REVIEW: The Problem of the Eukaryotic Genome Size
L. I. Patrushev1* and I. G. Minkevich2
1Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; E-mail: patrush@mx.ibch.ru2Skryabin Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, Pushchino, Moscow Region, Russia
* To whom correspondence should be addressed.
Received August 13, 2007; Revision received August 19, 2008
The current state of knowledge concerning the unsolved problem of the huge interspecific eukaryotic genome size variations not correlating with the species phenotypic complexity (C-value enigma also known as C-value paradox) is reviewed. Characteristic features of eukaryotic genome structure and molecular mechanisms that are the basis of genome size changes are examined in connection with the C-value enigma. It is emphasized that endogenous mutagens, including reactive oxygen species, create a constant nuclear environment where any genome evolves. An original quantitative model and general conception are proposed to explain the C-value enigma. In accordance with the theory, the noncoding sequences of the eukaryotic genome provide genes with global and differential protection against chemical mutagens and (in addition to the anti-mutagenesis and DNA repair systems) form a new, third system that protects eukaryotic genetic information. The joint action of these systems controls the spontaneous mutation rate in coding sequences of the eukaryotic genome. It is hypothesized that the genome size is inversely proportional to functional efficiency of the anti-mutagenesis and/or DNA repair systems in a particular biological species. In this connection, a model of eukaryotic genome evolution is proposed.
KEY WORDS: C-value paradox, genome size, genome evolution, coding sequences, non-coding sequences, gene protectionDOI: 10.1134/S0006297908130117
Abbreviations: AP sites, apurinic sites; BER, base excision repair; cDNA, complementary DNA; LINE, long interspersed elements; LTR, long terminal repeat; MAR/SAR-NS, matrix-/scaffold-attachment regions; Mb, megabases; 5′-mC, 5′-methylcytosine; MMR, mismatch repair; NE, nuclear envelope; NIR, nucleotide incision repair; NPC, nuclear pore complex; NS, nucleotide sequences; nt, nucleotide; rDNA, ribosomal DNA; RNP, ribonucleoprotein; ROS, reactive oxygen species; SINE, short interspersed elements; SNP, single nucleotide polymorphism; TE, transposable element.
The term “genome” was proposed by H. Winkler in 1920 to
describe a combination of genes included in a haploid set of
chromosomes of a single biological species [1].
Already at that time, it was emphasized that, unlike genotype, the
concept of genome is a characteristic of a species as a whole rather
than of an individual. Intensive investigation of genomes during the
last fifty years has noticeably changed the concepts of their structure
and function. As the complete primary structure of the human genome was
established along with quite a number of animal, plant, and microbial
genomes, humankind entered the “post genomic era”. Now the
term “genome” includes the combination of DNA of a haploid
set of chromosomes that are included in a single cell of germ line of a
multicellular organism. In this case it is also necessary to consider
genetic potential of DNA of all extrachromosomal genetic elements of an
organism, which are also constantly transmitted from one generation to
another through the maternal line, control functions of the nuclear
genome, and define many phenotypic features [2]. In
our opinion, close interweaving of functions of genes localized both in
the cell nuclei and organelles allows one to consider their genomes as
a unified system of genes comprising the full genome of a living
organism.
Instability of the primary structure of the genome was an intriguing discovery in this trend of investigations. It appeared that the genome is not a static place for storage and first steps of realization of genetic information, but its structure is highly dynamic. In the human genome the enormous number of allele variants of genomic nucleotide sequences (NS) (10-15 millions SNP (single nucleotide polymorphism)) were found, including structural polymorphisms like deletions, inserts, inversions, translocations, copy number variations (CNV) of genomic NS, as well as the possibility of programmed structural genome rearrangements in ontogeny [3]. The same is also characteristic of other studied organisms. Recently the pangenome concept for bacteria has been formulated according to which the genome of some bacterial species is represented by the core genome, whose nucleotide sequences are identified in absolutely all bacterial strains of a given species, as well as by its non-obligatory part that includes dispensable genes specific to particular bacterial isolates [4, 5]. Attempts to spread the pangenome concept to the world of plants have been undertaken [6], and it may happen that organization (arrangement) of genes in the form of pangenome is a widespread phenomenon. Khesin was the first in Russia who paid attention to the problem of genome instability and contributed much to its development [7, 8].
The phylogenetic genome instability is quite clearly reflected in the fact that total DNA content in a haploid set of chromosomes (in the gamete nucleus) of different eukaryotic species, designated by “C” symbol (the size of their genome), differs over 200,000-fold [9-12]. Among vertebrates, amphibians (especially salamanders) and lungfishes have the largest genomes of ~120 pg (1 pg DNA corresponds to ~109 bp) DNA (for comparison, the size of the human genome is 3.5 pg). In terrestrial plants, giant genomes are found in members of Liliaceae family (Fritillaria assyriaca - about 127 pg). The lack of correlation between the organism genome size and phenotypic complexity was termed as the C-value paradox [13]. In this case, the main part of DNA of large eukaryotic genomes is represented by NS, not coding proteins and RNA. In particular, the fraction of coding gene parts in the human genome makes up only ~3% [14].
