. In conclusion, note that this section deals with a supercomplex consisting of more than 30 subunits with molecular weight of ~700 kDa.
CONTROL OF COMPOSITION OF ENZYMATIC COMPLEXES IN THE
MITOCHONDRIAL INNER MEMBRANE
Respiratory complexes and components of the ATP synthasome include proteins encoded by both the nuclear and mitochondrial genome, and mitochondria contain an enzymatic and regulatory system that is responsible for replication of mitochondrial DNA and expression of the information encoded in it which is different from the nuclear system [32]. Immediately after the discovery of mitochondrial DNA, a question arose about communications between the nuclear and mitochondrial genomes separated in cellular space. In particular, it was unclear how a constant protein composition of the respiratory chain complexes (e.g. of cytochrome c oxidase) could be reached if some subunits of these complexes were expressed in the cytosol and others in the mitochondria. Despite intensive studies, these questions for a long time remained without answer. However, some ways for solving this problem appeared during recent years (for references see [33]).
Expressions of the nuclear and mitochondrial genomes were shown to be mutually coordinated. Transcription of genes of many respiratory chain proteins encoded in the mammalian cell nucleus are directly or indirectly regulated by factors NRF-1 and NRF-2 [34]. These factors, in turn, are controlled by co-activator proteins of the PGC-1 family [35]. Activities of the PCG-1 proteins are modulated by various extra-and intracellular signals [36]. In particular, they react to increase in Ca2+ concentration in the cytosol, which occurs on inhibition of the respiratory activity of mitochondria. Activated by PGC-1α and mediated by NRF-1 transcription of genes encoding the respiratory chain proteins results in increase in their amount in the mitochondrial inner membrane [36].
In addition to Ca2+, many factors can influence the correlation of subunits in the enzymatic complexes of the mitochondrial inner membrane. In principle, supplies of these proteins can depend not only on induction of their genes but also on special features of processing of the corresponding transcripts, formation of mRNAs, their stabilization and translation in the cytosol and mitochondria, etc. Here under consideration are multistage catalytic processes resulting in products (e.g. mRNAs) with uncertain half-lives. The next point is the transport of protein precursors into mitochondria and their addressing to different mitochondrial compartments under conditions of coordinated involvement of translocases and chaperones [3, 37, 38]. Final stages of formations of the respiratory complexes are controlled by a set of proteases localized in all mitochondrial compartments.
Mitochondrial Proteases: Localization in Mitochondria, Features, and Role in Topogenesis of Mitochondrial Proteins
Features and functions are currently known in detail for three mitochondrial proteases from S. cerevisiae: PIM1, m-AAA (Yta10/Yta12p), and i-AAA (Yme1p) (Fig. 1). These proteases play a key role in formation of mitochondrial proteins, elimination of deficient proteins, and also of proteins deprived of their partners in protein complexes. Moreover, at least one of these enzymes, i-AAA, is involved in the general system of regulation of mitochondrial morphogenesis [39].
It should be pointed out that yeast mitochondria contain specific proteases (such as MoP112, Oma1) responsible for selective regulatory functions during the vital activity of the yeast cells. To realize these functions, these proteases require some factors, such as protein ABC carriers Mdl1 and prohibitins Phb1p and Phb2p. The protein transporter Mdl1 is responsible for export of polypeptides with molecular weights within the limits ~2100-600 Da with involvement either of m-AAA or i-AAA protease, depending on which one is present in the compartment [40]. Prohibitins Phb1p and Phb2p are attached to the mitochondrial inner membrane by their N-terminal region, whereas the C-terminal region is exposed to the intermembrane space. These prohibitins regulate the protease activity of m-AAA [41]. But with accumulation of new data, the mitochondrial proteolytic apparatus controlling protein quality is likely to prove even more complicated.Fig. 1. Proteolytic system of the mitochondrion (adapted from [39]). ATP-dependent proteases: matrix-embedded PIM1/Lon protease; i-AAA and m-AAA proteases submerged into the inner membrane with catalytic sites turned, respectively, to the intermembrane space and matrix; inter-oligomeric Clp protease includes peptidase and ATPase subunit identified in organelles of higher eucaryotes but not in S. cerevisiae. ATP-independent peptidases: matrix processing peptidase (MPP), intramembrane peptidase (IMP), and mitochondrial intermediate peptidase (MIP). OM, outer membrane; IM, inner membrane; M, matrix; IMS, intermembrane space.
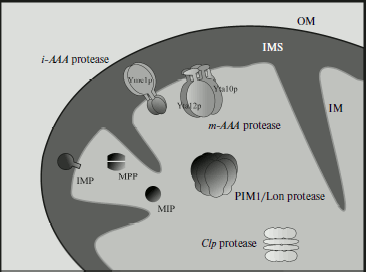
Molecular organization and compartmentalization of mitochondrial proteases. The Lon-like serine protease PIM1 is localized in the mitochondrial matrix, and its purified form is a ~800-kDa complex with heptameric stoichiometry [39]. The enzyme subunits (Pim1p) are synthesized in the cytosol as a precursor containing 1133 amino acid residues. In the presence of ATP, PIM1 is oligomerized; this process can be inhibited by non-hydrolyzable nucleotide analogs.
