REVIEW: IgA-Specific Proteins of Pathogenic Bacteria
T. N. Kazeeva1 and A. B. Shevelev1,2*
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; fax: (495) 954-2732; E-mail: shevelev@inbi.ras.ru2Chumakov Institute of Poliomyelitis and Viral Encephalitis, Russian Academy of Medical Sciences, Leninsky District, 142782 Moscow Region, Russia; fax: (495) 439-9321
* To whom correspondence should be addressed.
Received March 4, 2008; Revision received March 31, 2008
Data on structure and specificity of bacterial IgA receptors (IgA-binding M-like proteins Arp4 and Sir22 from hemolytic streptococci of serogroup A, β-antigen from hemolytic streptococci of serogroup B, and SSL family proteins from Staphylococcus aureus) are surveyed in this review. The principal conclusion derived from comparison is the fact that all bacterial receptors bind the same site in the IgA molecule overlapping with the binding site of endogenous human IgA receptor CD89. We assume that this site, consisting of spatially close amino acid strands Leu257-Gly259 in domain Cα2 and Pro440-Phe443 in domain Cα3, is subject to conformational rearrangement induced by the binding of antigen in the IgA active site.
KEY WORDS: IgA, IgA-binding proteins, CD89DOI: 10.1134/S0006297909010027
Abbreviations: CHα (Cα), constant domain of heavy (α) chain of IgA; Fab, variable antigen-binding superdomain of Ig; Fc, constant superdomain of Ig; FcαR, receptor specific for Fc-superdomain of IgA; GAS, GBS, group A and B streptococci, respectively; IgA, IgG, and IgM, immunoglobulins A, G, and M, respectively; NIP, 3-nitro-4-hydroxy-5-iodophenylacetate (hapten); S-IgA, secretory form of IgA.
The function of immunoglobulins A (IgA), which are the main type of
secreted antibodies, is traditionally reduced to the binding of
antigens located on the outer side of the basal epithelium membrane [1, 2]. For this reason, the
effector mechanisms of purposeful elimination of targets opsonized by
IgA have been studied insufficiently as yet. The character of
conformational rearrangements of IgA caused by antigen binding in their
active site is also unknown. However, the presence of specialized
IgA-binding proteins in many pathogenic bacteria suggests both the
presence of such rearrangement in IgA and existence of mechanisms of
immune protection of humans and other animals that response to these
rearrangements. It is particularly important that blocking by
IgA-binding proteins usually does not affect the antigen-binding
ability of antibodies. Numerous cases of horizontal transfer of genes
of IgA1-specific proteases between long-diverged groups of
microorganisms, in particular, the genera Haemophilus and
Neisseria, Streptococcus and Gemella, and
Prevotella and Capnocytophaga, confirm the adaptive
significance of IgA-specific functions of microorganisms in
implementation of their pathogenicity and survival in a host organism
[3, 4]. The character of
production of IgA-proteases proves to be stringently confined to
enhanced virulence of strains; at the same time, nonpathogenic
representatives of the above genera may have no IgA-protease genes.
These data emphasize the adaptive significance of IgA recognition
ability of strains, and the only universal consequence of such
recognition for all of the known models is transmission of a signal
about the binding of antibody to antigen [5]. We
have tried to generalize and compare the data on peculiarities of
recognition of ligands by IgA-specific proteins from pathogens such as
hemolytic streptococci and Staphylococcus aureus. The goal of
the analysis of structural peculiarities of interaction between these
proteins and IgA was to reveal the dependence of the binding process on
the presence of antigen in the active site of the antibody and to
assess the physiological significance of IgA-specific proteins with
respect to interactions of a pathogen with the immune mechanism of a
macrohost.
By selection of factual material, we pursued the comparison of peculiar features of IgA-specific receptors, maximally different in structure and origin, covering all classes of bacterial IgA receptors known to date. Thus, particular attention is focused on the IgA-binding M-like proteins of hemolytic streptococci of serogroup A, β-antigen of hemolytic streptococci of serogroup B, and proteins of the SSL family of St. aureus. At the same time, the problems of genetic polymorphism within each class of proteins have not been discussed. Data on IgA-specific proteases of Gram-positive and Gram-negative pathogens are also excluded from the analysis.
The data included in the review concern the receptors of pathogenic microorganisms binding mainly human IgA. Unfortunately, we have failed to confirm the published data obtained in a purposeful search for animal IgA receptors. The testing of IgA binding in organisms other than humans was performed only for more exact defining of the secondary specificity of ligands binding by specific receptors of human IgA.
IgA-BINDING M-LIKE PROTEINS OF STREPTOCOCCI FROM GROUP
A
The group A streptococci (GAS) are etiological agents of some human diseases with a rather heterogeneous nosology (angina, impetigo, cellulitis, scarlet fever, bacteremia, toxic shock syndrome, necrotic fascitis, etc.). Secondary infection by Streptococcus pyogenes is the major cause of severe diseases with autoimmune mechanism of pathogenesis, such as rheumatoid arthritis, rheumatic affliction of the heart, and acute glomerulonephritis [6].
IgA-binding proteins were found in GAS in the course of serological studies of immunodominant protein antigens, the most significant of which are M-proteins [7]. IgA receptors are connected with this functionally important group of GAS proteins also in the structural respect, so we will characterize M-proteins in more detail.
M-Proteins associated with the cell wall are concurrently the most important factor of pathogenicity and an immunodominant hypervariable antigen determining the capability of humoral response to a particular GAS strain [8-10]. Physiological functions of M-proteins are rather diverse and not quite clear to date. However, there is no doubt that these proteins make a decisive contribution to protection of bacteria from nonspecific cell response and phagocytosis during primary infection [11-13]. In contrast, in the presence of preexistent specific antibodies against serologically compatible M-protein, the pathogen is effectively eliminated both by macrophages and by non-phagocytizing cells of the immune system [14].
