Production of Biologically Active Human Myelocytokines in Plants
A. S. Zvereva1*, L. E. Petrovskaya2, A. V. Rodina3, O. Y. Frolova1, P. A. Ivanov1, L. N. Shingarova2, T. V. Komarova4, Y. L. Dorokhov4, D. A. Dolgikh2, M. P. Kirpichnikov1,2, and J. G. Atabekov1
1Faculty of Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 938-0601; E-mail: zvereva@gmail.com; anna.zvereva@genebee.msu.ru2Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: (495) 330-6983; E-mail: lpetr65@yahoo.com
3Moscow Institute of Medical Ecology, Simferopolsky Bulvar 8, 117638 Moscow, Russia; fax: (499) 113-4827; E-mail: allrodina@yandex.ru
4Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 938-3181; E-mail: dorokhov@genebee.msu.ru
* To whom correspondence should be addressed.
Received March 24, 2009; Revision received May 27, 2009
An effective system for expression of human granulocyte and granulocyte macrophage colony-stimulating factors (hG-CSF and hGM-CSF) in Nicotiana benthamiana plants was developed using viral vector based on tobacco mosaic virus infecting cruciferous plants. The genes of target proteins were cloned into the viral vector driven by actin promoter of Arabidopsis thaliana. The expression vectors were delivered into plant cells by agroinjection. Maximal synthesis rate was detected 5 days after injection and was up to 500 and 300 mg per kg of fresh leaves for hG-CSF and hGM-CSF, respectively. The yield of purified hG-CSF and hGM-CSF was 100 and 50 mg/kg of fresh leaves, respectively. Recombinant plant-made hG-CSF and hGM-CSF stimulated proliferation of murine bone marrow and human erythroleucosis TF-1 cells, respectively, at the same rate as the commercial drugs.
KEY WORDS: granulocyte colony-stimulating factor, granulocyte macrophage colony-stimulating factor, viral vector, agroinjectionDOI: 10.1134/S0006297909110029
Abbreviations: CFU-C, colony-forming units in culture; crTMV, tobacco mosaic virus infecting cruciferous plants; ED50, dose necessary for 50% stimulation of proliferation; FCS, fetal calf serum; GFP, green fluorescent protein; hG-CSF, human granulocyte colony-stimulating factor; hGM-CSF, human granulocyte macrophage colony-stimulating factor; IPTG, isopropyl-β-D-thiogalactopyranoside; phG-CSF, rhG-CSF synthesized in plants; phGM-CSF, rhGM-CSF synthesized in plants; PMSF, phenylmethylsulfonyl fluoride; rhG-CSF, recombinant hG-CSF; rhGM-CSF, recombinant hGM-CSF.
In recent years the development of new methods for cost-effective
production of pharmaceuticals has become increasingly important. Use of
plant expression systems for recombinant protein production has a
number of advantages such as inexpensive cultivation, possibility of
simultaneous production of a large amount of raw material and
comparatively easy storage and transportation. In addition, the
technology of human protein production in plants is usually safe
because there are no common pathogens. Present knowledge demonstrates
that recombinant proteins expressed in plants are biologically active
and can be used as pharmaceuticals [1, 2].
Earlier, we developed a system for expression of foreign proteins in Nicotiana benthamiana leaves using phytoviral vectors [3]. Application of plant viral vectors challenges such gene expression methods as use of stably transformed or transiently modified plant tissues. Due to extremely high rate of viral RNA replication, high copy number of foreign genes transcripts is achieved in cytoplasm of infected cells. Therefore, the efficiency of a viral expression system is two degrees higher comparing with stably transformed plants [1]. In addition, viral infection in plants is commonly fully developed after 1-2 weeks, while development, control, and cultivation of transgenic plants take about 2 years.
Using viral expression systems, such proteins as somatotropin [4], hepatitis B viral antigen [5], the antigens of tuberculosis [6] and plague [7] bacteria, and some monoclonal antibodies [8] have been produced in plants.
Granulocyte colony-stimulating factor (hG-CSF) regulates formation and maturation of functionally active neutrophilic granulocytes [9]. Recombinant hG-CSF (rhG-CSF) is widely used in oncology since chemotherapy often leads to the depletion of neutrophils. In addition, hG-CSF activates various infection protective systems of an organism as well as being applied in stimulation of stem cell release from bone marrow [10].
Granulocyte macrophage colony-stimulating factor (hGM-CSF) regulates proliferation and maturation of myeloid cell precursors as well as functions of mature neutrophils, eosinophils, and monocytes. It is used in clinics for treatment of myelodysplastic syndromes, myelosuppression resulting from chemotherapy and bone marrow transplantation, aplastic anemia, etc. [11].