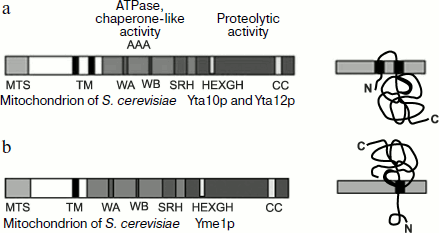
Protease m-AAA consists of two proteins: Yta10p/Afg3p and Yta12p/Rca1p (further complex YTA10-12) with 53%-identical primary structures. Yta10p (761 residues) and Yta12p (825 residues) are encoded in the nucleus and addressed into mitochondria through NH2-terminal presequences. Both proteins are “anchored” in the inner mitochondrial membrane by two hydrophobic segments in the NH2-terminal third of the protein that are separated by a hydrophilic loop of ~10 kDa size. The NH2-terminal parts of Yta10p and Yta12p, as well as their massive COOH-terminal domains (~520 residues) carrying ATP- and metal-binding sites are exposed to the matrix.
Yta10p and Yta12p form a ~1000-kDa complex in the mitochondrial inner membrane. The integrity of the complex requires the presence of nucleotides but not hydrolysis of ATP (Fig. 2a) [42].
Protease i-AAA is encoded by the gene YME1 (YTA11) and contains as a subunit polypeptide Yme1, which includes 747 residues and is 32% homologous to Yta10p and Yta12p (Fig. 2b) [42]. Yme1p contains an N-terminal mitochondrial addressing signal. In contrast with Yta10p and Yta12p, Yme1p crosses the mitochondrial inner membrane only once, whereas the 55-kDa catalytic domain is exposed to the intermembrane space. This domain includes a Zn-binding region HEXXH, which is specific for metalloproteases. It seems that Yme1p is not the only component of protease i-AAA, which is a high molecular weight complex of ~1000 kDa. Stability of this complex does not depend on the presence of nucleotides and mutations in the ATP-binding motif of Yme1p.Fig. 2. Domain structure of AAA-protease subunits (adapted from [42]). a) Protease m-AAA; b) protease i-AAA. TM, transmembrane segment; AAA, ATPase domain; CC, coiled-coil domain; MTS, mitochondrial addressing signal; HEXGH, metal-binding site; WA and WB, A and B Walker motifs; SRH, second region of homology.
Protease Oma1 (overlapping activity with m-AAA) is an ATP-independent protease that carries no separable N-terminal sequence and is not processed by proteolysis during the import from the cytosol. Protease Oma1 is an inner membrane protein with a supposed homo-hexameric structure. The active site of the protein contains the motif HEXXH corresponding to proteases S2P [43].
Protease Mop112 is a 112-kDa metallopeptidase containing a motif HXXEH. This metallopeptidase is supposed to have a region with an amphiphilic helix mediating its Δψ-dependent addressing to mitochondria. The addressing region in this protease is inseparable. Mop112 is imported into the intermembrane space from the cytosol [44].
Targets for ATP-dependent mitochondrial proteases. Incomplete polypeptide chains and proteins beyond protein ensembles are the main targets of ATP-dependent mitochondrial proteases. Earlier only the total contribution of ATP-dependent proteolysis of the products of mitochondrial translation was estimated in vitro and in vivo under conditions of disturbed cytosolic translation [45-47]. Now a correlation has been found between the abovementioned proteases and their specific targets in mitochondria.
Thus, according to the PIM1 localization in the matrix, it acts mainly on proteins exposed to the mitochondrial matrix, such as α-, β-, and γ-subunits of F1-ATPase, β-subunit of the matrix processing peptidase (Mas1p or MPP, see above), and some mitochondrial ribosomal proteins [48]. The m-AAA complex with its protease site turned to the mitochondrial matrix degrades integral proteins of the inner membrane: subunits I and III of cytochrome c oxidase (complex IV), cytochrome b (complex III), and also subunits 6, 8, and 9 of ATPase in the inner membrane [49]. More likely, protein can be assigned to substrates of PIM1 and m-AAA based on their general conformation and not their specific nature [50].
In the case of the i-AAA complex with the protease site oriented to the intermembrane space, subunit II of complex IV was identified as a substrate [51]. As shown by X-ray crystallographic analysis [10], a significant part of this subunit is also exposed to the intermembrane space.
As differentiated from the three previous proteases, which are not only proteolytic enzymes but also perform in mitochondria various ATP-dependent regulatory functions (see above), protease Oma1 seems to possess only proteolytic activity. It is an integral protein of the mitochondrial inner membrane with its active site localized in the transmembrane domain close to the matrix. Substrates of this protease are polytopic membrane proteins, in particular, Oxal that is involved in the interaction of the ribosome with the growing polypeptide chain and its incorporation into the mitochondrial inner membrane [43].
Some proteins that are mainly products of mitochondrial translation are rejected under the influence of i‑AAA, and this results in detachment of large protein fragments directed to the intermembrane space, where they are converted to oligopeptides by protease Mop112.
The mechanisms of protein degradation in the intermembrane space remain unclear. The only example is iso-1-cytochrome c, which is about 95% of the isocytochrome pool in aerobically growing derepressed cells. Under conditions of partial anaerobiosis or glucose repression, iso-1-cytochrome c is rapidly degraded under the influence of proteasomes. In fact, the apo-iso-1-cytochrome c degradation decreases in pre2-2 and pre1-1 mutants and also in ubc1, ubc4, and ubc5 mutants of ubiquitin-conjugating enzymes. These and other data suggest that mitochondrial proteins can be degraded in the cytosol [52].