In the structural respect, M-proteins are multidomain formations with extracellular N-terminal domains variable in length, sequence, and physiological functions, while their C-terminal domains integrated in the plasma membrane are conserved. M-Proteins immobilized on bacterial surface are dimerized via noncovalent interaction of two α-helical segments anchored in the membrane.
Hypervariability of M-proteins significantly complicates their classification. It has been suggested to divide the sequence into segments A-D, each containing one or several tandem structural motifs [13]. The type of motif occurring in a certain segment is designated by an Arabic figure. The highest degree of polymorphism is typical of segment A, located immediately after the leader peptide in the M-protein sequence. The adjacent segment B is less variable (Scheme 1).
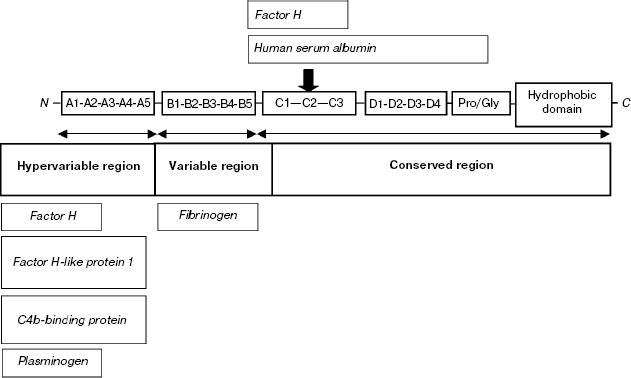
Scheme 1
The antigenic distinctions of hypervariable segments of M-proteins are used for serological classification of GAS proposed by Lansfield [15]. It was established that the antiphagocytic activity of M-proteins is due to the binding of individual proteins of the complement system from blood plasma [16]. Indeed, the hypervariable segments of M-protein and the conserved segment C are characterized by the ability to bind factor H of the complement and related blood proteins [17-20]. Segment C of some isotypes of M-protein can bind serum albumin [21]. Segments A and B of some types bind the C4BP component, which is the key regulator of the classical pathway of complement activation known for its ability to suppress phagocytosis [22-25]. Other M-proteins bind fibrinogen using segment B, thereby also decreasing the efficiency of phagocytosis [26, 27].
The GAS proteins significantly different from true M-proteins in the number of segments but containing the repeating motifs of the same types are called M-like proteins. They bind a broader spectrum of blood proteins than the M-proteins. For example, the M-like protein PAM reacts with plasminogen [28], while proteins Arp, H, Mrp, Sir, and Enn react with IgG and IgA [29, 30]. Like the true M-proteins, M-like proteins form a dimer coiled-coil structure, which is stabilized mostly by to the interaction of transmembrane domains containing heptade tandem repeats of amino acids. It has been proved that the dimer structure is necessary for the protein for forming a receptor [31]. Both M-proteins and M-like proteins are absolutely devoid of disulfide bonds. Thus, their spatial structures coincide by all significant characteristics. However, there is an important difference between them in the functional respect: M-like proteins do not inhibit the phagocytosis [32].
The group of M-like proteins of Str. pyogenes has been the only known structural type of IgA receptors in GAS, and up to 50% of all of the known strains of Str. pyogenes express M-proteins binding the Fc-fragments of human IgA [33]. The following representatives of IgA-specific M-like GAS proteins have been characterized most completely: Arp binding almost only IgA, protein H binding IgG, and Sir22 binding both IgA and IgG of all subclasses. Arp can bind some of monoclonal IgG, but the binding constant is so low that it cannot be determined [34].
The first M-like GAS protein studied because of the ability to bind human IgA was Arp. The structural gene of protein Arp4 was isolated from a GAS strain of serotype M4. The expression of this gene in Escherichia coli resulted in a functional product with molecular weight of 42 kD [35].
The Arp4 structure is based on the group of repeating motifs typical of segment C of M-proteins. Besides, it contains a unique N-terminal region highly divergent of the respective regions of other Str. pyogenes M-like proteins, e.g. Arp60, Sir22, ML2.2, and Enn4 [36].
The first attempts for mapping the IgA binding site within Arp4 were carried out by the authors of work [37]. By means of CNBr cleavage of protein, they established that the unclassified N-terminal region of 52 amino acid residues completely maintained the IgA-binding activity typical of the full-size protein. The residual part of the extracellular half of Arp4 composed mainly by repeats of type C was completely devoid of this activity.
Comparison of the IgA-binding protein sequences from Str. pyogenes (Arp4, Arp60, Sir22, ML2.2, and Enn4) resulted in identification of a still shorter peptide (29 amino acid residues) providing a ligand recognition [36]. This segment comprises the most conserved part of the unclassified N-terminal region of Arp4 (invariant residues share 41% when compared with the abovementioned five M-like proteins). The authors experimentally showed that even short deletions within the identified 29-residue region abolish IgA-binding ability of the protein. On the contrary, the transfer of this region to the PAM protein, which is similar to Arp4 in size and organization but has no specificity to IgA, confers the specificity on this protein.
A synthetic peptide of 33 amino acids corresponding to the isolated IgA-binding site of Arp4 did not exhibit the receptor properties by itself. In contrast, a 50-residue synthetic peptide with the active site flanked by 10 natural amino acid residues from each side showed IgA-binding ability identical to the fragment of native protein obtained by CNBr cleavage (the binding constant in both cases was 10–9 M) [38]. It should also be noted that the authors confirmed localization of the site using synthetic 15- and 16-residue peptides spanning various parts of the 29-residue sequence. These peptides were tested for ability to compete with AprA for IgA binding. The peptides from the C-terminal part of the IgA-binding site were shown to inhibit binding to IgA. This observation suggests that just the C-terminal half of the 29-residue region (residues 58-72) makes the major contribution to binding, although it may not functionally substitute for the whole active site.
The present data convincingly prove that Arp4 contains the single IgA binding site, and C-repeats make no contribution to recognition of this ligand. Such localization of the ligand-binding site significantly differs from localization of IgG-binding domains of the surface proteins of streptococci and staphylococci. For example, the IgG-binding sites in the best known proteins A and G are formed due to repeating tandem motifs. The same organization is found also in FcRA76 (the IgG-binding protein of Str. pyogenes), which contains repeating motifs of type A [39].