We have developed a method for expression of human hG-CSF and hGM-CSF in N. benthamiana plants. The genes hG-CSF and hGM-CSF were cloned into a viral vector based on the tobacco mosaic virus genome infecting cruciferous plants (crTMV). The vector was introduced into plants by the agroinjection method. Nicotiana benthamiana leaves were inoculated using a suspension of agrobacteria transformed with crTMV genome constructions containing target protein (hG-CSF or hGM-CSF) genes. To stimulate productivity and increase the replication rate of the viral vector, the experiments were carried out under conditions of co-agroinjection of a mixture of agrobacteria carrying viral vector genome and agrobacteria containing viral p19 gene of tomato bushy stunt virus. Protein p19 is a suppressor of antiviral response of the host plant cells, i.e. a suppressor of gene silencing of viral RNA. Co-agroinjection of p19 gene increases the stability of the viral vector RNA [12]. This expression system is shown to increase the synthesis of both target proteins to 0.1-1.0 g/kg of fresh leaves. We elaborated a method of target protein extraction and purification from plant leaves that provides the biologically active rhG-CSF and rhGM-CSF.
MATERIALS AND METHODS
Strains Escherichia coli XL-1 Blue (Stratagene, USA) and SG13009[pREP4] (Qiagen, Germany) as well as Agrobacterium tumefaciens GV3101 strain from the collection of the Department of Virology (Moscow State University) were used in the present research. Oligonucleotides were synthesized by Sintol (Russia). Recombinant DNA was cloned in E. coli XL-1 Blue using standard methods [13]. Restrictases, DNA ligase, Taq- and Pfu-polymerases from Fermentas (Lithuania) and enteropeptidase purified in the Laboratory of Protein Engineering of the Institute of Bioorganic Chemistry (Russian Academy of Sciences) were used [14].
Cloning of the target genes into expression vectors. For amplification of the hG-CSF gene by PCR, the DNA of pGGF8 plasmid [15] as a template and primers G-CSF-NcoI-p 5′- ctagccatggatgacaccattaggtcctgcttcg and G-CSF·6His-m 5′-ctagtctagattagtgatggtgatggtgatggggctgggcaaggtggcgtag were used. Under the conditions recommended by producer of the enzyme, PCR was carried out using Taq/Pfu (100 : 1) mixture of DNA polymerases. The resulting PCR product 1 containing the hG-CSF gene with six histidine codons at the 3′-end was cloned into the crTMV:GFP viral vector based on crTMV described earlier [3]. The GFP (green fluorescent protein) gene in this vector was replaced with hG-CSF at NcoI-XbaI restriction sites.
The hGM-CSF gene was amplified under the same conditions on DNA template of pFGM17 plasmid [16] using the following primers: GM-CSF-p 5′-gacgacgacaaggcacctgctagatctcaaa and GM-CSFSalI-m 5′-GATCgtcgacTTACTCCTGCACGGGTTCCC (PCR product 2). The hexahistidine sequence together with an enteropeptidase cleavage site (PCR product 3) was obtained by PCR using the following primers: Ent-p 5′-gatctgtacaatgcaccatcaccatcatcatccagatctg and Ent-m 5′-gtagtaggtctagacccatggctgctgctgttccgtg. Ent-m and GM-CSF-p primers contain overlapping regions, therefore PCR products 2 and 3 were joined through an additional PCR round using Ent-p and GM-CSF-SalI-m primers.
The resulting PCR product containing the hGM-CSF gene with the enteropeptidase cleavage site and six histidine codons at the 5′-end (6His·Ent·GM-CSF) was cloned into the crTMV:GFP vector in place of the GFP gene at the BsrGI-SalI restriction sites. The structure of the resulting plasmid was confirmed by restriction analysis and sequencing (Genome Multiple Access Center, Russia).
Extraction of rhG-CSF from E. coli. An rhG-CSF producing strain was obtained by cloning the hG-CSF synthetic gene into pQE30 plasmid (Qiagen) from the pGGF8 plasmid [15] at the BamHI-SalI sites. Escherichia coli SG13009[pREP4] cells transformed by pQE30:hG-CSF plasmid were incubated in LB medium containing ampicillin (100 mg/liter) overnight at 37°C. The overnight culture was diluted with the same medium, incubated to A600 = 0.7, isopropyl-β-D-thiogalactopyranoside (IPTG) was added to the final concentration 1 mM, and the culture was incubated for 4 h at 37°C. The biomass was suspended in buffer A (50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 100 mM NaCl), and the cells were disrupted by ultrasonication. After centrifugation, the pellet was washed with buffer A containing 0.5% Triton X-100 and 2 M urea and diluted with B buffer (20 mM sodium phosphate, pH 7.4, 6 M guanidine chloride) and kept for 16 h at room temperature. The rhG-CSF·6His was isolated using metal affinity chromatography on Ni2+-NTA agarose (Qiagen). Supernatant of dissolved inclusion bodies was applied onto the column equilibrated with B buffer. Gradually increasing imidazole concentration (from 10 to 500 mM) in the same buffer, rhG-CSF was eluted. The resulting rhG-CSF carrying 10 extra amino acid residues (including the six histidine residues) at the N-terminal end came as a result of cloning into the pQE30 vector.
