Aggregation of Frog Rhodopsin to Oligomers and Their Dissociation to Monomer: Application of BN- and SDS-PAGE
S. A. Shukolyukov
Sechenov Institute for Evolutionary Physiology and Biochemistry, Russian Academy of Sciences, pr. M. Toreza 44, 194223 St. Petersburg, Russia; fax: (812) 552-3012; E-mail: tinycapers@mail.ru
Received January 15, 2009
After solubilization of frog rod outer segments (ROS) with mild detergents (digitonin, n-dodecyl-β-D-maltoside, Chaps, Triton X-100) and subsequent one-dimensional blue native polyacrylamide gel electrophoresis (1D BN-PAGE), the position of rhodopsin (Rh) on the gradient gel does not match the monomer with molecular weight of 40 kDa but appears self-associated into aggregate of Rh (RhA) with molecular mass varying in different detergents from 85 to 125 kDa. Short-term treatment (~2 h) of the excised BN-PAGE strip containing RhA by denaturing detergent mixture (10% SDS + 1 mM dithiothreitol (DTT)) followed by 2D SDS-PAGE revealed dissociation of the RhA into opsin monomer and unidentified proteins. Long-term treatment (~2 days) of RhA that included extraction, denaturation, concentration, and electrophoresis induced, along with dissociation of RhA into opsin monomer + unidentified proteins, also formation of opsin dimers, trimers, and higher oligomers owing to a secondary aggregation of opsin. Direct solubilization of the ROS by harsh SDS + DTT detergent mixture followed by 1D SDS-PAGE revealed only opsin monomer that upon heating disappeared, transforming into higher oligomers owing to secondary aggregation. The data show that degree of Rh oligomerization depends on specific conditions in which it stays. In the native state in the photoreceptor membrane as well as in mild detergents frog Rh exists mainly as dimers or higher oligomers. After solubilization with denaturing detergents, RhA can dissociate into monomers that then spontaneously self-associate into higher oligomers under the influence of various factors (for example, heating).
KEY WORDS: frog rhodopsin, oligomerization, dissociation, BN-PAGE, SDS-PAGEDOI: 10.1134/S0006297909060029
Abbreviations: 6AA, 6-aminohexanoic acid; BN-PAGE, blue-native polyacrylamide gel electrophoresis; CBB, Coomassie brilliant blue G-250; Chaps, 3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate; DIG, digitonin; DM, dodecyl-β-D-maltoside; DOC, deoxycholate; DTT, dithiothreitol; GPCRs, G-protein-coupled receptors; 1D- and 2D-PAGE, 1- and 2-dimension polyacrylamide gel electrophoresis; Mapp, apparent molecular mass; MSF, methylsulfonyl fluoride; Rh, rhodopsin; RhA, rhodopsin aggregate; ROS, rod outer segments; TX, Triton X-100.
Rhodopsin (Rh), a G-protein-coupled receptor (GPCR) that is densely
packed in the disc membranes of rod outer segments (ROS), was
originally proposed to function exclusively as a monomer
with Mapp ~ 39-40 kDa that is free to move in the
lipid bilayer [1-4]. The free
mobility of Rh and other components of the transduction system is
necessary to ensure interaction of any Rh molecule that has absorbed a
quantum of light with many molecules of the amplifier cascade. However,
a growing body of evidence indicates that different kinds of GPCRs
exist in membranes as dimers and higher oligomers, acquiring sometimes
the state of heterooligomers [5, 6]. This equally concerns the receptor Rh: new
morphological (transmission electron and atomic force microscopy) and
biochemical approaches (experiments on chemical cross-linking of Rh and
its limited proteolysis) have shown that in mouse and bovine disk
membranes of ROS Rh can really form dimers and higher oligomers which
are then organized into paracrystallic arrays [7-11]. Rhodopsin of cool-blooded animals and, in
particular, amphibians, in this respect only has started to be studied.
So, earlier using high-speed dichroic microspectrophotometry we found
that depending on still unclear reasons, the percent of freely
diffusible Rh in ROS of frogs, toads, and salamanders can fluctuate
from 0 to 100%, which apparently reflects its ability to be involved in
various degrees of oligomerization [12].