The study of Arp4 specificity demonstrated the protein to bind both serum and secretory IgA of both subclasses. However, the binding constant of Arp4 was 5.6·10–8 M for serum IgA1 and IgA2 and about tenfold higher for secretory Ig of the same isotypes [35].
Besides Arp4, one more structurally related streptococcus surface protein, Sir22, has been well characterized. The first works on the study of Sir22 were carried out with a protein of GAS serotype M22 [40]. Sir22 consists of a conserved signal peptide, a central region constituted by repeating motifs, and a series of conservative C-terminal domains. Like in Arp4, the central part is constituted by repeats of type C. In different types of M-like proteins, the number of C-repeats varies from two to four. Protein Sir22 contains as few as two repeats and therefore has lower weight than the most other proteins of this group, e.g. Arp4. The N-terminal part of mature Sir22 (41 amino acid residues) is unique. Like other M-like proteins, Sir22 exists as a coiled-coil dimer. In contrast to Arp4, it can bind at least four different proteins of human blood plasma: IgA, IgG, C4BP, and serum albumin. The results of mapping of the IgA, IgG, and C4BP binding sites have been described in work [41] (Scheme 2).
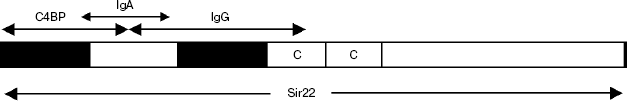
Scheme 2
Experiments were based on the technique of expression of the cloned Sir22 gene and its modified derivatives in E. coli, rather than synthetic peptides as in the case of Arp4. Recombinant proteins were separated by denaturing electrophoresis in polyacrylamide gel and stained by the Fab (variable antigen-binding superdomain of Ig)- and Fc (constant superdomain of Ig)-fragments of nonspecific IgA and IgG (by Western blotting, without preliminary renaturing of receptor protein). As a result, it has been established that Sir22 effectively binds the Fc-fragments of both classes of immunoglobulins, and both IgA subtypes are recognized with the same efficiency. On the contrary, Sir22 showed no affinity to any of the Fab-fragments of the same immunoglobulins. The binding constants of full-size immunoglobulins with Sir22 were 7.0·108, 2.4·108, and 7.8·108 M–1 in the reactions of Sir22/serum IgA, Sir22/secretory IgA, and Sir22/IgG, respectively [40].
Individual domains discriminated within Sir22 were able to bind a ligand of only one type, including IgA. Experiments on deletion analysis of the Sir22 gene showed that the region homologous to the active IgA-binding site of Arp4 (29 amino acids) partially overlapped the minimal region that determined the binding of C4BP in Sir22. In spite of this fact, IgA and C4BP do not compete for binding to Sir22. The minimal IgA-binding active site partially overlaps also with the underlying potential domain containing the IgG binding site. However, in contrast to the binding to C4BP, the bindings of full-size Sir22 to IgA and to IgG are mutually exclusive. All together, this suggests that the IgA binding site is located in a concave formed by the outer surfaces of neighboring domains; one of them contains the site for C4BP recognition and the other contains the site for IgG recognition. Due to closeness of the binding sites of such huge molecules as IgA and IgG, they prove to be competitors for the binding to Sir22. In addition to the above three active sites located within the two hypothetical globular domains, the serum albumin binding site was mapped in the central region of Sir22 constituted by tandem motifs of type C [41].
As derived from deletion mapping of Sir22, an artificial peptide Sap with the ability for selective reception of IgA was synthesized on the basis of its sequence. Like the Arp4-based peptide, this peptide was 50 amino acids in length and overlapped the supposed IgA-binding site of Sir22 with the adjacent 10 amino acid residues from each side. On the assumption that Sir22 can function as the receptor only in a form of a coiled-coil type dimer, an additional Cys residue capable of linking an inter-chain disulfide bond was introduced into the C-terminus of the synthesized peptide. Thus, the peptide included residues 35-83 (by the Sir22 numeration) and the C-terminal Cys (Scheme 3) missing in the parental protein [42].

Scheme 3
The peptide was soluble in water at physiological pH and ionic strength. Denaturing electrophoresis under non-reducing conditions showed that the peptide in solution is a dimer formed by disulfide bonds. For studying the effect of Sap dimerization on IgA binding, a perfectly analogous peptide without the additional Cys was synthesized, and the binding properties of both peptides were compared by radioimmunoassay. Only the peptide with the C-terminal Cys residue demonstrated tight binding with IgA immobilized on a carrier (the equilibrium binding constant was 0.1 µM), this confirming the functional significance of dimerization of Sir22-type receptors [42]. The sensitivity of Sir22 receptor ability to isotypic affiliation of the ligand and the presence of a secretory component in its structure was studied in the same work. Secretory and serum IgA1 and IgA2 were shown to bind with nearly the same efficiency. This conclusion contradicts the data of an earlier work [38] demonstrating that streptococcal proteins bound secretory antibodies with lower affinity than serum antibodies. However, the peptide was applicable for detection of S-IgA (secretory form of IgA) chains separated by denaturing electrophoresis.
The question of Sir22 sensitivity to the presence of an antigen in the active site of the bound IgA remains obscure, though its ability to recognize both free IgA and their immune complexes is undisputed. It was demonstrated that the Sap peptide can also be used for specific detection of IgA bound with antigen (immune complexes of IgA) and for affinity purification of free IgA [42]. Using 5 mg of Sap immobilized on a Hi-Trap matrix pre-activated with N-hydroxysuccinimide, the authors obtained up to 2 mg of purified IgA from 1 ml of serum. Analysis of the IgA preparation purity by ELISA showed that the eluates obtained as a result of one-stage chromatography on Sap-Hi-Trap contained ≤0.45% IgG and ≤0.33% IgM as admixtures. Sap showed the same affinity to polymer and monomer serum IgA.