Agroinjection. Strain GV3101 of the A. tumefaciens was transformed by pA10523 and pA12633 plasmids separately and then incubated overnight at 28°C in LB medium containing 50 mg/liter rifampicin, 50 mg/liter kanamycin, and 25 mg/liter gentamycin. The overnight culture was centrifuged at 3000g for 5 min. The pellet was resuspended in agroinjection buffer containing 10 mM MgSO4 and 10 mM MES-NaOH (pH 5.5). The agrobacterial suspension was diluted with agroinjection buffer to A600 = 0.4. Before infiltration, equal amounts of agrobacteria containing target protein gene (hG-CSF or hGM-CSF) and agrobacteria containing p19 protein gene known as suppressor of post-transcriptional transgene silencing were mixed. N. benthamiana leaves were infiltrated with such suspension using a syringe without a needle. After infiltration, the plants were grown at 16 h long daylight at 22°C.
Expression analysis. To analyze the expression, the injection zones (2 mg) were ground with celite (Serva, Germany), and 40 µl of loading buffer (60% glycerol, 5 mM β-mercaptoethanol, 10% SDS, 250 mM Tris-HCl, pH 6.8) was added. The resulting extracts were heated for 5 min at 95°C. The total amount of protein was measured using a Protein Assay Kit (Bio-Rad, USA), and 50 µg protein was applied to each well of an electrophoresis gel. As a control, a known amount of recombinant protein from E. coli was applied on the gel. The proteins were fractionated using electrophoresis in 15% polyacrylamide gel using the Laemmli method [17] and then transferred to Hybond-P membrane (Amersham, Sweden). After transfer, the membrane was incubated with blocking solution of 5% skim milk (Difco, USA). To detect hG-CSF, primary murine antibodies purified using affinity chromatography and secondary antibodies conjugated with peroxidase (Sigma, Germany) were used. Primary goat antibodies to hGM-CSF (R&D Systems, USA) and secondary peroxidase-conjugated antibodies (R&D Systems) were used to detect hGM-CSF. The reaction products were visualized by the chemiluminescence method using the ECL system (Amersham). To estimate quantitatively the expression, the blot was scanned and band intensities were compared using TotalLab 1.10 software (Nonlinear Dynamics Ltd., Great Britain).
To analyze preparations of total soluble protein after electrophoresis, the polyacrylamide gel was stained with 0.05% Coomassie R-250 solution (Serva) in a mixture of 2% trichloroacetic acid and 20% ethanol for 1 h and then washed by boiling in distilled water.
Extraction and purification of proteins from plant leaves. On the fifth day after infection, 5 g of leaves were homogenized in 15 ml of buffer H (100 mM Hepes-NaOH, pH 7.4, 10 mM EDTA, 100 mM NaCl, 5 mM dithiothreitol, 25 mM sucrose, 0.4 mM phenylmethylsulfonyl fluoride (PMSF)) (Amresco, USA). The extraction was carried out as described by Yamamoto et al. [18]: homogenate was centrifuged at 10,000g for 10 min, and the resulting pellet was suspended in 15 ml of buffer S (50 mM Tris-HCl, pH 8.0, 6 M guanidine hydrochloride, 20% glycerol, 1 mM β-mercaptoethanol, 10 mM imidazole) and incubated for 1 h at room temperature. The sample was centrifuged at 12,000g for 20 min to remove cell debris and insoluble material. The supernatant was added to 2 ml of Ni2+-NTA agarose (Qiagen), carefully mixed for 1 h at room temperature, and then loaded onto a column. The column was washed with 40 ml of S buffer and then with 120 ml of buffer S with decreasing gradients of guanidine hydrochloride (from 6 to 0 M), glycerol (from 20 to 0%), and β-mercaptoethanol (from 1 to 0 mM). Then the column was equilibrated with buffer L (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 5% sorbitol) containing 10 mM imidazole, eluted with buffer L containing 40 mM imidazole, and then the protein was eluted by increasing the concentration of imidazole in buffer L to 250 mM. The eluate was analyzed by electrophoresis in 15% polyacrylamide gel and stained by Coomassie R-250. Fractions containing the largest amount of protein were dialyzed against buffer containing 10 mM Tris-HCl, pH 8.0, 50 mM NaCl and kept at 4°C.
Hydrolysis by enteropeptidase. The hexahistidine tag was cleaved by enteropeptidase [14]. Three units of the enzyme per mg protein were added, and the mixture was incubated for 16 h at room temperature. The rate of hydrolysis was monitored by electrophoresis in 15% polyacrylamide gel by the Laemmli method. To inactivate the enzyme, PMSF was added to final concentration 0.4 mM, and then chromatography on Ni2+-NTA agarose was repeated.