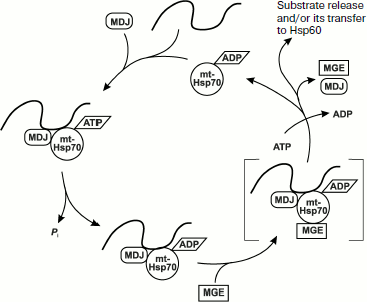
Regulation of proteolysis of mitochondrial proteins. Proteins imported into the mitochondrial matrix are folded or included in protein complexes. If these processes occur with errors, the proteins are subjected to the action of Lon-like PIM1 protease, the content of which must correspond to the number of errors determined by the functional state of mitochondria in the cell. In model systems, deficient proteins are shown to aggregate and accumulate in the matrix [53, 54] in the absence of mitochondrial chaperone mt‑Hsp70 (Ssc1p, analog of DnaK in E. coli). This chaperone is associated with co-chaperone Mdj1p (analog of DnaJ in E. coli), which is necessary for mt-Hsp70 to stimulate the ATPase activity and with protein Mge1p (analog of GrpE in E. coli) for realizing nucleotide exchange on it. This chaperone system provides for either converting proteins imported into mitochondria to the native shape (folded) or making them available for degradation under the influence of protease PIM1. This is illustrated by the scheme (Fig. 3) [55]. Mitochondria contain a system of Hsp78 and Mcx1 chaperones of the family Clp/Hsp100, and this system is involved in degradation of proteins [56]. These proteins are homologous to bacterial proteins in active complexes ClpB and ClpX, the first of which performs proteolysis and the other reactivation of aggregated polypeptides. A similar situation is observed in mitochondria of higher eucaryotes. The Hsp78 from yeast has no proteolytic activity but manifests an ATP-dependent unfolding activity, which seems to prepare a substrate for degradation under the influence of matrix protease PIM1. The function of Mcx1 remains unclear despite its belonging to the family of Clp/Hsp100 chaperones [57].
Proteins of the mitochondrial inner membrane. Protease PIM1 and chaperone mt-Hsp70 are shown to function separately in the control of the protein folding “quality”. Another picture is observed for the YTA10-12 (m-AAA) complex (Fig. 2a) that combines features of chaperones and proteases possessing, on one hand, NTPase motifs WA and WB and, on the other hand, a metalloprotease site of the HEXXH type [58]. In this case, production of a stable complex from Yta10p and Yta12p requires ATP or any of its non-hydrolyzable analogs (ATPγS, AMP-PNP, ADP, GTP). Proteins of the mitochondrial inner membrane are hydrolyzed under the influence of YTA10-12 only in the presence of ATP, and this occurs very rapidly; therefore, when proteins are degraded in mitochondria in organello only its association with low molecular weight intermediate products of proteolysis can be observed.Fig. 3. A hypothetical model of nucleotide-regulated binding of unfolded proteins with mt-Hsp70 and their dissociation with involvement of co-chaperones MDJ and MGE (adapted from [55]). Mt-Hsp70–ATP and MDJ interact with a newly imported unfolded polypeptide producing a MDJ–mt-Hsp70–ATP complex. The binding of the substrate promotes hydrolysis of ATP, and this converts mt-Hsp70 to a form with high affinity for the substrate mt-Hsp70–ADP. The substrate dissociates as a result of the interaction of MGE with the complex. It seems that the function of MGE is replacing ADP by ATP in the complex with mt-Hsp70. The replacement/release of ADP from the complex with mt-Hsp70 leads to dissociation of the complex and release of the substrate with a possibility of its subsequent binding with Hsp60 for folding.
The complex is supposed to occupy a cavity in the inner membrane and promote degradation of membrane proteins in hydrophilic loops. But according to another viewpoint, proteolysis by the YTA10-12 complex occurs not only in water-soluble loops, but rather can involve hydrophobic domains that act as “membrane anchors” that are also subjected to proteolysis [58].
The role of ATP in mechanisms of proteolysis by YTA10‑12 is also unclear. First, it was suggested as a working hypothesis that ATP should be hydrolyzed for initiating the YTA10-12 complex, which could undergo several association–dissociation cycles during the proteolysis. Second, as it has been shown for some chaperones every act of ATP hydrolysis in YTA10-12 may be necessary for the transfer of the substrate from one subunit of the complex to another to ensure its repeated binding and cleavage. Third, the energy of ATP hydrolysis can be spent during the proteolysis of the substrate to increase its sensitivity to the proteolytic attack [58].
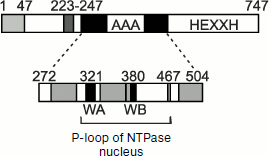
Whereas the proteolytic site of YTA10-12 is exposed to the matrix, proteolysis by the membrane complex Yme1p (i-AAA) occurs in the intermembrane space and is associated, in particular, with removal of an unbound (free) subunit Cox2p of cytochrome c oxidase (see above). The structure of i-AAA and its position in the membrane are characterized by the presence of a special domain that acts as a “sensor” of an unfolded polypeptide chain. This domain has been detected by introduction of deletions into the protein molecule gene and study on their influence on the features of Yme1p [59]. Figure 4 shows that the region between amino acid residues 272 and 504 contains NTPase Walker motifs WA and WB and has no active site of metalloprotease HEXXH inherent in the Yme1p complex. The sensor domain is localized near the transmembrane region of Yme1p (residues 223-247) and is exposed to the intermembrane space (Figs. 2b and 4). Synthesis in E. coli of the Yme1p fragment from residues 250 to 525 (and even from residues 250 to 313) shows that this peptide can act as a chaperone and induce disaggregation of model proteins. Thus, proteolysis of proteins incorporated into the mitochondrial inner membrane from the intermembrane space requires them to be unfolded, and such a condition is more likely provided for by the interactions of the proteins with the Ymel complex and, possibly, with other accessory components [60].