Although the total size of eukaryotic genomes does not correlate with their phenotypic complexity, the evolutionary transition from pro- to eukaryotes and further from unicellular to multicellular organisms is accompanied by an increase in total number of genes in their genomes [15]. In this case, there are attempts to explain great differences in phenotypic complexity of higher eukaryotes with approximately equal (~104) number of genes (N) (N-value paradox [16]) by unique combinatorics of association of universal exons of their genes (and protein domains) in phylogeny and during gene expression [17]. Such data withdraw paradoxical features from the abovementioned phenomenon and transfer it into the category of not solved problems such as the problem of functional significance of non-coding NS. As a result, the term “C-value enigma” is introduced into the modern literature instead of “C-value paradox” [18]. Factors that define the genome size of particular biological species as well as functions of most non-coding eukaryotic NS are still unknown.
The aim of this review is to consider the C-value enigma within the context of present-day data about eukaryotic genome structure and mechanisms of functioning and its constant interactions with intracellular medium such as nucleoplasm and cytoplasm. An original model is presented that points to a new aspect of this problem of general biological significance.
EUKARYOTIC GENOME: STRUCTURAL ASPECT
Two aspects, structural and functional, can be distinguished in investigations of the genome as for any other macromolecular complex of a living organism. The genome structural organization at all levels, which will be briefly considered here, assures the fulfillment of all the main functions associated with storage, protection, reproduction, and realization of genetic information encoded in genomic NS. The size of the eukaryotic genome, the main subject of our review, is defined by the length of its individual NS that form genes and intergenic regions of the genome. Exhausting information concerning the concrete genomic NS, which is a congealed mold of the functioning dynamic genome, can be obtained from modern databases. The comparison of NS of whole genomes by the present-day genomic techniques makes possible detection of traces of their constant change and conclusion concerning molecular mechanisms that are the basis of such processes.
Nucleotide Sequences of the Genome
Previously studying NS of eukaryotic genome was based on analysis of peculiarities of re-association kinetics of genomic DNA short fragments (up to 500 bp) after their denaturing and following slow annealing on lowering of the temperature of the reaction mixture [19]. This method gave the first ideas concerning the great complexity of primary structure of such genomes. Recent complete genome sequencing of several eukaryotic species has made it possible to compile a more concrete concept on the peculiarities of their primary structure.
Highly repetitive sequences. Satellites. Satellite NS, whose content in a eukaryotic genome can reach 5-50% of total DNA, are very long (several hundreds of kilobase (kb)) DNA regions with short blocks (5-200 bp) repeated in tandems (“head-to-tail”) [20]. These NS got their name because they accompanied the main optical density peak as a shoulder (satellite) during total eukaryotic DNA centrifugation in a CsCl density gradient. The correct homogeneous base composition of satellite DNA, determined by the presence of numerous of short repeats, changed its buoyant density, which was easily detected during centrifugation. As a rule, these NS are characteristic of constitutive (constantly present) non-transcribed heterochromatin. Typical representatives of such DNA in the human genome are α-satellites. These NS of total length 105-106 bp formed by the main repetitive unit of 171 bp are localized mainly in centromeric regions of each chromosome and contain binding sites of centromere-associated proteins.
Human β and γ satellite NS of total length up to 0.25-0.50 megabases (Mb) are formed by GC-rich repeats of 68 and 220 bp, respectively. They are characteristic of telomeric (terminal) and some pericentromeric (located near centromeres) chromosome regions. Other members of these NS families, in particular, a family with main repeating unit of 48 bp and the Sn5 family are found in the human genome [21].
Microsatellites. Highly polymorphous microsatellite DNA formed by tandem repeat unit of 1-4 bp in length are arranged in blocks up to 200 bp that are spread over the genome. Unlike satellite and minisatellite DNA, microsatellites are, as a rule, transcribed. Homopolymeric microsatellites of the (A)n/(T)n type, which can be retrotransposon remnants, are often present in animal genomes. On the contrary, homopolymers of the (G)n/(C)n type are very rare in animals. Dinucleotide minisatellites of the CA/GT or CT/GA types are most frequent in animals, on average in every 20-50 kb. The AT-rich repeats, especially specific of the chromosome centromeric regions, are also highly represented in animal genomes. Tri- and tetranucleotide microsatellites are rare in animals. The microsatellite length and their total number in the genome correlate with the genome size [22].