Biological activity of hG-CSF was determined by the rate of stimulation of cell proliferation of murine bone marrow. For this assay, 4·105 cells in 100 µl medium were applied into 1 well of a 24-well plate (four wells per variant) in 1.12 ml medium of the following composition: 600 µl fetal calf serum (FCS), 120 µl concentrated nutrient medium (see further), 200 µl medium containing colony-stimulating factor, 100 µl erythropoietin (50 U/ml; Boehringer Mannheim, Germany), 0.8% methyl-cellulose (ICN, USA). Concentrated nutrient medium contained 2% glutamine, 0.075% bovine serum albumin (Gibco, USA), 10 µM β-mercaptoethanol, 20 mM Hepes-NaOH, 100 U/ml penicillin, and 50 µg/ml streptomycin. As a positive control, 200 µl mixture of conditioned media of WEHI 3B and L929 lines (2 : 1 v/v) [19, 20] and 200 µl hG-CSF (Neupogen; F. Hoffmann-La Roche, Switzerland) in 20 ng/ml concentration were used. As testing samples, 200 µl of purified hG-CSF from plants in various concentrations were used. After seven days of cultivation (37°C, 5% CO2), colony number for each variant was counted using an inverted microscope (Nikon, China).
Biological activity of hGM-CSF was determined by stimulation of proliferation of TF-1 cell line of human erythroleucosis [21]. TF-1 cells (No. CRL-2003; ATCC, USA) were cultivated in RPMI 1640 medium supplemented with 10 mM Hepes-NaOH, 1 mM sodium pyruvate, 4.5 g/liter glucose, 1.5 g/liter sodium bicarbonate, 2 ng/ml recombinant hGM-CSF (rhGM-CSF) [16], 10% FCS, 100 U/ml penicillin, and 100 µg/ml streptomycin. Before the experiment, the cells were washed with serum-free RPMI 1640 medium. Then the cells were applied into 96-well plates (Corning, USA) at 15,000 per well in RPMI 1640 medium containing 2% FCS, and hGM-CSF was applied in concentrations from 0.4 pg/ml to 10 ng/ml. The cells were incubated for 72 h, and then 3H-labeled thymidine (1 µCi per well; Izotop, Russia) was added. After 18 h, the cells were collected on filters using an automatic cell collecting device, and radioactivity was measured using a liquid scintillation counter (Rackbeta, USA; LKB, USA). Stimulation index was calculated in the cells containing hGM-CSF and without it as the ratio of radioactivities and presented in percent. Inclusion of 3H-labeled thymidine into the control (non-stimulated) cells was taken as 100%. Biological activity of hGM-CSF (ED50) was determined as hGM-CSF concentration at which 50% of the maximal proliferation stimulation was observed.
RESULTS AND DISCUSSION
In recent years transgenic plants producing a wide range of hormones, cytokines, growth factors, and enzymes having potential pharmacological application have been created in a number of biotechnological centers [1, 2]. Recombinant proteins produced in plants are shown to have the same biological activity with the protein analogs produced in other expression systems. Despite very effective promoters, the level of foreign gene expression in plants, however, is usually low. Thus, the amount of human serum albumin in transgenic tobacco tissues was 0.02% of total protein [22]. Even lower values were obtained for erythropoietin (0.003%) and β-interferon (0.001%) [23, 24]. The low productivity of the target proteins in the transformed plants might be caused by the plant defense reaction – gene silencing [25].
To increase the level of target protein accumulation in plants, transient expression of transgenes using phytoviral vectors is also used. Use of viral vectors together with agroinjection is one of the most promising methods of gene expression [26]. Proteins were earlier demonstrated to show the most effective expression using tobacco mosaic virus (TMV). For instance, the amount of GFP protein synthesized using TMV30B vector was 10% of total soluble protein of infected N. benthamiana leaves [27]. A vector based on TMV was also used for production of HIV replication inhibitor α-trichosanthin in N. benthamiana with yield of 2% of total soluble protein [28].
As mentioned earlier, vectors based on crTMV with very high productivity were used in the present work [29]. The technology of viral vector agroinjection is optimal for inexpensive and rapid expression of target proteins since the higher protein concentration in cells simplifies the purification process. This is particularly significant in the case of pharmaceutical proteins requiring high level of purification.
Human colony-stimulating factors hG-CSF and hGM-CSF were chosen for expression in plants using crTMV. At present, imported recombinant cytokines, which, along with granulopoiesis (hG-CSF, hGM-CSF) and monocytopoiesis (hGM-CSF) stimulation, possess immunomodulatory effect, are registered and available for sale in Russia. Protein hG-CSF proved to be effective in prevention and treatment of purulent septic infections in surgery, including oncosurgery, as well as treatment of patients with refractory chronic infections. High prices of colony-stimulating factor pharmaceuticals significantly restrict their wide application. Use of viral vectors for hG-CSF and hGM-CSF synthesis in plants might significantly lower production cost and, consequently, make the treatment of various diseases including oncological ones less expensive.