Similar results were obtained during purification of S-IgA from human saliva on the Sap-Hi-Trap. S-IgA obtained by purification on the column with Sap were as pure as commercial S-IgA preparations. At the same time, the yield of S-IgA was far from the theoretical maximum because a significant part of S-IgA was not bound with the Sap-sorbent. These immunoglobulins did not react with the immobilized Sap when re-applied to the column. This provides indirect evidence of heterogeneity of the IgA population revealed by the reaction with this peptide. However, the low yield of purified product was not caused by the selectivity of Sap binding towards one of the IgA subclasses, because the ratio of S-IgA1 to S-IgA2 in the intact saliva and purified S-IgA was the same. Similar results were observed on testing of a Sap-based sorbent for isolation of S-IgA from human milk [43, 44]. Note that it was found that IgA preparations obtained by chromatography contained the same set of noncovalently bound minor proteins as IgA in the initial biological material [45].
The technologically significant peculiarity of Sap is its high capacity to spontaneous recovery of spatial structure under physiological conditions. It does not need a special procedure for refolding after solid-phase synthesis and retains the ligand binding ability after boiling [38].
The model of human recombinant antibodies against the NIP (anti-3-nitro-4-hydroxy-5-iodophenylacetate) hapten was used for detailed comparative studies of the efficiency of recognition of immune complexes IgA1, IgA2, and IgG1 by protein G and Sap peptide [42]. The immune complexes were formed by application of recombinant antibodies of different classes to the carrier activated by the NIP–BSA conjugate. Then the complexes were stained by enzymatically labeled Sap peptide or G protein. As expected, Sap with approximately equal efficiency bound the IgA1 and IgA2 but not IgG complexes. The affinity of protein G, which selectively bound IgG1 only, and Sap peptide to the specific immune complex proved to be the same. This experiment demonstrates the possibility of using Sap derivatives in laboratory test systems for different purposes, e.g. as a tool for enzyme immunoassay. However, since the values of Sap affinity to free IgA and their immune complexes were not compared in the work, it does not answer the question about the sensitivity of Sap and especially full-size Sir22 to the expected conformational rearrangements of IgA as a result of their interaction with an antigen.
The Sap peptide is selective only towards human IgA. It completely lacks affinity to mouse IgA and weakly binds bovine IgA. This observation correlates with the similarity of the amino acid sequences of IgA of these animals: the CH2- and CH3-domains of human and bovine IgA are much more similar to each other than to the homologous elements of mouse [46].
β-ANTIGEN OF Streptococcus agalactiae
Group B streptococci (GBS) are not related to the group A streptococci in taxonomic respect; however, they are often considered as a single group of pathogens of epidemically significant infections—hemolytic streptococci. This is due to the common feature used at their identification: the ability to induce hemolysis of sheep erythrocytes during cultivation on solid media. GBS representatives most important epidemiologically are pneumococci—Str. pneumoniae causing pneumonia, meningitis, overwhelming sepsis, and otitis [47]. However, their surface or secretory IgA-binding proteins are not described in the literature. However, GBS are known to have species Str. agalactiae possessing, like GAS, a surface IgA-binding protein—β-antigen of 125 kD. In the structural respect, this protein is not similar to the GAS M-proteins. Its name comes from serological studies of immunodominant protein antigens contained in the biomass of Str. agalactiae. Along with β-antigen, they are represented by surface proteins—Rib and α-antigen (members of the structural Alp family). In the cell wall of Str. agalactiae, α and β antigens are physically connected and form the so-called C-antigen [48]. There are arguments in favor of the dominant role of C-antigen in internalization of GBS strains causing meningitis. However, this role of C-antigen is not associated with its ability to interact with IgA [49].
A peculiar feature of β-antigen used for separation of surface GBS proteins is sensitivity to trypsin. By its primary structure, β-antigen is related to the Hic protein of pneumococcus, which binds factor H and thereby protects the pathogen from opsonization and subsequent phagocytosis [50]. Experiments with animal models showed that the antibodies against GBS surface proteins confer as high protection as antibodies against capsular polysaccharide [48]. In view of the high polymorphism and low immunogenicity of GBS capsular polysaccharides untypical of GAS, these proteins draw attention as a basis for a vaccine production. At the same time, the functional similarity of GBS C-antigen to GAS M-proteins with the total lack of structural similarity indicates their significant roles in protection of the pathogen from attacks of the immune system and maintenance of its transportation within the macrohost organism.
β-Antigen interacts with the two components of the human immune system: Fc-IgA and complement factor H. However, in contrast to GAS M-proteins, it has no affinity to IgG and IgM. When characterizing the selectivity of β-antigen as a receptor, one should note that, as distinct from GAS M-proteins, it binds with high affinity only the serum human IgA of both subclasses or their isolated Fc-fragments but is practically unable to react with secretory IgA.
Expression of β-antigen during cultivation on solid media was observed in about a half of GBS strains. It is typical of all Str. agalactiae strains of serotype Ib studied with regard to this parameter, less typical of serotypes Ia, II, and V, and does not occur in the strains of serotype III [51, 52]. Nagano et al. showed that the level of β-antigen expression in invasive Str. agalactiae strains of serotypes Ia and Ib is higher than in noninvasive strains. Note that the ability of some strains for β-antigen production is unequivocally coupled with the presence of α-antigen in the particular strain. In contrast, α-antigen is often expressed independently (as shown by the example of serotype Ia strains). The same works [51, 52] report the existence of single Str. agalactiae strains that express an alternative surface form of β-antigen incapable of IgA binding or a truncated soluble form of β-antigen.
The sequence of GBS β-antigen, in contrast to the sequences of GAS M-proteins, is moderately variable in the N-terminal region and practically invariant in the C-terminal region. The N-terminal signal peptide of 37 amino acids and the typical murein-binding C-terminal domain enriched in LPXTG motif, which are necessary for immobilization on the cell wall, were identified in the sequence of the protein-precursor of β-antigen. β-Antigen is the only protein released into the medium as a result of incubation of Str. agalactiae cells at enhanced pH values. This observation shows the noncovalent nature of β-antigen linkage with the cell wall and provides an easy method for its purification.