Minisatellites. Minisatellite DNA are composed of repeated NS of 5-50 bp form intermediate size blocks (up to 104 bp) and are localized in different chromosome regions. Two main types of human minisatellite NS are known. The first type includes hypervariable minisatellites with the main repeat unit GGGCAGGANG where N is any nucleotide (nt). Such NS are localized in ~1000 genome sites and the length of their blocks is highly polymorphous in different individuals. These NS are considered as preferable sites of homologous recombination. Human telomeric NS with the repeated unit TTAGGG make up the second minisatellite family (block length in such repeats is 10-15 kb).
Macrosatellites. As follows from their name, macrosatellite NS of DNA are characterized by a large repetitive unit size (>1 kb). They are found, in particular, in avian W chromosomes and feline and human genomes [23].
Moderately repetitive sequences. Moderately repetitive sequences of the eukaryotic genome are represented by gene families and numerous mobile genetic elements (transposons). Since native transposons also contain particular genes providing for their survival in the genome, separation of moderately repetitive NS to two abovementioned groups is conventional and emphasizes the fact that the functional significance of transposons for the eukaryotic genome is still not quite clear [24-27].
Mobile genetic elements are the DNA NS able to change their position in the genome, i.e. to perform acts of transposition. Although at the present time there is no officially accepted transposon classification, they are divided into two large classes on the basis of molecular mechanisms used by mobile genetic elements for transposition within a genome: (1) retroelements and (2) DNA transposons.
DNA transposons are typical of bacterial genomes and are quite widespread in eukaryotic genomes. As a rule, their transposition follows the “cut and paste” mechanism with involvement of transposase, a well studied member of the recombinase class [28]. The realization of this mechanism results in duplication of a short NS in the site of transposon integration. In this case, a new site of the mobile element integration is usually located close to the old one, and as a result transfer of DNA transposons was named “local hopping” [29].
Unlike DNA transposons, retroelements use for their mobilization mechanisms in which an important role belongs to reverse transcription, i.e. DNA synthesis on RNA template by reverse transcriptase. Retroelements, in turn, are divided into two large groups on the basis of structural peculiarities and replication mechanisms: (i) LTR (long terminal repeats)-containing retroelements including retrotransposons and endogenous retroviruses [30], and (ii) retroelements without LTR and consisting of long (LINE) and short (SINE) interspersed elements [31, 32]. In these mobile genetic elements, the transposition event requires transcription of their genomic NS, reverse transcription of formed mRNA, and cDNA (complementary DNA) integration into a new genetic locus (the “copy and paste” mechanism).
Structurally and by replication mechanisms retrotransposons resemble exogenous retroviruses. Their structural peculiarity is the presence at their ends of LTR containing NS involved in regulation of their transcription. Besides, retrotransposons contain genes that provide for their replication and do not contain the capsid protein genes present in retrovirus genomes.
LINE elements, called long retroposons, contain the same genes as retrotransposons but have no LTR. Nevertheless, they contain promoters of RNA polymerase II performing transcription of corresponding genes. Since retrotransposons and LINE elements have everything necessary for transposition in the genome, they are called autonomous transposons.
SINE elements (short retroposons) are not autonomous, and for transposition they require the presence of protein products of the autonomous transposon gene expression. They contain near the 5′ end the internal promoter of RNA polymerase III that performs their transcription.
It should be emphasized that after each event of retroelement transposition its initial NS remains at the old place in the genome, and the corresponding copy emerges in a new genetic locus. Thus, the number of copies of transposon in the genome is doubled. This is one of the powerful mechanisms of eukaryotic genome enlargement, which is especially important for the problem being discussed.
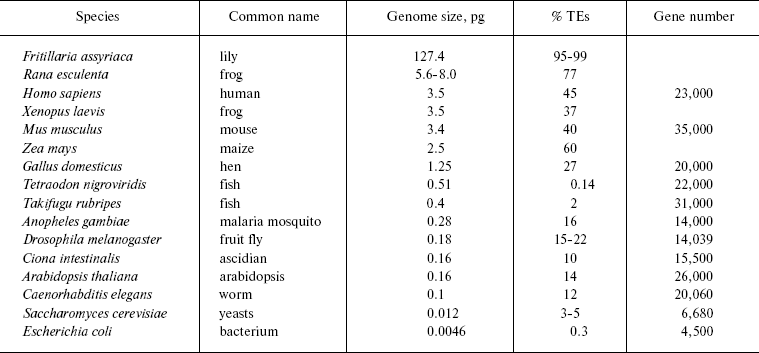
The table summarizes data on the content of mobile genetic elements in genomes of different taxonomic groups. Such high representation of transposons is indicative of their key role in the evolution of eukaryotic genomes. On the other side, the ubiquitous spreading of transposons allows one, together with Orgel and Crick as well as with Georgiev, to consider them not as molecular parasites, but as genomic endosymbionts [33, 34]. Unfortunately, it is still not clear what advantages are gained by the genome, the cell, and the organism as a whole from such symbiosis.