Up to the present, hG-CSF expression in plants was achieved only using cell cultures of transgenic plants with low product output (105 µg/liter) [30], and hGM-CSF expression was achieved only using transgenic tobacco seeds [31] with output 0.03% of soluble protein, and using other cell cultures of transgenic plants [32-34]. High expression rate was achieved using a vector based on potato X virus, and hGM-CSF yield in leaves was up to 20 mg per g of fresh leaves. However, isolation and characterization of the purified recombinant cytokine preparation was not shown in the work [35].
To express colony-stimulating factors in plant cells, we have cloned hG-CSF and hGM-CSF genes of target proteins into a vector based on crTMV (Scheme). To make purification easier, their 3′- and 5′-end regions, respectively, contain sequences encoding six histidine residues. In some cases, the hexahistidine tag is known to affect negatively the structure and biological activity of recombinant proteins [36]. In addition, the extra amino acids are undesirable if used as pharmaceuticals since it can promote pathologic immune response of the organism. Therefore, in the case of hGM-CSF, the enteropeptidase cleavage site was integrated between the gene sequence and histidine codons to study the possibility of hexahistidine fragment cleavage. As a result, pA10523 and pA12633 plasmids containing crTMV polymerase gene, transport protein gene, target gene, and 3′ NTR, were cloned into binary vector pCambia 1300. Expression of this cassette was driven by the actin promoter from Arabidopsis thaliana and nopaline synthase gene terminator from A. tumefaciens.
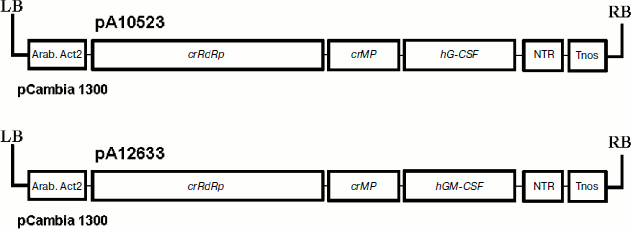
To deliver the viral vectors into plant leaves, a method of agroinjection of A. tumefaciens cell cultures transformed with pA10523 and pA12633 constructs was used. To suppress the effect of posttranslational gene silencing (PTGS), an equal number of bacteria containing p19 gene of tomato bushy stunt virus – PTGS suppressor – was added to the suspension. The PTGS inhibition effect is due to the ability of p19 protein to form complexes with small interfering RNAs, which blocks the system of PTGS signal transduction [37].
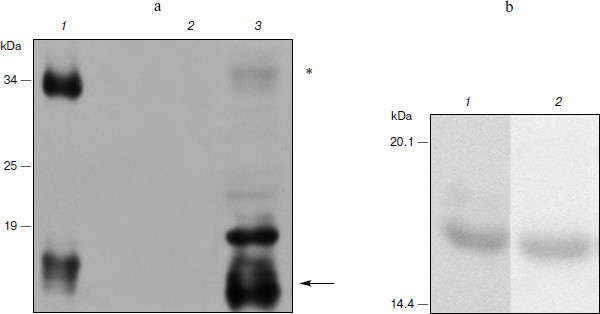
Expression of hG-CSF in N. benthamiana leaves. To estimate the efficiency of hG-CSF gene expression in plants, protein was extracted from leaves and recombinant cytokine yield was measured by protein electrophoresis and Western blot. At the same time, we compared the band intensity corresponding to phG-CSF (rhG-CSF synthesized in plants) and a known amount of E. coli rhG-CSF. To determine the time of maximal protein accumulation in leaves, the amount of phG-CSF in infected leaves was determined every 24 h after agroinjection. It was shown that 5 days after the infection, accumulation of phG-CSF reached a maximum of 500 mg/kg of fresh leaves weight (Fig. 1a). An additional band with molecular mass of 34 kDa detected with anti-hG-CSF antibodies seems to be dimerized protein. The reason might be that the high level of expression in the heterologous system leads to misfolding of some recombinant protein molecules. Their exposed hydrophobic regions tend to form the aggregates.
Ni2+-NTA agarose chromatography was used to extract proteins from plant cells. After extraction and purification of phG-CSF, one band with molecular mass 17 kDa corresponding to the target protein with the hexahistidine tag was detected by electrophoresis (Fig. 1b). The yield of the purified protein was 100 mg/kg of fresh leaves, which significantly exceeds the yield obtained in earlier studies [30].Fig. 1. Expression of hG-CSF in N. benthamiana leaves. a) Lanes: 1) positive control, rhG-CSF produced in E. coli; 2) negative control, non-inoculated leaf; 3) Western blot of extract from leaf agroinjected with pA10523 construct. On the left, protein molecular weight markers in kDa (Fermentas). The arrow shows a protein band corresponding to phG-CSF. Putative hG-CSF dimers are shown with an asterisk. b) Electrophoresis using the Laemmli method of hG-CSF produced in E. coli (1) and of purified phG-CSF (2).