Comparison of WA and WB regions of the YTA10-12 complex with the 250-525 segment of Yme1 (Fig. 2, a and b) shows that the first can also act as a chaperone folding and unfolding proteins newly imported into mitochondria. This is evidenced by an experiment with the double mutant Δyta10E559QΔyta12E614Q deprived of protease sites. This mutant is capable of producing respiratory chain components, as distinguished from a similar mutant containing protease sites [61].Fig. 4. Domain structure of chaperone Yme1: 1-47, mitochondrial addressing sequence; 223-247, transmembrane segment; 275-504, AAA domain containing Walker motifs A and B (WA and WB) (typical AAA regions are shown in gray color); 504-747, proteolytic domain with metal-binding site HEXXH; 272-504, region functioning as a chaperone (adapted from [59]).
What is the further fate of products of proteolysis by different mitochondrial proteases? Mass spectrometry data show that, depending on ATP and temperature, mitochondria release about fifty peptides encoded in mitochondria and nucleus. They are generated by ATP-dependent proteases of the matrix and inner membrane. Pulse-labeling experiments in logarithmically grown yeast have revealed that these peptides are produced as a result of degradation of ~10% of the pre-existing and newly imported proteins [62].
It has been mentioned that synthesis of respiratory chain subunits encoded in the nucleus and consequently the respiratory ability of the cells are determined by transcription factors. On the other hand, synthesis of the respiratory chain proteins encoded in the mitochondrial genome is modulated by expression of certain nuclear genes. These genes, as well as genes needed for biogenesis of the respiratory chain, form a regulon. In particular, this regulon is stimulated under conditions of protease i-AAA deficiency (e.g. with the Δyme1 mutation) because of appearance of deficiency of the mitochondria-generated peptides and increase in the induction of nuclear genes with functions associated with expression of mitochondrial genes. This can be a new signaling pathway from the nucleus to mitochondria [63].
It is also obvious that proteases play an important role in the “quality control” of proteins in all mitochondrial compartments. In particular, cells containing proteolytically inactive proteases manifest phenotypes identical to phenotypes of the corresponding null-mutant cells. All mitochondrial proteases are synthesized in the cytosol under control by the nucleus on the transcription level. On the other hand, proteases PIM1 and YTA10-12 are themselves promoting synthesis of some respiratory chain proteins (e.g. those encoded by COX1 and COB, see above) contributing to stability of their pre-mRNAs and indirectly controlling their splicing [39].
Deficiencies of mitochondrial proteases can lead to accumulation of deficient mitochondrial proteins and their aggregation. The protein aggregation is prevented by a special system of chaperones in mitochondria, among which mt-Hsp70 (Ssc1p) and Hsp78 (hClcX in human) are universal. Chaperonins hsp60 and hsp10 (GroEL and GroES) also could be mentioned, but they are not very efficient in stimulation of folding of proteins synthesized in E. coli cells (in vivo) and yeast (in vitro) [64].
Protein Ssc1p whose functions are associated with the transport protein Tim44 and supported by the co-chaperone Mdj1p and nucleotide exchanger Mge1p (see above) is universal, whereas Ssq1p is specialized for import and assembly of mitochondrial Fe/S clusters (Cyt b2 and Cyt bc1) with involvement of protein Yfh1. This nucleus-encoded mitochondrial protein is involved in iron homeostasis. Ssq1p manifests typical features of a chaperone on the ATP-dependent binding of an unfolded substrate, but it cannot bind with either Tim44 or Mdj1p. It seems that an imbalance between Yfh1 and chaperone Ssq1p results in deficiency of Yfh1 associated with degradation of the latter [65, 66].
Obviously, not all chaperones functioning in mitochondria and proteases coupled with them are known at present. In some cases, proteases are functioning as chaperones that can be exemplified by the yeast protease i-AAA [59].
Deletion of the protease genes YTA10(AFG3) or YTA12(RCA1) in yeast disturbs synthesis of respiratory enzymes (the causes have been mentioned above) and the PIM1(LON) mutation suppresses this deletion, which allows the yeast to grow on a non-fermented substrate, i.e. synthesize components of the respiratory chain and F1Fo-ATPase [67]. This can be partially associated with overlapping of the Yta10p and Yta12p functions with those of Pim1p in stabilization and splicing of pre-mRNAs for respiratory enzymes (encoded by COX1, COB, etc.).
The YTA10(AFG3) deletion affects the growth of yeast on respiration substrates (the pet phenotype) not because of the inactivation of protease Yta10 but because of its non-involvement in formation of the respiratory ensemble. The destruction of YTA10(AFG3) can be suppressed with the polytopic transmembrane protein Oxa1, which is a component of a translocase exporting nucleus-and mitochondria-encoded proteins from the matrix across the inner membrane (see above). The suppression of Oxa1 is manifested by synthesis of cytochrome b, cytochrome aa3, and other products of mitochondrial translation. By contrast, in the case of oxa1 mutation the yeast are deprived of these respiratory proteins and also of ability to assemble F1Fo-ATPase. Similar effects are found for the membrane protein Mba1, which is involved in the assembly of the respiratory chain complexes [68].