To determine the state of frog Rh, we have applied the relatively new method of BN-PAGE (blue-native polyacrylamide gel electrophoresis) [13, 14]. This method is widely used for isolation in native state of various membrane proteins of animals, plants, and bacteria, and also for studying mitochondrial respiratory chain complexes which are in most cases high-molecular-weight aggregates [13-17]. In brief, the principles of the method are as follows. When solubilized with mild detergents, membrane proteins bind the anionic dye Coomassie brilliant blue G-250 (CBB), which imposes a negative charge on the protein and prevents their secondary aggregation. The colored membrane complexes or individual proteins visible by eye in the presence of proteolysis inhibitors migrate in BN-PAGE to the anode and are separated in gradient gel according to their size owing to a molecular sieve effect (see reviews [15, 16]). No detergents are required in gels or cathode buffer once CBB has occupied protein surfaces. It is important to note that after solubilization with mild detergents, contact with CBB and electrophoretic run, the subunits of the membrane complexes or individual proteins separated in the gradient gel maintain their native properties, including enzymatic activity. This makes it possible to reveal them directly in the gel using specific color reactions [18, 19].
In the present work, we applied BN-PAGE in combination with SDS-PAGE for studying of state of frog Rh. We investigated the following questions: to what degree is frog Rh aggregated after its extraction from ROS with mild detergents; whether extracted Rh reflects that state characteristic in the native photoreceptor membrane and whether it is possible to induce aggregate dissociation or to change the degree its aggregation using various influences (for example denaturation, heating).
MATERIALS AND METHODS
All chemicals used in the paper were purchased from Sigma-Aldrich (USA), and partially from Fluka, Merck, Serva, and Ferak (Germany).
Grass frogs Rana temporaria were used as experimental animals. The animals were kept in a refrigerator at 4°C in small volume of regularly replaced water without food. On the eve of an experiment of 5-25 animals were placed in a small plastic box with water and held 3-4 h at a room temperature under the conditions of usual light exposure of the laboratory. Then animals were transferred to a lightproof box and adapted for darkness until the next morning in the presence of a small volume of water. All subsequent operations, including isolation and treatment of ROS and BN-PAGE of extracts from dark-adapted ROS, were carried out under dim red light. Ten to twenty-five frogs were decapitated, eyes were excised, and retinas were washed in Ringer solution (in mM): NaCl, 110; KCl, 2.5; CaCl2, 1; MgSO4, 1; NaHCO3, 5; EDTA, 0.05; Glc, 10; Hepes (Na salt), 10. The saline had pH 7.5-7.6. Then the retinas transferred to a mixture of 0.25 M sucrose–10% (w/v) glycerol and stored at –80°C in lightproof Eppendorf tubes until the moment of isolation ROS.
For ROS isolation, the retinas were thawed and placed in 2 ml of sucrose solution (d = 1.17 g/ml) prepared in Ringer and containing 1 mM methylsulfonyl fluoride (MSF). The retinas were homogenized in homogenizer with a Teflon pestle. The homogenate was poured out, the homogenizer was washed with 2 ml of the same sucrose solution, and both solutions were combined. The combined homogenate was divided between four transparent plastic centrifuge tubes (~1 ml) and 1 ml sucrose solutions (d = 1.13 g/ml and d = 1.11 g/ml) and 1 ml of Ringer were layered. All solutions contained of 1 mM MSF. The plastic tubes were centrifuged in a bucket-rotor of a Beckman ultracentrifuge for 1 h at ~100,000g. After centrifugation the interphase between sucrose layers of 1.13 and 1.11 g/ml, containing floating ROS, was sucked away, diluted 1 : 1 with Ringer containing 1 mM MSF, and centrifuged for 30 min at ~100,000g. The ROS pellet was washed two times with 50 mM imidazole buffer, pH 7.0, suspended in a mixture of 50 mM imidazole buffer–10% (w/v) glycerol, divided into aliquots (0.2 ml, 150-450 µg of protein), and then stored at –80°C or used immediately.