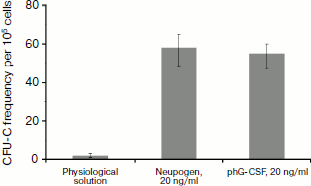
To determine the biological activity of the phG-CSF, murine bone marrow cells were treated with purified phG-CSF, and then newly formed neutrophil colonies were counted. Proliferation of murine myeloblasts is a standard test for hG-CSF biological activity, since the murine and human proteins are cross-reactive [38, 39]. As a positive control, hG-CSF (Neupogen) (Hoffmann-La Roche) was used. It has been demonstrated that control hG-CSF and phG-CSF have similar biological activities (Fig. 2). Consequently, the purification methods that we used do not affect the biological activity of hG-CSF.
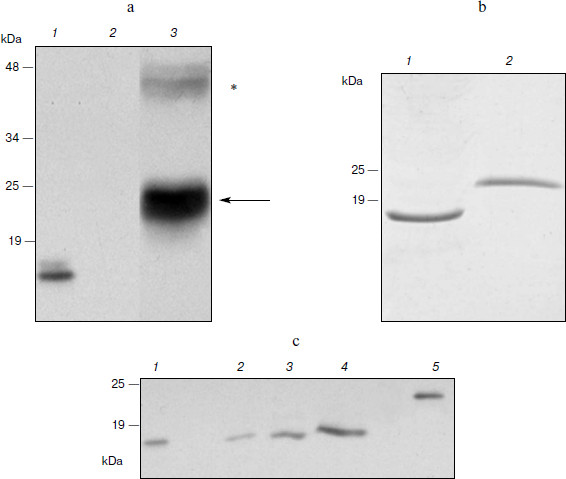
Expression of hGM-CSF in N. benthamiana. Expression of the hGM-CSF gene in the pA12633 plasmid, the rate of target protein accumulation, phGM-CSF extraction, and purification were carried out similarly to pA10523 and phG-CSF. According to Western blot, phGM-CSF yield in leaves was up to 300 mg/kg of fresh leaves on the fifth day after infection (Fig. 3a). An additional band with molecular mass of 45 kDa detected with anti-hGM-CSF antibodies also seems to be protein dimers. Using Laemmli PAGE, one band, corresponding to estimated molecular mass of hGM-CSF with the hexahistidine tag and enteropeptidase cleavage site (24 kDa; Fig. 3b), was found. After purification, the protein yield was 50 mg/kg of fresh leaves.Fig. 2. Biological activity of phG-CSF.
To obtain recombinant hGM-CSF with the native amino acid sequence, conditions of protein treatment by enteropeptidase were chosen and preparative protein hydrolysis was carried out. To isolate hydrolysis products, Ni2+-NTA agarose chromatography was repeated. At this time, hydrolysis products containing the hexahistidine sequence (uncleaved hybrid protein and hexahistidine tag) as well as extrinsic proteins possessing nonspecific affinity to Ni2+-NTA were retarded by the resin. As a result of hydrolysis and re-purification, recombinant phGM-CSF with molecular mass similar to that of the rhGM-CSF isolated from E. coli producent was obtained (Fig. 3c).Fig. 3. Expression of hGM-CSF in N. benthamiana leaves. a) Western blot: 1) a sample of rhGM-CSF produced in E. coli (positive control); 2) a non-inoculated leaf (negative control); 3) a leaf agroinjected with pA12633 construct. Protein molecular weight markers (in kDa) are shown on the left. Protein bands corresponding to phGM-CSF are shown with arrows. Putative phGM-CSF dimers are shown by an asterisk. b) Laemmli electrophoresis of hGM-CSF produced in E. coli (1) and of purified phGM-CSF (2). c) Western blot of phGM-CSF samples after enteropeptidase treatment (2-4), purified phGM-CSF before enteropeptidase treatment (5), and hGM-CSF produced in E. coli (1).