Transposon and gene content depending on genome size (after [24, 369])
Gene families consist of many genes characterized by high homology of coding (and in some cases non-coding, i.e. intron) NS. It is supposed that their origin is based on duplication of precursor genes or whole genetic loci. In the aggregate, genes combined by the community of their origin via duplication of precursor genes are called paralogs [35]. Genes with identical NS, organized in clusters or spread along the genome, provide the cell with gene expression products for increased requirements. Such eukaryotic genes include specifically histone genes, genes of rRNA, tRNA, and small nuclear RNA (snRNA). Besides, functioning of the multigene family genes regulates formation of systems of different signal recognition. In this connection, the largest known gene family of vertebrates consists of the olfactory receptor genes and pseudogenes (see below). In man and mouse, their number is 800 and 1400, respectively, and pseudogenes make up 60 and 25%, respectively. Genes of the major histocompatibility complex (MHC) and variable parts of immunoglobulins are represented by large families.
Unique sequences. The content of unique NS that appear only once in eukaryotic genome varies in different organisms and comprises from 15 to 98% of total DNA. Although many structural genes get into the unique NS fraction, most of them are non-coding. The well-known example of unique NS are the introns, whose total size usually exceeds that of exons of corresponding genes. An intron is a transcribed gene segment whose sequence is absent from mature RNA and is eliminated from precursor RNA by different mechanisms. Mature RNA consists of the exon sequences of the gene.
Exons and introns. The mosaic exon-intron gene structure has been found in members of most taxonomic groups of modern biological species [36]. In accordance with the present-day classification, introns are divided to four main classes differing by mechanisms of mRNA elimination from precursors [37]. The possession of the fourth class introns, excised from RNA molecules by splicing (sometimes they are called spliceosomal introns), is the exclusive prerogative of eukaryotic organisms.
The intron density in eukaryotic genomes (the number of introns per gene) differs by more than 1000 times in different species [38]. In particular, ~140,000 introns (8.4 introns/gene) were found in the human genome, the richest in introns, whereas the genome of the microsporidium Encephalitozoon cuniculi contains only 13 introns (0.0065 intron/gene). Moreover, as in the case of the eukaryotic genome size as a whole, there is no correlation between density of introns, their localization within the genome, and phylogenetic characteristics of biological species.
It was noted that human “housekeeping” genes, i.e. those expressed in cells of most tissues, are shorter in coding and non-coding parts compared to the “luxury” genes of specialized tissues, which is, for example, not characteristic of Arabidopsis thaliana and Drosophila melanogaster [39]. The real factors responsible for compactness of housekeeping genes, including, in particular, the intron-less ones, such as histone genes and most receptor genes coupled with G-proteins [40], are still unknown.
Pseudogenes comprise another class of unique NS of eukaryotic genes. The DNA NS that are most often inactive copies of original genes, altered by mutations, are called pseudogenes [41]. One such NS variety is composed of processed pseudogenes free of the precursor gene introns. It is supposed that processed pseudogenes emerge via integration into the genome of cDNA that resulted from reverse transcription of a corresponding mRNA after complete splicing of the latter has occurred. In this case, the incorporation of cDNA can be provided for by genes of autonomous retrotransposons. The pseudogene content in members of various taxonomic groups significantly differs. The highest number of pseudogenes (3600-19,000) is found in humans and they are spread over the whole genome; their number in individual chromosomes correlates with the chromosome size [42].
Unlike bacteria, whose pseudogenes undergo rapid degeneration, eukaryotes are characterized by a lower pressure of selection towards elimination of pseudogenes. The question concerning the role of pseudogenes in the genome is the subject for discussions. In some cases, their functional significance has been demonstrated [41]. There are indications in favor of possible involvement of pseudogene antisense RNA in transcription regulation. The participation of pseudogenes in human and other animal immune response is supposed, and it is already proved for birds. Pseudogenes are necessary for formation of genes of human olfactory receptors. Many eukaryotic pseudogenes are highly conserved and actively transcribed, while some can be activated by point mutations.
Mosaic pattern of the warm-blooded animal genome by GC-composition: isochores. The presence in a eukaryotic genome of discrete, extended (hundreds of kilobase pairs (kb)), highly homogeneous by GC-composition regions called isochors is evidently one of the fundamental principles of its organization at the level of primary structure [43]. Isochores are divided into several “light” and “heavy” families (they are numbered according to increase in GC content): L1, L2, H1, H2, and H3 (L - light, H - heavy). The metaphase chromosome banding patterns (cross-striation after staining) correlates well with quantitative and qualitative content of isochores belonging to particular families [44].