Four of the five C-terminal residues of β-antigen are basic, and the hydrophobic region ahead of them serves for anchoring in the membrane [53]. In contrast to many other surface proteins of pathogenic streptococci, β-antigen contains no elongated tandem repeats in the domains responsible for ligand binding. Only the region of a hinge between the ligand-binding and murein-binding domains is represented by the tandem repeat XPZ of variable length. The repeat has a period of three residues: the first residue is Pro, the second residue carries any positive or negative charge, and the third residue is uncharged.
The analysis of the spatial structure of recombinant β-antigen obtained in E. coli showed that the N-terminal half of its molecule consists of two or more globular domains designated as A and B, and both of them are necessary for specific recognition of IgA [29]. The minimal functional IgA-binding site can be formed by 73 amino acid residues located within domain A (Scheme 4). Works [54, 55] demonstrated that the key role in this site belonged to a 6-residue block, MLKKIE, which was conserved in all strains of Str. agalactiae exhibiting IgA-binding activity.
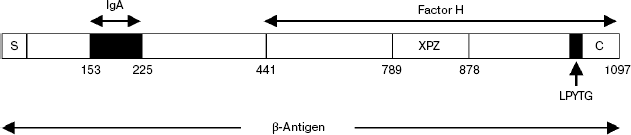
Scheme 4
Study of Str. agalactiae strain polymorphism with the sampling of 20 clinical isolates showed that the values of their specific activity in the binding of both ligands of β-antigen, factor H and IgA, strongly correlate with each other [56]. This observation suggests that Str. agalactiae has no alternative IgA receptors. In the same work, a unique natural isolate of Str. agalactiae exhibited an ability for interaction only with factor H but not with IgA. This isolate of serotype Ib was found in cerebrospinal fluid. It produced a noticeable amount of native β-antigen without IgA-binding activity. Analysis of the sequence of the β-antigen cloned gene (bac) from this source revealed the presence of a MLKKIE block; however, the C-terminal part of the previously described IgA-binding 73-residue minidomain was impaired due to deletion of positions 214-264 (51 amino acid residues). This deletion spanned the last 12 amino acid residues of the IgA-binding domain (positions 214-225). This example shows that the loss of the IgA-binding activity of β-antigen by the strain does not compromise its viability in vivo and does not abolish the virulence in general. These data do not contradict the hypothesis that IgA and complement factor H binding sites do not overlap.
IgA-BINDING PROTEINS OF STAPHYLOCOCCI
Staphylococci pathogenic for humans, joined in the species St. aureus, are the most typical pathogens of posttraumatic purulent processes. They draw special because of hospital, in particular, post-natal infections. In contrast to streptococci, staphylococci do not constitute a significant epidemic menace; therefore the development of vaccination and serodiagnostic methods for addressing them is not relevant. However, the treatment of diseases induced by staphylococci is often long-term and coupled with complications. Staphylococci, along with Mycobacterium tuberculosis, are extremely liable to development of mutations of multiple drug resistance, which imparts relevance to the development of specific therapeutic remedies [57, 58].
Staphylococci, although not related to streptococci, exist in similar conditions and often encounter the immune response on the mucous membranes of humans and other mammals. That is probably why many strains of St. aureus, like streptococci, produce IgA-binding factors, the most known of which are proteins of the SSL family [6]. Like IgA-specific proteins of streptococci of groups A and B, SSL proteins have the properties of dominant antigens and are even ranked among superantigens. The striking peculiarity of SSL, as compared with GBS M-proteins and β-antigen, is the absence of affinity to the cell wall of the producer strain [6]. The same picture is observed also when IgG-specific receptors of both groups of pathogens are compared. M-proteins of streptococci are associated with the cell wall, while protein A of staphylococci is secreted in a soluble form. This suggests that the disposition of staphylococci to release of immunity factors into the environment is determined by localization of this pathogen in physically close nidi of pyogenic infection, whereas streptococci often encounter moving fluxes of large volumes of fluids, e.g. saliva and other secretory fluids of mucous membranes as well as blood.
In contrast to streptococcal proteins, SSL have no pronounced species specificity to IgA but for all that maintain high affinity to the ligand. SSL7, also known as an enterotoxin-like factor of staphylococcus SET1, binds human monomer IgA1 and IgA2 with binding constant of 1.1 nM. Practically the same affinity is typical of its binding with serum IgA of primates, pigs, rats, and horses and secretory IgA from human, cow, and sheep milk. The SSL proteins inhibit the binding of IgA with the neutrophil receptor CD89: both with the native protein present on the cell surface and with its isolated extracellular domain [6, 59].
Besides IgA binding, SSL is related to SAG M-proteins and SBG β-antigen by the ability to bind components of the complement, although all of them block the cascade of activation of this bactericidal mechanism in different points. The M-proteins of streptococci bind the regulator of classical pathway of the complement (C4BP), β-protein binds factor H, and SSL bind factor C5 [60].
Such surprising uniformity of the functions of considered bacterial IgA receptors for independent origin unambiguously points to existence of their common target as an IgA-dependent effector mechanism.
BINDING SITES OF PATHOGEN PROTEINS IN IgA MOLECULE
Similarity of the spectra of recognized ligands in all three structural types of considered IgA receptors (GAS M-proteins, GBS β-antigen, and SSL proteins of staphylococci) substantiates relevancy to the question about localization of their recognition sites in the IgA molecule. In consideration of independent origin of all three types of receptors, the grouping of the sites for their binding on ligand surface may be evidence of the functional importance of this region with respect to transmission of a signal about antigen recognition from immunoglobulin to the effector system.
The activation of phagocytosis and the respiratory burst of neutrophils through the surface receptor CD89 have been considered up to now as the only known IgA-dependent effector system of target elimination [61-63]. Therefore, the question about the competition for IgA binding to CD89 has arisen from the very beginning of research for the mapping of the sites of binding to bacterial receptors.