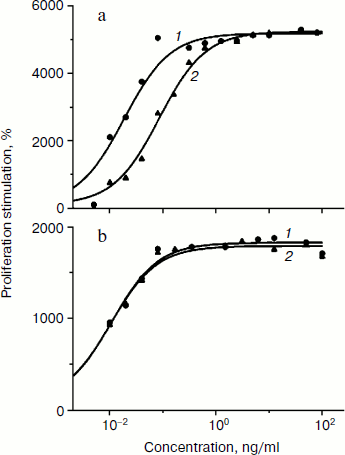
Biological activity of phGM-CSF was determined by the ability to stimulate the proliferation of TF-1 human erythroleucosis cells. Recombinant rhGM-CSF isolated from E. coli with activity similar to that of recombinant hGM-CSF (Sargramostim Leukine; Immunex, USA) produced in yeast was used as a control [16]. The rate of proliferation stimulation was determined by inclusion of 3H-labeled thymidine. Protein activity ranges are demonstrated to slightly differ (Fig. 4a). Maximal proliferation stimulation is achieved when 1 ng/ml phGM-CSF is added, and the dose necessary for 50% stimulation (ED50) is 86 pg/ml, which is about five times higher than that of the control preparation (18 pg/ml). However, ED50 values of phGM-CSF and rhGM-CSF are practically the same (11 and 10 pg/ml, respectively) (Fig. 4b) when the hexahistidine tag is cleaved by enteropeptidase (Fig. 3c). This result might be explained by the fact that the presence of a quite long charged N-terminal peptide (hexahistidine tag and enteropeptidase cleavage site) of phGM-CSF molecule somehow interrupts formation of the native protein spacial structure necessary for interaction with the cell receptor. After enteropeptidase treatment, the native amino acid and, presumably, spacial structures of the cytokine are restored, and, as a result, colony-stimulating ability of the protein increased to the control rate.
Thus, we have obtained viral vectors producing recombinant cytokines hG-CSF and hGM-CSF, which have as effective colony stimulating activity as analogous pharmaceuticals currently used in medicine. The synthesis rate achieved using the viral expression system is quite high – 300-500 mg/kg of fresh leaves. Further increase might be possible through optimization of codon usage of the target genes and careful selection of cultivation and purification conditions. The elaborated method of protein isolation from plant leaves provides pure biologically active cytokines. This makes the usage of viral-based systems for expression of recombinant cytokines in plants a fascinating alternative to existing methods.Fig. 4. Biological activity of phGM-CSF. a) Activity of phGM-CSF (curve 2) compared with rhGM-CSF (curve 1). b) Activity of phGM-CSF treated by enteropeptidase and purified (curve 2) compared with rhGM-CSF (curve 1). The curves are drawn to fit the equation SI = 100 + (SIlim – 100)/(1 + ED50/[M]), where SI is stimulation index, SIlim is limiting value of SI, and [M] is myelocytokine concentration. Values of ED50 (18, 86 (a) and 11, 10 (b) for curves 1 and 2, respectively) and SIlim were determined by nonlinear regression.
We thank N. I. Drize and I. N. Shipunova (National Research Center for Hematology, Russian Academy of Medical Sciences) for testing the biological activity of phG-CSF, M. E. Gasparyan for providing the enteropeptidase enzyme, and E. A. Sukhacheva (Institute of Bioorganic Chemistry, Russian Academy of Sciences) for providing the murine anti-rhG-CSF antibodies.
This work was supported by state contract 02.512.11.2103 “Expression of Pharmaceutical Proteins in Plants Using Viral Vectors”.
REFERENCES
1.Giddings, G., Allison, G., Brooks, D., and Carter,
A. (2000) Nature Biotechnol., 18, 1151-1155.
2.Giritch, A., Marillonnet, S., Engler, C., van
Eldik, G., Botterman, J., Klimyuk, V., and Gleba, Y. (2006)
PNAS, 103, 14645-14646.
3.Dorokhov, Y., Skurat, E., Frolova, O., Gasanova,
T., Smirnov, A., Zvereva, S., Ivanov, P., Ravin, N., Zamchuk, L., and
Atabekov, I. (2004) Dokl. Biokhim. Biofiz., 394,
30-32.
4.Gils, M., Kandzia, R., Marillonnet, S., Klimyuk,
V., and Gleba, Y. (2005) Plant Biotechnol. J., 6,
613-620.
5.Mechtcheriakova, I., Eldarov, M., Nicholson,
L., Shanks, M., Skryabin, K., and Lomonossoff, G. (2006) J.
Virol. Meth., 131, 10-15.
6.Dorokhov, Y., Sheveleva, A., Frolova, O., Komarova,
T., Zvereva, A., Ivanov, P., and Atabekov, J. (2007)
Tuberculosis, 87, 218-224.
7.Santi, L., Giritch, A., Roy, C., Marillonnet, S.,
Klimyuk, V., Gleba, Y., Webb, R., Arntzen, C., and Mason, H. (2006)
PNAS, 103, 861-866.
8.Hiatt, A., and Pauly, M. (1989) Nature,
342, 76-78.
9.Basu, S., Dunn, A., and Ward, A. (2002) Int. J.
Mol. Med., 10, 3-10.
10.Isola, L., Scigliano, E., Skerrett, D., Shank,
B., Ross, V., Najfeld, V., and Fruchtman, S. (1997) Bone Marrow
Transplant., 20, 1033-1037.