Immediately before experiment a ROS aliquot was thawed, Rh was bleached 15 min with a lamp (15 W) equipped with a green optical filter (λmax 520 nm) or used without bleaching. Sample solubilization with dodecyl-β-D-maltoside (DM), digitonin (DIG), TX (Triton X-100), or 3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate (Chaps), gradient gel (8-26%, w/v) preparation, 1D- and 2D-BN-PAGE, as well as treatment of the protein bands and their staining were done as described by Wittig et al. [14] for isolation and the characterization of the mitochondrial respiratory complexes I-V. EDTA and 6-aminohexanoic acid (6AA) (1 and 2 mM, respectively) were added to the solubilization buffer and gels to inhibit proteolysis. SDS-PAGE was carried out as described by Laemmli [20]. A vertical electrophoretic chamber Mini-Protean 3 Cell (BioRad, USA) with 10 × 8 cm glasses, 1 mm thickness spacer, and 5.6 ml volume of gradient gel were used. The power supply was a Power Pac HC (250 V/3 A/300 W; BioRad), and a J5 Jule gradient former (Jule, USA) was used to prepare a linear gradient. To determine the molecular mass of unknown proteins in BN-PAGE, purified molecular mass markers (Sigma) β-lactoglobulin (36.8 kDa), ovalbumin (45.0 kDa), BSA (66.2 kDa), and their corresponding dimers and oligomers were used. For denaturing SDS-PAGE along with above mentioned molecular mass markers, the kit of markers (β-lactoglobulin, 18.4 kDa; restriction endonuclease Bsp981, 25 kDa; lactate dehydrogenase, 35 kDa; ovalbumin, 45 kDa; BSA, 66.2 kDa; β-galactosidase, 116.2 kDa) from Fermentas (Lithuania) was used. Of note, relative mobility (Rf) of protein markers (β-lactoglobulin, ovalbumin, and BSA) from the two sources differed somewhat under similar denaturing conditions. After sample loading onto the gel (5-10 µl, 10-30 µg of protein), the current was turned on and kept for 30 min at 5 mA per gel, then the electrophoresis was continued with current and voltage limited to 20 mA and 250 V, respectively. BN-PAGE was run for 1.5-2.0 h in a refrigerator at 0 to 4°C, and the buffer was changed as described by Wittig et al. [14]. For further study, after 1D BN-PAGE, strips of gradient gel with visible stained protein were cut and placed between glasses on a gradient gel for 2D BN-PAGE, a comb with the required number of teeth was applied, and empty places were filled with 4% mixture for spacer-gel. After polymerization, the comb was removed and corresponding protein markers or their mixture were added to wells. The sandwich was subject to 2D BN-PAGE with cathode buffer supplemented (in addition to 0.02% CBB) with 0.05% deoxycholate (DOC) and TX. After 1.5-2.0 h of separation, the upper strip of bleached gel was removed, and the remaining gel was fixed, stained, and washed [21]. In case of 2D SDS-PAGE, the gel strip cut after 1D BN-PAGE was transferred for 15 min into denaturing mixture of 10% SDS and 1 mM dithiothreitol (DTT) and then rinsed with water and placed between glasses on denaturing 12-13% gel for 2D SDS-PAGE. All manipulations with comb and corresponding markers were done as above. SDS-PAGE was carried out at room temperature for 1.5 h. Gels were fixed, stained, and destained as described by Schagger [21]. Protein in the samples was determined by the method of Lowry et al. [22] after its solution in 1 N NaOH. The standard protein was BSA. Statistically processed results were expressed in terms of average ± standard error, and the number of measurements is specified in parentheses.