It was found that NS of the abovementioned families differ both by GC composition and other structure-function characteristics. In the human genome, the most GC-rich isochores have the highest density of gene arrangement (“genome core”), and isochores with low GC content are characterized by low gene concentration (“genome desert”) [45, 46]. GC-rich isochores are replicated early in the cell cycle, while GC-poor ones do this late [47]. The most GC-rich isochores are most often located in telomeric zones of metaphase chromosomes, whereas a different localization is specific of the least GC-rich isochores [48, 49]. In this case, GC-rich isochores are less densely packed in chromatin fibrils compared to GC-poor ones [48, 50]. All this suggests the existence of clear association between functional compartmentalization of interphase nuclei and structural organization of the eukaryotic genome, i.e. with its primary structure.
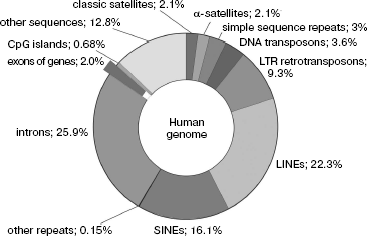
Coding and non-coding sequences. Our analysis of peculiarities of the eukaryotic genome primary structure makes possible, in accordance with the long-standing tradition, dividing it into two functionally nonequivalent parts of different size, namely, to coding and non-coding NS. Figure 1 shows the share of different class NS in the total size of the human genome; it is seen that genes occupy only its small part.
Traditionally, coding NS are considered to mean the genomic DNA regions (gene parts) that contain information on the protein and nucleic acid primary structures realized during their expression. However, this also includes NS of genes of mobile genetic elements, which, as mentioned above, can comprise the bulk of genomic NS. Moreover, present-day investigations of the eukaryotic transcriptome using biochip technologies show that up to 1/3 of all NS can be transcribed in large eukaryotic genomes [51, 52].Fig. 1. Content of different nucleotide sequences in the human genome. Protein coding sequences (~20,000 genes) represent less than 1.5% of the genome.
In modern studies, large-scale genome sequencing and comparative analysis of primary structures precede gene identification in the abovementioned sense. The criteria used for estimation of NS functionality are gradually changed, and their evolutionary conservativeness, indicative of the pressure of natural selection aimed at the maintenance of their structure and, as a result, of their function, is regarded as of paramount importance [53]. In particular, it was found that extended conserved genome regions are spread beyond genes. The comparison of the mouse and human genomes has shown that in the latter ~5% NS are under pressure of selection and only 1/3 of them belongs to the protein encoding genes [54].
Evidently, our concepts of coding and non-coding NS will change. It is reasonable to assume that at the present time insufficient attention is paid to experimental investigation of the role of non-coding NS in spatial location of exons and whole genetic loci in interphase nuclei. In particular, while carrying out their skeletal function (see below and [9]), non-coding NS could form unique intranuclear microcompartments influencing expression of genes, present in these microcompartments, and their protection against chemical mutagens. In this case, non-coding NS could incorporate the code translated by an interphase nucleus into spatial structure of the genome. However, now in this work the term coding NS means the genomic DNA sequences not included in transposons and containing information concerning proteins and RNA with known or supposed functions.
Spatial Organization of the Genome
Genomic DNA of all eukaryotic organisms, whose total length for a single genome copy can reach several meters, is localized within a highly ordered nucleoprotein complex called chromatin [55-57]. This allows it to go in, to be reproduced, and retain functional activity in the cell nuclei whose diameter does not exceed several micrometers. Accordingly, proteins providing for spatial DNA packing in individual chromosomes can be considered as peculiar DNA chaperones [58].
Morphological features of interphase nucleus. The cell nucleus is an intracellular organelle providing for the main processes associated with the storage, transformation, realization, reproduction, and maintenance of integrity of genetic information contained in DNA molecules. The present-day data have strengthened the concept of the interphase nucleus as a highly ordered cytogenetic system [59, 60]. The volume of an animal somatic cell nucleus is ~600-1500 µm3.
Nuclear envelope, forming the interface between nuclear and cytoplasmic compartments, plays the main role in maintenance inside the nucleus of unique biochemical conditions necessary for functioning of the cellular genetic apparatus. It consists of external and internal nuclear membranes that are perforated by 103-104 nuclear pore complexes (NPC) that provide for transport in both directions of high- and low-molecular-weight compounds necessary for the cell nucleus functioning and/or being products of its vital activity. The external membrane interacts with ribosomes and is a part of the cellular rough endoplasmic reticulum. The internal membrane establishes contacts with chromatin and is marked by specific membrane proteins [61]. The two membranes are joined with each other at the border of each NPC.
Nuclear lamina, a thin layer (~20 nm) of hydrophobic proteins is adjacent to the internal membrane from the side of the nucleoplasm. Chromatin loops can specifically interact with these proteins. Although the lamina is a component of nuclear matrix, it has a specific protein composition, including the best-characterized A-type and B-type lamins that are present in the highest amounts [61]. Mutations in their genes are accompanied by global spatial rearrangements in heterochromatin, alterations in DNA transcription and replication, in cytoskeleton and cell survival, as well as in development of severe human laminopathies.