The main contribution to the mapping of the sites of recognition of bacterial receptors on the IgA surface was made in works [46, 64, 65]. The authors used the system of recombinant expression of the genes of monoclonal human antibodies against NIP hapten in CHO cells of Chinese hamster. This system up to now has been the only tool for preparative purification of immune complexes of monoclonal human IgA. It permits the study of the structure–function organization of immunoglobulins by the method of site-directed mutagenesis and molecular hybrids. CHO cells transfected by expression construction with the CD89 gene (human FcαR (receptor specific for Fc-superdomain of IgA)) and human neutrophils purified from donor blood were a test system for estimation of efficiency of the binding of recombinant IgA derivatives with endogenous receptor. The efficiency of binding of exogenous bacterial receptors was assessed by the ability to suppress the binding of mutant IgA derivatives to the surface CD89 in competitive test. It should be noted that all experiments were performed with the immune complexes of monoclonal IgA with the NIP hapten.
The following hypothesis was verified in the first stage of work. Since the CD89 binding region in the IgA molecule coincided with the binding site of staphylococcal protein A in the IgG3 molecule. The latter pair was chosen because the protein A–IgG3 complex was shown to have a high-resolution spatial structure. The analysis of this structure showed that the most energy for the binding to protein A is provided by two destructured IgG regions, three and four amino acid residues, localized in domains Cγ2 and Cγ3, respectively. The comparison of IgA and IgG spatial structures revealed that homologous regions were present also in the IgA molecule and corresponded to Leu257-Gly259 in Cα2 and to Pro440-Phe443 in Cα3. Each of the residues in the above blocks was in turn replaced by Arg. As a result, it was established that five out of the seven tested residues were necessary for the interaction with CD89. Substitutions Gly259Arg and Pro440Arg had no effect on the binding to receptor.
The same model was used for the study of the structural bases of IgA species specificity. Comparison of human and mouse IgA sequences has shown that mouse IgA, which do not recognize human CD89, differ from human immunoglobulins only in two residues in the same loops that in the previous experiment. Based on this observation, the authors obtained human IgA1 with point substitutions of L441A and M442N simulating mouse antibodies. This mutant protein also completely lost the ability for binding to human CD89.
In a third experiment, for assessment of contribution of domains Cα2 and Cα3 to the binding with endogenous receptor, hybrid antibodies against NIP were constructed with replacement of some of the IgA domains by IgG domains, e.g. γ1γ2α3 and α1α2γ3. Analysis of the properties of these hybrid antibodies showed that none of them was able to bind to the CD89 receptor. This is evidence of the key role of both domains in formation of a complex with the receptor.
Taking into account the data on location of the sites of IgA and CD89 interaction, it was possible to map also the sites of IgA binding with the receptors of streptococci and staphylococci [46]. The contribution of IgA domains Cα2 and Cα3 with IgA-binding proteins was analyzed using the same chimeric IgA/IgG as in the previous experiment, namely γ1γ2α3 (constant domains Cγ1, Cγ2, Cα3) and α1α2γ3 (constant domains Cα1, Cα2, Cγ3). The ability of the proteins to bind with the three natural IgA receptors from streptococcus and IgA-binding Sap peptide was assessed by ELISA [65].
Since the full-size Sir22 is equally able to bind both IgA and IgG, hybrid antibodies cannot give an unambiguous result on the mapping of binding sites of this protein. Therefore, a synthetic analog of the IgA-binding domain of Sir22 (Sap peptide) was employed in the experiment along with the latter. Other proteins, for which IgG is not a ligand (Arp4 and β-antigen), have been used only as full-size recombinant products.
A high affinity towards γ1γ2α3 antibodies was established for all three IgA-binding molecules, slightly less than towards the wild type IgA1. On the contrary, α1α2γ3 was incapable of binding. Thus, one can conclude that the energy for IgA binding with exogenous receptors is mostly provided by domain Cα3. However, this does not exclude completely the role of domain Cα2, because the spatial structures of domains Cγ2 and Cα2 are much of the same type and, probably, Cγ2 to a certain extent can functionally substitute for Cα2 in the course of binding.
Point mutagenesis allowed characterization of the sites of IgA binding to streptococcus receptors in more detail (Arp4, Sir22, β-antigen, and Sap peptide were studied). It was shown that maintenance of the amino acid sequence of loop Pro-Leu-Ala-Phe (440-443) on the surface of Cα3-domain was more critical for the binding with bacterial receptors compared with that in loop Leu-Leu-Gly in the Cα2-domain (residues 257-259). The equilibrium constant of IgA binding with the bacterial receptors, measured by indirect ELISA, increased 30-100 times on substitutions in loop 440-443 and only 3 times on substitutions in loop 257-259. Point substitutions Leu257 and Pro440 made approximately equal contributions to the interaction with all types of receptors. Phe443 also made a certain contribution to the interaction with Arp4 and Sir22 but was insignificant for the binding to β-antigen.
Thus, it has been established that the site of binding with CD89 in the IgA molecule is represented by two blocks of amino acids located in the spatially close loops of domains CH2 and CH3. The sites of streptococcal receptors and Sap peptide are located almost identically, partially overlapping the site for CD89, which preferentially captures the loop of domain CH3.
The experimental data showed that all of the studied streptococcal IgA-binding proteins effectively block the interaction of antibodies with CD89. Thus, it can be suggested that this mechanism is realized also in vivo, thereby canceling the effector functions mediated by IgA.
Based on X-ray structure analysis [66], the site of interaction with the staphylococcal protein SSL7 in the IgA molecule was found to be more similar to the site for FcαRI than to streptococcal proteins. SSL7 directly interacts also with residues Leu257-Leu258 in domain Cα2 and with the Pro-Leu-Ala-Phe loop (440-443) in domain Cα3. This conclusion is particularly unexpected, because all three groups of bacterial IgA receptors have different secondary specificity to the binding site. GAS M-like proteins do not discriminate secretory and serum IgA but have a strict specificity to the human IgA; GBS β-antigen recognizes only serum human IgA, and SSL proteins interact with the serum and secretory IgA of human and other mammals with equal efficiency.