RESULTS AND DISCUSSION
Rhodopsin is a well-known membrane protein that is the dominant protein in ROS: it occupies ~50% of the membrane area in disks and accounts for 80% from all proteins of ROS, and its concentration being about 3 mM [23-25]. After solubilization of ROS with four mild detergents, BN-PAGE revealed in all of the solubilizates roughly similar pattern of protein bands with prevalence of one major band, which is clearly Rh (Fig. 1). The nature and properties of other minor protein bands in this paper will not be considered. Small differences in Rh position on the gels between extracts obtained with different detergents attract attention. Rhodopsin in the Chaps extract is closest to the anode, but in the DIG extract it is closest to the cathode, thus probably demonstrating lesser and greater size, respectively. Because Rh solubilized with different detergents has a different ratio of dimers and oligomers [11, 26], it appears that micelles formed with DIG have more of the larger oligomers than those formed with the other detergents. Accordingly, because of the larger DIG micelles, they stop sooner in the gradient gel owing to the molecular sieve effect and are situated closer to the cathode than micelles of others detergents. We found no distinction in movement and content between the DIG extracts of dark-adapted and bleached ROS (data not shown).
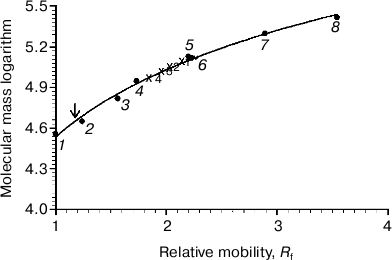
On comparing the position of Rh on the gel with that of protein markers, it is obvious that Rh is far for the position of a protein with Mapp ~ 39-40 kDa, but more likely corresponds to a high-molecular-weight Rh aggregate. The calibration curve representing the logarithm of molecular mass of the standard protein markers as a function of Rf of the band in the gel was used to calculate the molecular mass of the Rh aggregate. The Rh aggregates solubilized with different detergents have molecular masses within (85-125) ± 10 kDa (9), i.e. it represents a mixture of dimers and trimers (Fig. 2). In spite of the fact that low accuracy of the BN-PAGE is not discussed in methodological papers [13-16], the method nevertheless allows discrimination of monomeric form of the membrane protein from its oligomers.Fig. 1. 1D BN-PAGE of extracts obtained by solubilization of ROS with mild detergents. Lanes: 1) molecular mass isomers of marker BSA (monomer, dimer, and trimer) (Sigma); 2) molecular mass isomers of marker ovalbumin (monomer, dimer) (Sigma); 3) two charge isomers of marker β-lactoglobulin (Sigma); 4-7) ROS protein extracts obtained with 20% (w/v) Chaps, DIG, TX, and DM, respectively. Arrows show RhA bands. The volume of loaded markers and analyzed samples was 10 µl, the quantity of loaded protein being 15-40 µg. Data of one typical experiment are presented.
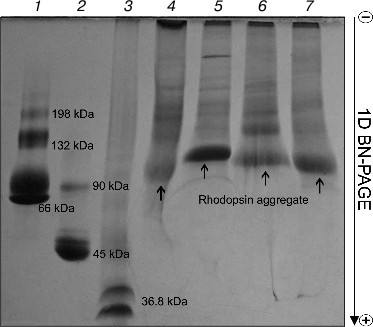
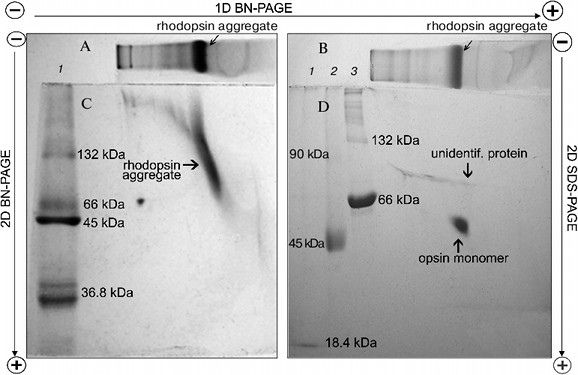
To dissociate Rh aggregates and detect their constituents, 2D BN-PAGE under more severe conditions (besides CBB the cathode buffer contained a mixture of 0.05% DOC and TX) was employed. However, it only has worsened the band resolution: Rh aggregate diffusely moved in the gel, thus causing a “smearing” effect (Fig. 3C). Such behavior shows that intermolecular interactions between aggregate components under these conditions are still not completely broken. As a further approach, we used short-term treatment (~2 h) with denaturing mixture (10% (w/v) SDS + 1 mM DTT) of the RhA, which was located on strips of gel after 1D BN-PAGE. The subsequent 2D SDS-PAGE has shown that such short-term denaturation induces RhA to dissociate into opsin monomer with Mapp ~ 40 kDa and accompanying proteins of unknown nature (Fig. 3D). Unfortunately, it is impossible to tell whether these proteins are impurities, which are extracted together with RhA, or they carry out some function (for example, regulatory) in the RhA.Fig. 2. Dependence between relative electrophoretic mobility and molecular mass logarithm of protein markers and RhA after BN-PAGE run. Arrow shows expected position for Rh with Mapp 40 kDa. 1) β-Lactoglobulin (36.8 kDa); 2) ovalbumin monomer (45 kDa); 3) BSA monomer (66 kDa); 4) ovalbumin dimer (90 kDa); 5) BSA dimer (132 kDa); 6) ovalbumin trimer (135 kDa); 7) BSA trimer (198 kDa); 8) BSA tetramer (264 kDa). X1-X4, position on the gel of RhA solubilized by DIG, TX, DM, and Chaps, respectively. Data of one typical experiment are presented.