Nuclear matrix is the second (after chromatin) nuclear component containing nucleic acids [62]. It is usually detected in the nucleus in the form of reticular, fibrillar, and RNP (ribonucleoprotein)-containing structures after removal of membranes (by nonionic detergents) and chromatin (by the incubation of nuclei with DNase), and hypertonic salt extraction of histones and DNA fragments. The core part of the nuclear matrix consists of branched filaments 10 nm in diameter, whose composition and molecular structure are still not quite clear. Filaments of internal nuclear matrix are associated with nuclear lamina and, in particular, they are involved in attachment to the latter of bases of 30 nm chromatin fiber loops [63]. DNA sequences 150-200 bp long providing for such interaction, designated as MAR/SAR-NS (matrix-/scaffold-attachment regions), were found.
The nucleolus is the best-studied nuclear subcompartment; it mainly provides for biogenesis of rRNA and ribosomal 40S and 60S subparticles, and it is usually detected in all eukaryotes. Simultaneously hundreds of biochemical processes take place in nucleoli of metabolically active animal and plant somatic cells [64, 65]. Among “non-canonical” functions of nucleoli, there are involvement in virus infection control, non-nucleolar RNA or RNP processing, influencing the telomerase functions and cell ageing, participation in cell cycle regulation, expression of tumor suppressors and oncogenes, signal transduction, etc. Most nucleolar proteins are retained in it for no more than 1 min, and the nucleolus proper exists in a stationary state in the form of a special intranuclear compartment owing to stronger interaction of some proteins with rDNA; these proteins form the intranucleolar area for different biochemical processes. Evidently, such state is also characteristic of other intranuclear subcompartments detected at morphological level [60, 66], as will be discussed below.
Other nuclear subcompartments. Numerous bodies are present in the nucleus that are not fixed intranuclear compartments but are indicative of genome activity in interphase, and often they exist in the nucleus for no more than a few minutes. A high content of specific proteins involved in various genetic processes is characteristic of subcompartments, which allows one to consider them as places of assembly of characteristic macromolecular complexes and their following functioning. The Cajal bodies are heterogeneous sets of subnuclear domains of different molecular composition and biological functions, the main being biogenesis of small nuclear and nucleolar RNP (snRNP and snoRNP, respectively) and of RNA telomerase (hTR) [67]. The so-called coiled bodies probably carry out a similar function [68]. PML bodies contain the promyelocytic leukemia protein (PML) and represent a site of viral genome efficient replication and transcription in infected cells [69]. OPT domains (Oct1/PTF/transcription domains), enriched with transcription factors, mark subnuclear structures of “transcription factories”. So-called speckles are the interchromatin granule clusters for accumulation of proteins involved in pro-mRNA splicing. Recently several new domains have been found in plant cell nuclei [66].
Euchromatin and heterochromatin. Beginning with the work by Heits (1928), two types of chromatin--euchromatin and heterochromatin--are distinguished in the interphase nucleus. Genomic DNA in heterochromatin is strongly condensed and as a rule (but not always) is not transcribed. Unlike this, euchromatic genome regions are less compact and more often, though not in all known cases, are occupied by expressed genes [70].
A fundamental feature of heterochromatin is its regulated ability of reverse spreading to extended adjacent euchromatin genome regions, which is often accompanied by euchromatin transition to heterochromatin (euchromatin heterochromatization) and inhibition of transcription of genes located in it (the phenomenon of epigenetic gene silencing) [71]. Heterochromatin is characterized by the presence of hypoacetylated and methylated histones. In this case, the character of their methylation makes possible the differentiation between the irreversibly formed (“constitutive”) and “facultative” heterochromatin. The latter can be decondensed, which is often accompanied by transcription activation [72]. Heterochromatization of specific genome regions is initiated by non-coding double-stranded (ds) or short interfering (si) RNA, coordinated by the work of histone deacetylases and methyl transferases, as well as by successive incorporation of specific proteins into chromatin. This results in changes in the chromatin spatial structure and, finally, in inhibition of transcription (and expression) of corresponding genes [55, 73]. Such (at first sight independent of the DNA primary structure) transcription regulation by changing the chromatin spatial structure is a typical eukaryotic mechanism of regulation of gene expression, recombination, and DNA repair.
Besides, gene-containing DNA regions, the main objects of heterochromatization in the genome, are repeated DNA sequences of chromosomes, such as satellite DNA and mobile genetic elements. There are indications that in some cases heterochromatization is also necessary for gene activation [74]. Mutations inhibiting heterochromatin formation change the internal spatial structure of nuclei, which is accompanied by change in regulated during ontogeny interaction of genetic loci via their cis-acting regulatory elements (see below) [75, 76].