Thus, there are complicated correlations between the “effective” concentration of mitochondrial proteolytic enzymes responsible for “quality control” of imported proteins on one hand, and the level of synthesis of these enzymes, contents of various chaperones, and different factors influencing the sorting of these proteins on the other hand.
GENERAL PRINCIPLES OF CONTROL OVER FORMATION OF STRUCTURES
RESPONSIBLE FOR RESPIRATORY FUNCTIONS OF MITOCHONDRIA
As follows from the previous sections, there are two fundamental principles of quality control of mitochondrial proteins. The first allows deficient proteins to be eliminated. In the second case, unbound (free) proteins are destroyed if there are disorders in stoichiometry of protein complexes.
This is realized with involvement of various proteases, chaperones, and different factors addressing proteins to their corresponding compartments. But a question arises how structures of higher order, such as mitochondria, are controlled? What principles are realized in addition to the abovementioned principle of quality control of proteins, which acts on a supermolecular (subcellular) level? Some aspects of this problem related to mitochondria will be elucidated in this section.
Effects of Vacuolar Protease Inhibitors on Contents of Cytochromes and Respiratory Complexes in Yeast
Yeast grown on fermentable and non-fermentable substrates in the presence of small amounts of synthetic inhibitors of vacuolar proteases A (yscA, PEP4) and B (yscA, PRB1) were more than 25 years ago found to contain 20-30% more cytochromes c, c1, b, and aa3, while the level of cell respiration was unchanged [69].
As presented in Fig. 5, during cell growth on galactose the cytochrome contents increased during the logarithmic growth phase and decreased during the stationary phase. The specific cell respiration (coupled or uncoupled) increased insignificantly. Addition of protease inhibitors (phenylmethylsulfonyl fluoride (PMSF) and pepstatin) into the medium in the middle of the logarithmic growth phase removed the decrease in the contents of cytochromes b, c1, c, and aa3 during the stationary phase and the specific cell respiration did not change during the subsequent growth of the cells. Therefore, it was suggested that in yeast cells grown on galactose a significant share of the cytochromes should not be involved in the cell respiration.
During yeast growth on glucose, the specific cell respiration increases in parallel with a monotonic increase in the cytochrome contents in the exponential growth phase (Fig. 6). Addition of protease inhibitors to the growth medium in the beginning of the process resulted in a progressing accumulation of cytochromes b, c1, c, and aa3. However, the inhibitors failed to induce changes in the respiratory activity of the cells. Under these conditions “surplus” cytochromes seemed to degrade so quickly that they could be detected only in the presence of protease inhibitorsFig. 5. Effects of protease inhibitors PMSF and pepstatin on contents of cytochromes b, c1, c, and aa3 in S. cerevisiae cells during their aerobic growth in galactose-containing medium (adapted from [74]). The left upper panel presents cell growth and cell respiration on stimulation and without stimulation by the uncoupler.
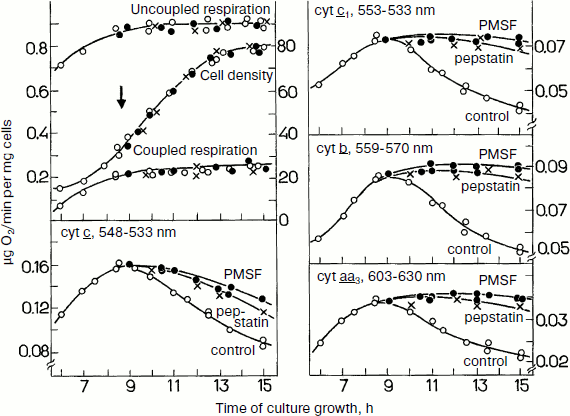
Experiments with the total mitochondrial fraction of yeast cells gave some information concerning the nature of the “surplus” cytochromes. Protease inhibitors seemed to stimulate accumulation or retention of some individual enzymatic complexes of the respiratory chain, which, in spite of the presence of potential activity, failed to contribute to the cell respiration and were degraded. As judged from values of the partial maximal enzymatic activities, the amount of “surplus” complexes correlated with the amounts of “surplus” cytochromes. Thus, the “surplus” cytochromes are likely to be components of non-functioning respiratory complexes.Fig. 6. Effects of protease inhibitors PMSF and pepstatin on contents of cytochromes b, c1, c, and aa3 in S. cerevisiae cells during their aerobic growth in glucose-containing medium (adapted from [69]). The left upper panel presents the cell growth and cell respiration on stimulation and without stimulation by the uncoupler.