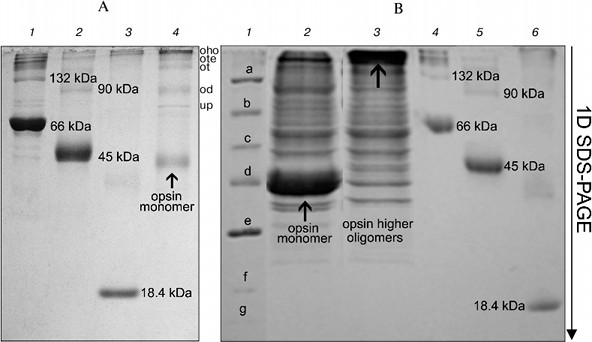
On the other hand, long treatment (2 days) of the RhA as described for mitochondrial complexes by Wittig et al. [14] and subsequent 1D SDS-PAGE revealed another picture. Such treatment, including extraction of RhA from the gel with 10% SDS + 1 mM DTT and its concentration and separation with 1D SDS-PAGE led to occurrence of not only opsin monomer and unidentified proteins, but also higher opsin oligomers owing to a secondary aggregation (Fig. 4a). One of the reasons for opsin aggregation in harsh SDS detergent could be the influence of unavoidable heating during 2-day manipulations with an extracts at room temperature. This possibility was checked experimentally. 1D SDS-PAGE has shown that after a direct solubilization of ROS proteins with a denaturing mixture, the extract contained only opsin monomers (ignoring other proteins), which after heating of an extract for 1 min at 90°C disappeared, transforming into higher opsin oligomers including those which could not penetrate into the gel and stayed on the top of the gel (Fig. 4b). Although this experiment is a rather crude model, it suggests that self-association of monomer molecules in harsh detergent extracts can occur during 2-day manipulation even at room temperature.Fig. 3. Scheme illustrating application of 1D BN-PAGE (gel A) + 2D BN-PAGE (gel C) and 1D BN-PAGE (gel B) + 2D SDS-PAGE (gel D) for separation of digitonin extract from ROS. Gel C: 1) mixture of native two charge isomers of β-lactoglobulin, ovalbumin monomer, and BSA monomer and dimer. Gel D: 1-3) denatured β-lactoglobulin, denatured ovalbumin monomer and dimer, and denatured BSA monomer and dimer, respectively. Volume of loaded markers was 5 µl, quantity of protein was 15-25 µg. On gels C and D, all markers are from Sigma. Data of one typical experiment are presented.