Hence, it can be stated that all three structurally unrelated groups of IgA receptors of streptococci and St. aureus, due to functional convergence, are aimed at recognition of the same structural element of IgA, occupying a small part of its vast surface. What makes this element unique, differentiating it from many other loops and elements of IgA secondary structure?
We suppose just in this point adjacent to the contact surface of domains CH2 and CH3, there is a place of a conformational shift induced by antigen binding in the active site of IgA. Thus, both CD89 and the exogenous receptors of pathogens located in this but no other point of IgA surface can differentiate free IgA from their complexes with the antigen. However, unfortunately, the authors of the published works did not perform the necessary controls for testing of this assumption. None of them compared the affinity of IgA receptors to free immunoglobulins and their complexes with the antigen.
Since all the above mentioned bacterial IgA-binding proteins are multifunctional receptors and, along with IgA, bind the complement components, it can be suggested that the complement and the IgA-dependent effector system of target destruction are concurrently operating systems, each capable of elimination of a pathogen on its own. Thus, a pathogen must suppress both of these systems in order to survive. It seems that this hypothetical IgA-dependent mechanism is based both on CD89 and on other, yet unidentified receptors recognizing the same site on the IgA surface. However, it is beyond question that the effective operation of such a system is possible only subject to discrimination of free IgA from the IgA complexes with antigen. Activation of phagocytosis or respiratory burst in neutrophils by free IgA, independent of the presence of antigen in an organism, not only would not promote protection from infection but would inevitably lead to a severe autoimmune affection of its own tissues and organs. Since the dependence of IgA CD89 activation on the presence of antigen has not yet been proved, we suggest the possibility of existence of alternative CD89 receptors that can react only to the immune IgA complexes but not to free antibodies of this class.
This review has been prepared in the framework of a grant of the President of the Russian Federation for support of young Russian scientists (MD-866.2008.4).
REFERENCES
1.Kerr, M. A. (1990) Biochem J., 271,
285-296.
2.Woof, J. M., and Mestecky, J. (2005) Immunol.
Rev., 206, 64-82.
3.Mistry, D., and Stockley, R. A. (2006) Int. J.
Biochem. Cell. Biol., 38, 1244-1248.
4.Takenouchi-Ohkubo, N., Mortensen, L. M., Drasbek,
K. R., Kilian, M., and Poulsen, K. (2006) Microbiology, 152
(Pt. 7), 2171-2180.
5.Kazeeva, T. N., and Shevelev, A. B. (2007)
Biochemistry (Moscow), 72, 485-494.
6.Langley, R., Wines, B., Willoughby, N., Basu, I.,
Proft, T., and Fraser, J. D. (2005) J. Immunol., 174,
2926-2933.
7.Kehoe, M. A. (1991) Vaccine, 9,
797-806.
8.Robinson, J. H., and Kehoe, M. A. (1992)
Immunol. Today, 13, 362-367.
9.Dale, J. B. (1999) Vaccine, 17,
193-200.
10.Batzloff, M. R., Pandey, M., Olive, C., and Good,
M. F. (2006) Immunol. Res., 35, 233-248.
11.Carlsson, F., Berggard, K., Stalhammar-Carlemalm,
M., and Lindahl, G. (2003) J. Exp. Med., 198,
1057-1068.
12.Persson, J., Beall, B., Linse, S., and Lindahl,
G. (2006) Public Lib. Sci. Pathog., 2, e47.
13.McArthur, J. D., and Walker, M. J. (2006) Mol.
Microbiol., 59, 1-4.
14.Fischetti, V. A. (1991) Sci. Am.,
264, 58-65.
15.Amezaga, M. R., and McKenzie, H. (2006) J.
Antimicrob. Chemother., 57, 443-449.
16.Jarva, H., Jokiranta, T. S., Wurzner, R., and
Meri, S. (2003) Mol. Immunol., 40, 95-107.
17.Sharma, A. K., and Pangburn, M. K. (1997)
Infect. Immun., 65, 484-487.
18.Kotarsky, H., Gustafsson, M., Svensson, H. G.,
Zipfel, P. F., Truedsson, L., and Sjobring, U. (2001) Mol.
Microbiol., 41, 817-826.
19.Kihlberg, B. M., Collin, M., Olsen, A., and
Bjorck, L. (1999) Infect. Immun., 67, 1708-1714.
20.Johnsson, E., Berggard, K., Kotarsky, H.,
Hellwage, J., Zipfel, P. F., Sjobring, U., and Lindahl, G. (1998) J.
Immunol., 161, 4894-4901.
21.Sandin, C., Carlsson, F., and Lindahl, G. (2006)
Mol. Microbiol., 59, 20-30.
22.Andre, I., Persson, J., Blom, A. M., Nilsson, H.,
Drakenberg, T., Lindahl, G., and Linse, S. (2006) Biochemistry,
45, 4559-4568.
23.Jenkins, H. T., Mark, L., Ball, G., Persson, J.,
Lindahl, G., Uhrin, D., Blom, A. M., and Barlow, P. N. (2006) J.
Biol. Chem., 281, 3690-3697.
24.Berggard, K., Johnsson, E., Morfeldt, E.,
Persson, J., Stalhammar-Carlemalm, M., and Lindahl, G. (2001) Mol.
Microbiol., 42, 539-551.
25.Morfeldt, E., Berggard, K., Persson, J.,
Drakenberg, T., Johnsson, E., Lindahl, E., Linse, S., and Lindahl, G.
(2001) J. Immunol., 167, 3870-3877.
26.Carlsson, F., Sandin, C., and Lindahl, G. (2005)
Mol. Microbiol., 56, 28-39.
27.Perez-Caballero, D., Garcia-Laorden, I., Cortes,
G., Wessels, M. R., de Cordoba, S. R., and Alberti, S. (2004) J.
Immunol., 173, 6899-6904.
28.Wistedt, A. C., Ringdahl, U., Muller-Esterl, W.,
and Sjobring, U. (1995) Mol. Microbiol., 18, 569-578.