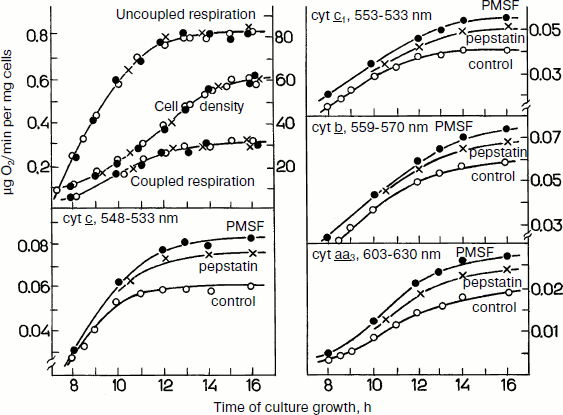
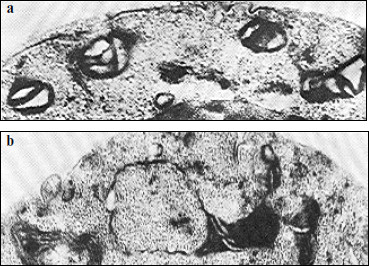
The nature of this phenomenon is still unclear. It might be caused by an inhomogeneous distribution of respiratory complexes in the mitochondrial inner membrane; in other words, there might be a disturbance in stoichiometry of the supercomplexes described above. Disorders in formation of respiratory chains because of changes in the membrane structure can also occur. Some authors suppose that large supramolecular structures or even complexes of respiratory chains can be formed within the membrane [28]. Some data suggest that deficient mitochondrial structures can characterize “young” cells during the pre-exponential growth phase on glucose [70]. Figure 7 exemplifies some variants of deficient mitochondria (“monsters”), which are specified either by a reduced matrix or by the presence of inner mitochondrial structures of unknown origin, etc.
Different variants of wrong interactions between membrane proteins and their complexes and also of local changes in the membrane structure have been analyzed in detail in [71] and are presented in Fig. 8.Fig. 7. Electron microphotographs of different mitochondrial structures of aerobically grown yeast S. cerevisiae (the author’s data). a) Promitochondrion-like structures in young cells, ×150,000. b) A magnified mitochondrion of wrong shape (“monster mitochondrion”) in young yeast cells, ×150,000.
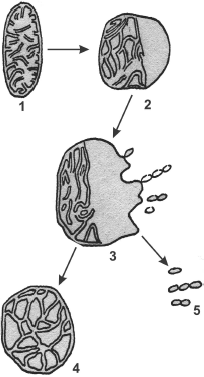
It has been mentioned above that deficient mitochondrial structures are probably eliminated with the involvement of vacuolar proteases A and B. This can be realized during macroautophagy processes, which are well studied in the case of mitochondria [72]. But microautophagy can be demonstrated only in model experiments. At first, swelling of a mitochondrion occurs. One can see a swollen mitochondrion in the electron microphotograph (Fig. 9) [70]. The further swelling of a mitochondrion results in a disruption of the inner and outer membranes and partial release of contents of the intermembrane space and cristae. Then the mitochondrion took the so-called condensed shape. The whole process is presented in Fig. 10 [73].Fig. 8. Possible mechanisms of elimination of deficient membrane components (adapted from [70]): (i) components are eliminated by direct lysis; (ii) components are separated with disruption of the attached protein; (iii) swelling of a membrane region with formation of a vesicle and subsequent release of components into the medium; (iv) invagination of a membrane part with formation of an intracellular vesicle.
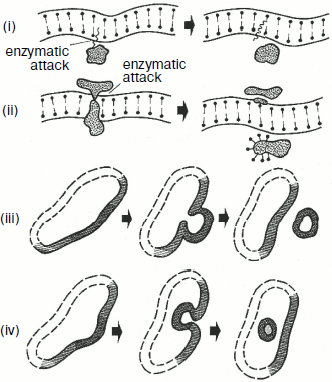
Fig. 9. Electron microphotograph of a swollen mitochondrion of aerobically grown S. cerevisiae, ×44,000 (the author’s data).
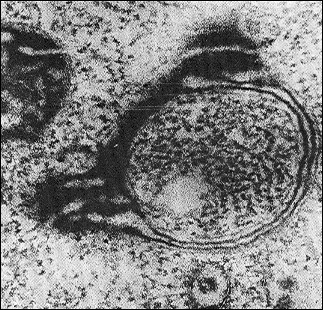
Fig. 10. A model of mitochondrial microautophagy (adapted from [70]): 1) normal mitochondrion; 2) initial stage of swelling; 3) swollen mitochondrion at the stage of released “vesicles”; 4) deformed mitochondrion; 5) free “vesicles” swimming in the cytosol.
CONCLUSIONS
The present review brings us to two conclusions. First, formation of the mitochondrial respiratory chain is accompanied by a surplus synthesis of cytochromes and “superfluous” parts of respiratory complexes, which possess catalytic activities but fail to contribute to the respiratory activity of mitochondria. Surplus components of the respiratory chain are eliminated with involvement of vacuoles, which is evidenced by the above-described effects of protease inhibitors. This phenomenon was earlier named the principle of “selection by performance criterion” [74].
Second, every type of disorder in oxidative phosphorylation is associated with destructions in “mature” mitochondria that, in particular, manifest themselves by swelling of mitochondria, decrease in the number of cristae, and partial digestion of mitochondria by the cells with involvement of vacuoles (the principle of “stabilization by functioning”) [74].
Obviously, these principles are speculative and require additional experimental studies. On the other hand, these principles are formulated to design a general line for further research. It should be emphasized that up to now virtually all works have been directed to elucidation of mechanisms of “quality control” of individual proteins and their complexes and did not concern principles of formation of cell organelles or their subcellular compartments.
This work was supported by the Russian Foundation for Basic Research (project Nos. 00-15-97942, 02-04-48204, and 05-04-48049).