Thus, our results have shown that after solubilization of frog ROS with mild detergents, the extracts contained Rh aggregates, mainly dimers and trimers. To what extent does this picture of multimerity obtained with BN-PAGE reflect the status of Rh in the native membrane? It apparently reflects this status. Successful application of BN-PAGE for the elucidation of other physiological problems supports this statement (see application of this method in [14]). Evidence in favor of Rh oligomers in native membrane is provided by data on its spontaneous self-association in dimers and higher oligomers after its reconstitution into asolectin liposomes [27], as well as after expression in culture of COS1 cells [28].Fig. 4. Formation of oligomers of denatured opsin at room temperature and after heating to 90°C. Gel A: 1) monomer, dimer, and higher oligomers of BSA; 2) monomer, dimer, and higher oligomers of ovalbumin; 3) β-lactoglobulin; 4) opsin monomer, unidentified protein (up), and dimer (od), trimer (ot), tetramer (ote), and higher oligomers of opsin (oho) formed due to secondary oligomerization. Volume and quantity of loaded protein are the same as in Fig. 3. All markers on gel A are from Sigma. Gel B: 1) mixture of denatured markers from Fermentas: β-galactosidase (a), BSA (b), ovalbumin (c), lactate dehydrogenase (d), restriction endonuclease (e), β-lactoglobulin (f), and lysozyme (g); 2) extract obtained by fast treatment of ROS with 10% SDS and 1 mM DTT; 3) same as lane 2 but extract before loading was heated to 90°C for 1 min; 4) monomer, dimer, and higher oligomers of BSA; 5) monomer, dimer, and higher oligomers of ovalbumin; 6) β-lactoglobulin. All markers in lanes 4-6 are from Sigma. Volume and quantity of loaded proteins are the same as in Fig. 3. Data of one typical experiment are presented.
It is necessary note two apparently different cases of Rh oligomerization: i) association of native molecules under physiological conditions in photoreceptor membrane in vivo as well as after solubilization with mild detergents; ii) association of denatured molecules under stringent solubilizing mixtures in vitro.
Data on Rh aggregation in vivo as a typical GPCR have appeared in the last ten years. The property of Rh to form higher oligomers after solubilization with denaturing mixtures in vitro has been known for a long time [29, 30] and it is apparently an important feature distinguishing its from other membrane proteins. The mechanism providing self-association of neighboring Rh molecules in vivo and in vitro is still not understood. It remains unclear how Rh oligomerization reflects on its diffusion and how various down-stream signaling components in the visual pathway, particularly, transducin and arrestin, interact with the most functional-reasonable oligomer, the Rh dimer [31, 32]. It is still obscure why with increased degree of oligomerization Rh increases its thermostability and efficiency in triggering the phototransduction cascade [11, 26]. Demonstration in ROS of micro-domains resistant to detergents (so-called “detergent-resistant membranes”), which were then identified as lipid “rafts”, and also existence of specific micro-domains characterized by lipid heterogeneity [34] complicated our understanding how dimer works and what is the role of higher oligomers in the act of phototransduction. For the sake of objectivity it should noted that papers demonstrating that Rh monomer is a real functional unit in native photoreceptor membrane still continue to appear [35, 36]. In what way further studies will develop and when ideas about a multimeric state of receptor Rh will be finally decided remains hard to predict.
This work was supported by the Russian Foundation for Basic Research (grant No. 09-04-00691-a) and by the Program OBS RAS (grant No. 1ОB_05).
REFERENCES
1.Blasie, J. K., and Worthington, C. R. (1969) J.
Mol. Biol., 39, 407-416.
2.Chabre, M. (1975) Biochim. Biophys. Acta,
382, 322-335.
3.Saibil, H., Chabre, M., and Worcester, D. (1976)
Nature, 262, 266-270.
4.Roof, D. J., and Heuser, J. E. (1982) J. Cell
Biol., 95, 487-500.
5.Milligan, G. (2008) British J. Pharmocol.,
153, Suppl. 1, 216-229.
6.Park, P. S., and Palczewski, K. (2005) Proc.
Natl. Acad. Sci. USA, 102, 8793-8794.
7.Fotiadis, D., Liang, Y., Filipek, S., Saperstein,
D. A., Engel, A., and Palczewski, K. (2003) Nature, 421,
127-128.
8.Fotiadis, D., Liang, Y., Filipek, S., Saperstein,
D. A., Engel, A., and Palczewski, K. (2003) FEBS Lett.,
564, 281-288.
9.Liang, Y., Fotiadis, D., Liang, Y., Filipek, S.,
Saperstein, D. A., Palczewski, K., and Engel, A. (2003) J. Biol.
Chem., 278, 21655-21662.