29.Navarre, W. W., and Schneewind, O. (1999)
Microbiol. Mol. Biol. Rev., 63, 174-229.
30.Akerstrom, B., Lindahl, G., Bjorck, L., and
Lindqvist, A. (1992) J. Immunol., 148, 3238-3243.
31.Cedervall, T., Johansson, M. U., and Akerstrom,
B. (1997) Biochemistry, 36, 4987-4994.
32.Husmann, L. K., Scott, J. R., Lindahl, G., and
Stenberg, L. (1995) Infect. Immun., 63, 345-348.
33.Lindahl, G., and Stenberg, L. (1990)
Epidemiol. Infect., 105, 87-95.
34.Stenberg, L., O’Toole, P., and Lindahl, G.
(1992) Mol. Microbiol., 6, 1185-1194.
35.Lindahl, G., and Akerstrom, B. (1989) Mol.
Microbiol., 3, 239-247.
36.Johnsson, E., Andersson, G., Lindahl, G., and
Heden, L. O. (1994) J. Immunol., 153, 3557-3564.
37.Akerstrom, B., Lindqvist, A., and Lindahl, G.
(1991) Mol. Immunol., 28, 349-357.
38.Johnsson, E., Areschoug, T., Mestecky, J., and
Lindahl, G. (1999) J. Biol. Chem., 274, 14521-14524.
39.O’Toole, P. W., Stenberg, L., Rissler, M.,
and Lindahl, G. (1992) Proc. Natl. Acad. Sci. USA, 89,
8661-8665.
40.Stenberg, L., O’Toole, P. W., Mestecky, J.,
and Lindahl, G. (1994) J. Biol. Chem., 269,
13458-13464.
41.Bessen, D. E. (1994) Infect. Immun.,
62, 1968-1974.
42.Sandin, C., Linse, S., Areschoug, T., Woof, J.
M., Reinholdt, J., and Lindahl, G. (2002) J. Immunol.,
169, 1357-1364.
43.Grubb, A., Mendez, E., Fernandez-Luna, J. L.,
Lopez, C., Mihaesco, E., and Vaerman, J. P. (1986) J. Biol.
Chem., 261, 14313-14320.
44.Vaerman, J. P., Hagiwara, K., Kobayashi, K., and
Rits, M. (1987) Immunol. Lett., 15, 67-72.
45.Janoff, E. N., Fasching, C., Orenstein, J. M.,
Rubins, J. B., Opstad, N. L., and Dalmasso, A. P. (1999) J. Clin.
Invest., 104, 1139-1147.
46.Pleass, R. J., Areschoug, T., Lindahl, G., and
Woof, J. M. (2001) J. Biol. Chem., 276, 8197-8204.
47.Nohynek, H. (2006) Adv. Exp. Med. Biol.,
582, 137-150.
48.Lindahl, G., Stalhammar-Carlemalm, M., and
Areschoug, T. (2005) Clin. Microbiol. Rev., 18,
102-127.
49.Auperin, T. C., Bolduc, G. R., Baron, M. J.,
Heroux, A., Filman, D. J., Madoff, L. C., and Hogle, J. M. (2005) J.
Biol. Chem., 280, 18245-18252.
50.Kong, F., Gowan, S., Martin, D., James, G., and
Gilbert, G. L. (2002) J. Clin. Microbiol., 40,
620-626.
51.Nagano, N., Nagano, Y., and Taguchi, F. (2002)
Infect. Immun., 70, 4643-4649.
52.Lachenauer, C. S., Baker, C. J., Baron, M. J.,
Kasper, D. L., Gravekamp, C., and Madoff, L. C. (2002) J. Infect.
Dis., 185, 368-374.
53.Jerlstrom, P. G., Chhatwal, G. S., and Timmis, K.
N. (1991) Mol. Microbiol., 5, 843-849.
54.Heden, L. O., Frithz, E., and Lindahl, G. (1991)
Eur. J. Immunol., 21, 1481-1490.
55.Jerlstrom, P. G., Talay, S. R., Valentin-Weigand,
P., Timmis, K. N., and Chhatwal, G. S. (1996) Infect. Immun.,
64, 2787-2793.
56.Nagano, N., Nagano, Y., Nakano, R., Okamoto, R.,
and Inoue, M. (2006) Microbiology, 152, 771-778.
57.Lowy, F. D. (1998) N. Engl. J. Med.,
339, 520-532.
58.Dinges, M. M., Orwin, P. M., and Schlievert, P.
M. (2000) Clin. Microbiol. Rev., 13, 16-34.
59.Williams, R. J., Ward, J. M., Henderson, B.,
Poole, S., O’Hara, B. P., Wilson, M., and Nair, S. P. (2001)
Infect. Immun., 68, 4407-4415.
60.Wines, B. D., Willoughby, N., Fraser, J. D., and
Hogarth, P. M. (2006) J. Biol. Chem., 281, 1389-1393.
61.Monteiro, R. C., and van de Winkel, J. G. (2003)
Annu. Rev. Immunol., 21, 177-204.
62.Van Egmond, M., Damen, C. A., van Spriel, A. B.,
Vidarsson, G., van Garderen, E., and van de Winkel, J. G. (2001)
Trends Immunol., 22, 205-211.
63.Woof, J. M. (2002) Biochem. Soc. Trans.,
30, 491-494.
64.Mattu, T. S., Pleass, R. J., Willis, A. C.,
Kilian, M., Wormald, M. R., Lellouch, A. C., Rudd, P. M., Woof, J. M.,
and Dwek, R. A. (1998) J. Biol. Chem., 273,
2260-2272.
65.Pleass, R. J., Dunlop, J. I., Anderson, C. M.,
and Woof, J. M. (1999) J. Biol. Chem., 274,
23508-23514.
66.Ramsland, P. A., Willoughby, N., Trist, H. M.,
Farrugia, W., Hogarth, P. M., Fraser, J. D., and Wines, B. D. (2007)
Proc. Natl. Acad. Sci. USA, 104, 15051-15056.