REFERENCES
1.Zorov, D. B., Isaev, N. K., Plotnikov, E. Yu.,
Zorova, L. D., Stelmashook, E. V., Vasileva, A. K., Arkhangelskaya, A.
A., and Khrjapenkova, T. G. (2007) Biochemistry (Moscow),
72, 1115-1126.
2.Suen, D. G., Norris, K. L., and Youle, R. J. (2008)
Genes Dev., 22, 1577-1590.
3.Neupert, W., and Herrmann, J. M. (2007) Annu.
Rev. Biochem., 76, 723-749.
4.Boekema, E. J., and Braun, H.-P. (2007) J. Biol.
Chem., 282, 1-4.
5.Luzikov, V. N. (1980) Regulation of Mitochondria
Formation [in Russian], Nauka, Moscow.
6.Shwerzmann, K., Cruz-Orive, L. M., Eggman, R.,
Sanger, A., and Weibel, E. R. (1986) J. Cell Biol., 102,
97-103.
7.Wallace, D. C. (2005) Ann. Rev. Genet.,
39, 359-407.
8.Poyton, R. O., and McEwen, J. E. (1996) Annu.
Rev. Biochem., 65, 563-607.
9.Fontanesi, F., Soto, I. C., and Barrientos, A.
(2008) IUBMB Life, 60, 557-568.
10.Tsukihara, T., Aoyama, H., Yamashita, E.,
Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono,
R., and Yoshikawa, S. (1996) Science, 272, 1136-1144.
11.Capaldi, R. A., Malatesta, F., and Darley-Usmar,
V. M. (1983) Biochim. Biophys. Acta, 726, 135-148.
12.Church, C., Goehring, B., Forsha, D., Wazny, P.,
and Poyton, R. O. (2005) J. Biol. Chem., 280,
1854-1863.
13.Zara, V., Palmisano, I., Conte, L., and
Trumpower, B. L. (2004) Eur. J. Biochem., 271,
1209-1218.
14.Cruciat, C.-M., Hell, K., Folsch, H., Neupert,
W., and Stuart, R. A. (1999) EMBO J., 18, 5226-5233.
15.Sazanov, L. A., Peak-Chew, S. Y., Fearnley, I.
M., and Walker, J. E. (2000) Biochemistry, 39,
7229-7235.
16.Grivennikova, V. G., and Vinogradov, A. D. (2003)
Usp. Biol. Khim., 43, 19-58.
17.Duarte, M., Sousa, R., and Videira, A. (1995)
Genetics, 139, 1211-1221.
18.Boyer, P. D. (1997) Annu. Rev. Biochem.,
66, 717-749.
19.Weber, J. (2007) Trends Biochem. Sci.,
32, 53-56.
20.Lau, W. C., Baker, L. A., and Rubinstein, J. L.
(2008) J. Mol. Biol., 382, 1256-1264.
21.Devenish, R. J., Prescott, M., and Rodgers, A. J.
(2008) Int. Rev. Cell Mol. Biol., 267, 1-58.
22.Tzagoloff, A., Barrientos, A., Neupert, W., and
Herrmann, J. M. (2004) J. Biol. Chem., 279,
19775-19780.
23.Wang, Z. G., White, P. S., and Ackerman, S. H.
(2001) J. Biol. Chem., 276, 30773-30778.
24.Green, D. E., and Hechter, O. (1965) Proc.
Natl. Acad. Sci. USA, 53, 318-325.
25.Schagger, H., and Pfeiffer, K. (2000) EMBO
J., 19, 1777-1783.
26.Cruciat, C.-M., Brunner, S., Baumann, F.,
Neupert, W., and Stuart, R. A. (2000) J. Biol. Chem.,
275, 18093-18098.
27.Schafer, E., Seelert, H., Reifschneider, N. H.,
Krause, F., Dencher, N. A., and Vonck, J. (2006) J. Biol. Chem.,
281, 15370-15375.
28.Schagger, H. (2001) IUBMB Life, 52,
119-128.
29.Luzikov, V. N. (1973) Subcell. Biochem.,
2, 1-31.
30.Ko, Y. H., Delannoy, M., Hullihen, J., Chiu, W.,
and Pedersen, P. L. (2003) J. Biol. Chem., 278,
12305-12309.
31.Chen, C., Ko, Y., Delannoy, M., Ludtke, S. J.,
Chiu, W., and Pedersen, P. L. (2004) J. Biol. Chem., 279,
31761-31768.
32.Shutt, T. E., and Gray, M. W. (2006) Trends
Gen., 22, 90-95.
33.Ryan, M. T., and Hoogenraad, N. J. (2007)
Annu. Rev. Biochem., 76, 701-722.
34.Scarpulla, R. C. (2008) Physiol. Rev.,
88, 611-638.
35.Wu, Z., Puigserver, P., Andersson, U., Zhang, G.,
Adelmant, G., Mootha, V., Troy, A., Cinti, S., Lowell, B., Scarpulla,
R. C., and Spiegelman, B. M. (1999) Cell, 98,
115-124.
36.Arny, Z., Wagner, B. K., Ma, Y., Chinsomboom, J.,
Laznik, D., and Spiegelman, B. M. (2008) Proc. Natl. Acad. Sci.
USA, 105, 4721-4725.
37.Kutik, S., Guiarad, B., Meyer, H., Wiedemann, N.,
and Pfanner, N. (2007) J. Biol. Chem., 179, 585-591.
38.Neupert, W. (1997) Annu. Rev. Biochem.,
66, 863-917.