Levels of genomic DNA compaction within chromatin. The compaction of eukaryotic DNA in interphase nuclei is regulated at least at three levels of intermolecular interactions providing for spatial organization of chromatin. They include intranucleosomal interactions (i), internucleosomal interactions (ii), and nucleosome interactions with the chromatin structural proteins (iii). All three levels of interactions, functionally associated with each other, first of all provide for regulated gene transcription [77].
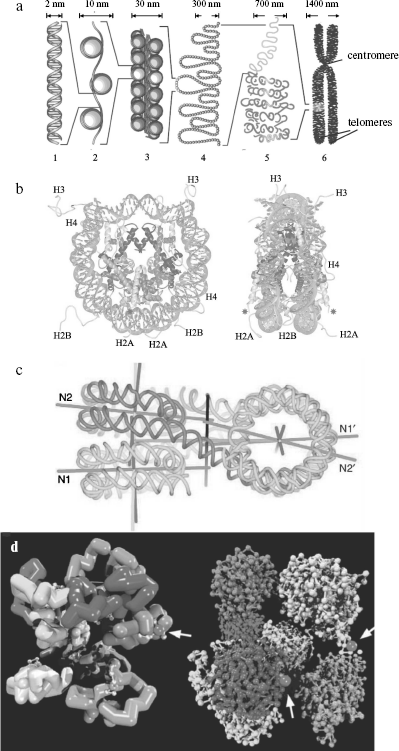
The nucleosome is the fundamental structural repeating unit of chromatin. The so-called core (main) part of it consists of four histone proteins (two dimers H2A-H2B and tetramer (H3)2-(H4)2), and a DNA molecule forms two turns around this protein octamer. Each nucleosome contains ~165 bp of DNA, and this parameter depends on the species of organism and cell type. Nucleosomes within chromatin are arranged one after another and are separated by short regions of so-called linker DNA of 10-80 bp. Inclusion of DNA into nucleosomes is accompanied by ~5-10 times reduction in its linear size (compaction) [78]. Recently spatial structure of in vitro reconstructed core particle of nucleosomes has been determined by X-ray analysis at resolution <2 Å (Fig. 2b) [79].
Covalent modifications of histones play an important role in transcription regulation (through changes in chromatin structure) and histone metabolism, and they exhibit a mediated effect on many cell processes including the cell cycle. Among these modifications there are reversible histone acetylation at Lys residues, phosphorylation (Lys, Ser), methylation (Arg), poly(ADP)-ribosylation (Glu), and ubiquitinylation and sumoylation (Lys) [28, 80, 81]. Besides, structural variants of histones found in nucleosomes are markers of specific genomic NS and DNA states [82, 83].Fig. 2. a) DNA compaction levels within chromatin; b) spatial structure of the nucleosome core particle obtained by X-ray analysis at 2 Å resolution [371]; c) spatial structure of a tetranucleosome forming a chromatin fiber of 30-nm diameter; d) a hypothetical structure of 1 Mb chromatin domain [96]. a) From left to right: 1) DNA double helix; 2) nucleosomal fiber; 3) solenoid; 4) chromatin loops; 5) a region of chromosome territory; 6) metaphase chromosome. The size of corresponding chromatin elements is shown above; only two of four telomeres are designated on a metaphase chromosome. b) Left: front view of a nucleosome; histones H2A, H2B, H3, and H4 are designated at their extruding end parts, DNA being shown as light gray at the periphery of the nucleosome; asterisks show sites of histone H4 ubiquitination in yeasts. Right: side-view of the same complex after turning by 90° around the vertical axis. c) Separate nucleosomes in the “solenoid” tetranucleosome are superimposed one above another (N1, N2 and N1′, N2′) forming two piles; two terminal nucleosomes are located at the top (in left part of the figure) and bottom (in right part of the figure), and two central nucleosomes - vice versa, linker DNA being crossed between them. Straight lines show symmetry axes of nucleosomal strand, its supercoiling, nucleosome piles, as well as (the supposed one) of the total 30-nm fiber. d) It is shown on the left that the chromosomal territory domain consists of 10 subdomains (loops) of 100 kb in length (with different shades of gray), the zigzag-like cylinders within them show chromatin 30-nm fibers that is broken from time to time by untwisted chains of nucleosomes of 10-nm diameter (white dots). On the right, each of 10 subdomains of 100 kb is represented by a zigzag-like chain of nucleosomes, where each nucleosome is shown by a separate dot; most subdomains are shown in a closed configuration except a domain in the right part of the figure penetrating into interchromatin compartments; black dots show protein complexes (nuclear bodies) involved in transcription or splicing (indicated by arrows).
It now appears that the genome primary structure may contain nucleosome code, i.e. information that relates the process of nucleosome formation to definite NS (so-called nucleosome positioning) that are periodically repeated along extended DNA regions. It is supposed that the existence of such a code could make it significantly easier for chromosomes to carry out their specific functions, namely, transcription control via interaction of protein factors with regulatory NS and substantial rearrangement of nucleosome structures proper [84, 85]. The nucleosome positioning along DNA molecules can also define fundamental features of DNA secondary structure, so-called DNA bend sites that are found on the average in each four nucleosomes [86]. The presence of bend sites correlates with regularities of eukaryotic genome segmentation, according to which the genome can be described as ~350 bp NS blocks (two nucleosomes) following one after another. Such blocks could result from the combinatorial fusion of short NS at early stages of evolution [87].