10.Filipek, S., Krzysko, K. A., Fotiadis, D., Liang,
Y., Saperstein, D. A., Engel, A., and Palczewski, K. (2004)
Photochem. Photobiol. Sci., 3, 1-12.
11.Jastrzebska, B., Fotiadis, D., Jang, G.-F.,
Stenkamp, R. E., Engel, A., and Palczewsk, K. (2006) J. Biol.
Chem., 281, 11917-11922.
12.Govardovskii, V. I., Korenyak, D. A., Zueva, L.
V., and Shukolyukov, S. A. (2008) Abst. “Visionarium
VII” at Tvarminne Zoological Station, University of Helsinki,
p. 4.
13.Schagger, H., and von Jagow, G. (1991) Analyt.
Biochem., 199, 223-231.
14.Wittig, I., Braun, H.-P., and Schagger, H. (2006)
Nature Protocols, 1, 418-428.
15.Eubel, H., Braun, H.-P., and Millar, A. H. (2005)
Plant Meth., 1, 1-13.
16.Krause, F. (2006) Electrophoresis,
27, 2759-2781.
17.Wang, Z.-J., Xu, X.-P., Fan, K.-Q., Jia, C.-J.,
and Yang, K.-Q. (2007) J. Biochem. Biophys. Meth., 70,
565-572.
18.Molnar, A. M., Alves, A. A., Pereira-da-Silva,
L., Macedo, D. V., and Dabbeni-Sala, F. (2004) Brazil. J. Med. Biol.
Res., 37, 939-947.
19.Sabar, M., Balk, J., and Leaver, C. J. (2005)
Plant J., 44, 893-901.
20.Laemmli, U. K. (1970) Nature, 227,
680-685.
21.Schagger, H. (2006) Nature Protocols,
1, 16-22.
22.Lowry, O. H., Rosenbrough, N. I., Farr, A. L.,
and Randall, R. J. (1951) J. Biol. Chem., 193,
265-290.
23.Robinson, W. E., Gordon-Walker, A., and Bownds,
D. (1972) Nature. New Biol., 235, 112-114.
24.Heitzmann, H. (1972) Nature. New Biol.,
235, 114.
25.Molday, R. S. (1998) Invest. Ophthalmol. Vis.
Sci., 39, 2491-2513.
26.Jastrzebska, B., Maeda, T., Zhu, Li., Fotiadis,
D., Filipek, S., Engel, A., Stenkamp, R. E., and Palczewski, K. (2004)
J. Biol. Chem., 279, 54663-54675.
27.Mansoor, S. E., Palczewski, K., and Farrens, D.
L. (2006) Proc. Natl. Acad. Sci. USA, 103, 3060-3065.
28.Kota, P., Reeves, P. L., Rajbhandary, U. L., and
Khorana, H. G. (2006) Proc. Natl. Acad. Sci. USA, 103,
3054-3059.
29.Shukolyukov, S. A. (1972) Biokhimiya,
37, 914-921.
30.Papermaster, D. S., and Dreyer, W. J. (1974)
Biochemistry, 13, 2438-2444.
31.Fotiadis, D., Jasterzebska, B., Filippsen, A.,
Muller, D. J., Palczewski, K., and Engel, A. (2006) Curr. Opin.
Struct. Biol., 16, 252-259.
32.Hanson, S. M., Gurevich, E. V., Vishnivetskiy, S.
A., Ahmed, M. R., Song, X., and Gurevich, V. V. (2007) Proc. Natl.
Acad. Sci. USA, 104, 3125-3128.
33.Nair, K. S., Balasubramanian, N., and Slepak, V.
Z. (2002) Curr. Biol., 12, 421-425.
34.Wang, Q., Zhang, X., Zhang, Li., He, F., Zhang,
G., Jamrich, M., and Wensel, T. G. (2008) J. Biol. Chem.,
283, 30015-30024.
35.Edrington, T. C., 5th, Bennett, M., and Albert,
A. D. (2008) Biophys. J., 95, 2859-2866.
36.Chabre, M., Deterre, P., and Antonny, B. (2009)
Trends Pharmacol. Sci.], 5 March 2009 [Epub ahead of print].