39.Van Dyck, L., and Langer, T. (1999) Cell Mol.
Life Sci., 56, 825-842.
40.Young, L., Leonhard, K., Tatsuta, T., Trowsdale,
J., and Langer, T. (2001) Science, 291, 2135-2138.
41.Steglich, G., Neupert, W., and Langer, T. (1999)
Mol. Cell. Biol., 19, 3435-3442.
42.Langer, T. (2000) Trends Biochem. Sci.,
25, 247-251.
43.Kaser, M., Kambacheld, M., Kisters-Woike, B., and
Langer, T. (2003) J. Biol. Chem., 278, 46414-46423.
44.Kambacheld, M., Augustin, S., Tatsuta, T.,
Muller, S., and Langer, T. (2005) J. Biol. Chem., 280,
20132-20139.
45.Wheeldon, L. W., Dianoux, A. C., Bof, M., and
Vignais, P. V. (1974) Eur. J. Biochem., 46, 189-199.
46.Kalnov, S. L., Novikova, L. A., Zubatov, A. S.,
and Luzikov, V. N. (1979) Biochem. J., 182, 195-202.
47.Kalnov, S. L., Novikova, L. A., Zubatov, A. S.,
and Luzikov, V. N. (1979) FEBS Lett., 101, 355-358.
48.Suzuki, C. K., Rep, M., van Dijl, J. M., Suda,
K., Grivell, L. A., and Schatz, G. (1997) Trends Biochem. Sci.,
22, 118-123.
49.Guelin, E., Rep, M., and Grivell, L. A. (1996)
FEBS Lett., 381, 42-46.
50.Savel’ev, A. S., Novikova, L. A., Kovaleva,
I. E., Luzikov, V. N., Neupert, W., and Langer, T. (1998) J. Biol.
Chem., 273, 20596-20602.
51.Weber, E. R., Hanekamp, T., and Thorsness, P. E.
(1996) Mol. Biol. Cell., 7, 307-317.
52.Pearce, D. A., and Sherman, F. (1997) J. Biol.
Chem., 272, 31829-31836.
53.Wagner, I., Arlt, H., van Dyck, L., Langer, T.,
and Neupert, W. (1994) EMBO J., 13, 5135-5145.
54.Kovaleva, I. E., Novikova, L. A., Nazarov, P. A.,
Grivennikov, S. I., and Luzikov, V. N. (2003) Eur. J. Biochem.,
270, 222-229.
55.Stuart, R. A., Cyr, D. M., Craig, E. A., and
Neupert, W. (1994) Trends Biochem. Sci., 19, 87-92.
56.Rottgers, K., Zufall, N., Guiard, B., and Voos,
W. (2002) J. Biol. Chem., 277, 45829-45837.
57.Von Janovsky, B., Major, T., Knapp, K., and Koos,
W. (2006) J. Mol. Biol., 31, 799-807.
58.Arlt, H., Tauer, R., Feldmann, H., Neupert, W.,
and Langer, T. (1996) Cell, 85, 875-885.
59.Leonhard, K., Sriegler, A., Neupert, W., and
Langer, T. (1999) Nature, 398, 348-351.
60.Graef, M., Seewald, G., and Langer, T. (2007)
Mol. Cell. Biol., 27, 2476-2485.
61.Arlt, H., Steglich, G., Perryman, R., Guiard, B.,
Neupert, W., and Langer, T. (1998) EMBO J., 17,
4837-4847.
62.Augustin, S., Nolden, M., Muller, S., Hardt,
O., Arnold, I., and Langer, T. (2005) J. Biol. Chem.,
280, 2691-2699.
63.Arnold, I., Wagner-Ecker, M., Ansorge, W., and
Langer, T. (2006) Gene, 367, 74-88.
64.Dubaquie, Y., Looser, R., Funfschilling, U.,
Jeno, P., and Rospert, S. (1998) EMBO J., 17,
5868-5876.
65.Voisine, C., Schilke, B., Ohlson, M., Beinert,
H., Marszalek, J., and Craig, E. A. (2000) Mol. Cell. Biol.,
20, 3677-3684.
66.Schmidt, S., Strub, A., Rottgers, K., Zufall, N.,
and Voos, W. (2001) J. Mol. Biol., 313, 13-26.
67.Rep, M., van Dijl, J. M., Suda, K., Schatz, G.,
Grivell, L. A., and Suzuki, C. K. (1996) Science, 274,
103-106.
68.Rep, M., Nooy, J., Guelin, E., and Grivell, L. A.
(1996) Curr. Genet., 30, 206-211.
69.Galkin, A. V., Tsoi, T. V., and Luzikov, V. N.
(1980) Biochem. J., 190, 145-156.
70.Luzikov, V. N., Zubatov, A. S., and Rainina, E.
I. (1973) J. Bioenerg. Biomembr., 5, 129-149.
71.Parry, G. (1978) Subcell. Biochem.,
5, 261-325.
72.Baba, M., Takeshige, K., Baba, N., and Ohsumi, Y.
(1994) J. Cell Biol., 124, 903-913.
73.Ziegler, D. M., Linnane, A. W., Green, D. E.,
Dass, C. M., and Ris, H. (1958) Biochim. Biophys. Acta,
28, 524-538.
74.Luzikov, V. N. (1985) Mitochondrial Biogenesis
and Breakdown, Plenum Publisher Corporation, New York-London.