11.Costello, R. (1993) Acta Oncol.,
32, 403-408.
12.Voinnet, O., Rivas, S., Mestre, P., and
Baulcombe, D. (2003) Plant J., 33, 949-956.
13.Sambrook, J., and Russell, D. (2001) Molecular
Cloning: A Laboratory Manual, CSHL Press.
14.Gasparian, M., Ostapchenko, V., Schulga, A.,
Dolgikh, D., and Kirpichnikov, M. (2003) Protein Exp. Purif.,
31, 133-139.
15.Shingarova, L. N., Kashyap, S. K., Petrovskaya,
L. E., Petrenko, L. A., Pustoshilova, N. M., Sinichkina, S. A., and
Korobko, V. G. (1998) Biotekhnologiya, 6, 24-35.
16.Petrovskaya, L. E., Kryukova, E. A., Kayushin, A.
L., Rodina, A. V., Moskaleva, E. Y., and Korobko, V. G. (2002)
Bioorg. Khim., 28, 440-446.
17.Laemmli, U. (1970) Nature, 227,
680-685.
18.Yamamoto, A., Iwata, A., Saitoh, T., Tuchiya, K.,
Kanai, T., Tsujimoto, H., Hasegawa, A., Ishihama, A., and Ueda, S.
(2002) Veter. Immunol. Immunopathol., 90, 169-177.
19.Mayer, P. (1983) Comp. Immunol.
Microbiol. Infect. Dis., 6, 171-187.
20.Hilton, D., Nicola, N., Gough, N., and Metcalf,
D. (1988) J. Biol. Chem., 263, 9238-9243.
21.Kitamura, T., Tange, T., Terasawa, T., Chiba, S.,
Kuwaki, T., Miyagawa, K., Piao, Y., Miyazono, K., Urabe, A., and
Takaku, F. (1989) J. Cell Physiol., 140, 323-334.
22.Sijmons, P., Dekker, B., Schrammeijer, B.,
Verwoerd, T., van den Elzen, P., and Hoekema, A. (1990) Nature
Biotechnol., 8, 217-221.
23.Edelbaum, O., Stein, D., Holland, N., Gafni, Y.,
Livneh, O., Novick, D., Rubinstein, M., and Sela, I. (1992) J.
Interferon Res., 12, 449-453.
24.Kusnadi, A., Nikolov, Z., and Howard, J. (1997)
Biotechnol. Bioeng., 56, 473-484.
25.Stam, M., Mol, J., and Kooter, J. (1997)
Annals Bot., 79, 3-12.
26.Marillonnet, S., Thoeringer, C., Kandzia, R.,
Klimyuk, V., and Gleba, Y. (2005) Nature Biotechnol., 23,
718-723.
27.Shivprasad, S., Pogue, G., Lewandowski, D.,
Hidalgo, J., Donson, J., Grill, L., and Dawson, W. (1999)
Virology, 255, 312-323.
28.Kumagai, M., Turpen, T., Weinzettl, N.,
Della-Cioppa, G., Turpen, A., Donson, J., Hilf, M., Grantham, G.,
Dawson, W., and Chow, T. (1993) PNAS, 90, 427-430.
29.Marillonnet, S., Giritch, A., Gils, M., Kandzia,
R., Klimyuk, V., and Gleba, Y. (2004) PNAS, 101,
6852-6857.
30.Hong, S., Kwon, T., Lee, J., Jang, Y., and Yang,
M. (2002) Enzyme Microb. Technol., 30, 763-767.
31.Sardana, R., Alli, Z., Dudani, A., Tackaberry,
E., Panahi, M., Narayanan, M., Ganz, P., and Altosaar, I. (2002)
Transgen. Res., 11, 521-531.
32.James, E., Wang, C., Wang, Z., Reeves, R., Shin,
J., Magnuson, N., and Lee, J. (2000) Protein Exp. Purif.,
19, 131-138.
33.Kwon, T., Kim, Y., Lee, J., and Yang, M. (2003)
Biotechnol. Lett., 25, 1571-1574.
34.Wang, M., Goldstein, C., Su, W., Moore, P., and
Albert, H. (2005) Transgen. Res., 14, 167-178.
35.Zhou, F., Wang, M., Albert, H., Moore, P., and
Zhu, Y. (2006) Appl. Microbiol. Biotechnol., 72,
756-762.
36.Woestenenk, E., Hammarstrom, M., van den Berg,
S., Hard, T., and Berglund, H. (2004) J. Struct. Funct. Genom.,
5, 217-229.
37.Voinnet, O., Rivas, S., Mestre, P., and
Baulcombe, D. (2003) Plant J., 33, 949-956.
38.Nicola, N. (1987) Int. J. Cell Cloning,
5, 1-15.
39.Scheerlinck, J. (1999) Veter. Immunol.
Immunopathol., 72, 39-44.