Another level of periodicity in eukaryotic genomic DNA that is potentially able to influence chromatin structure is represented by regions with destabilized DNA double helix, which facilitates its melting in these sites (so-called unpairing elements (UE) appear in each ~3 kb) [88]. UE and their clusters called BUR (base unpairing regions) mark loci of DNA interaction with specific proteins that provide for the nucleosome strand folding into higher order structures.
The 30 nm chromatin fiber. In the presence of linker histone H1 and/or divalent metal ions, the nucleosome string undergoes further compaction with formation of a structure formerly known as “solenoid”, a compact fiber of ~30-nm diameter (Fig. 2a). Spatial structure of reconstructed chromatin region containing four nucleosomes has been recently determined at 9 Å resolution to refine the solenoid structure [89] (Fig. 2c). It follows from the data that in a tetranucleosome individual nucleosomes interact in pairs with each other, while linker NS of DNA are crossed as a zigzag. The latter observation may indicate that the widespread concept of cylindrical shape of 30 nm chromatin fiber is a simplification. Histone H1 and some other proteins stabilize nucleosome structure, prevent their transposition (slipping) along DNA, and are involved in maintenance of the chromatin higher order spatial structure [89, 90].
The majority of chromatin in interphase nuclei is in a highly condensed state (the level of condensation ~10 times exceeds that in 10 and 30 nm chromatin fibers) [91-93]. In this case, interchromatin space, in which DNA is not found, occupies approximately half of the nuclear volume.
It is important to emphasize for further discussion that DNA within chromatin is located outside the core particle of the histone octamer, and thus it is accessible for low-molecular-weight reactive compounds like chemical mutagens. As a whole, intranuclear chromatin structure in interphase nuclei is highly dynamic and is far from the crystalline one revealed by X-ray analysis in the simplified model systems [94].
Chromosome territories. Individual chromosomes occupy in interphase nuclei more or less clearly distinguished regions called chromosome territories. This was first supposed at the turn of the XIX and XX centuries by K. Rable and by T. Bovery [95, 96]. Now the fact of the existence of chromosome territories is proved experimentally.
Models of chromosome territories. In the interchromosomal domains model (ICD), chromosome areas, represented by highly condensed chromatin, are separated from each other by a system of channels called interchromosomal domains [97, 98]. Transcribed genes are localized at the periphery of chromosome territories in the decondensed state in interchromosomal space (domains) whose branches may deeply penetrate chromosome territories, creating interchromatin compartments (interchromatin compartment model (CT-IC)) [99]. The latter originate from nuclear pores and are branched within chromosome areas, forming a network of channels terminating at the chromatin domains incorporating ~1 Mb DNA (Fig. 2d). These domains, in turn, consist of hypothetical loops (SL, small loop) ~0.1 Mb long subdomains joined by proteins in the center of the abovementioned 1 Mb domain with formation of a rosette of SL domains (multiloop subcompartment model (MLS)).
The 1 Mb chromatin domains, following one after another and distinguishable during the whole interphase, are characterized by spherical packing of chromatin SL loops that provide for over 300-fold DNA condensation; these domains are supposed to be one of higher levels of chromatin compaction in chromosome territories [100]. Besides, now a model of chromosome territories composed of giant loops (CT-GL model)--chromatin fibrils several Mb in length--is developed [101, 102].
An important organizing role of nuclear matrix in the chromosome territories models is emphasized [95, 96, 103]. According to some data, mammalian genomes in interphase nuclei can be organized in ~60,000 chromatin loops attached to nuclear matrix (MAR/SAR-NS). The mean size of such loops is ~70 kb [88].
Intranuclear arrangement of chromosome territories. The arrangement of chromatin and chromosome territories in interphase nuclei is ordered and evolutionarily conserved in animals and plants. In particular, chromatin whose DNA is replicated during early S phase of the cell cycle is mainly localized inside nuclei, while the late replicating chromatin is associated with the nuclear periphery and the nucleolus [104-107]. Isochores in interphase nuclei of some vertebrates are also non-randomly distributed. Human, pig, and chicken GC-richest NS are located closer to the nuclear center, while DNA with low GC content is located preferably at the nuclear periphery [48, 50, 108]. In addition, in cells of most tissues with spherical nuclei the gene-poor regions of chromosome territories, centromere regions of chromosomes, and intercalated heterochromatin (present as interspersions in euchromatin genome regions) tend to be localized at the periphery of interphase nuclei near the nuclear lamina. Chromosomes with high gene content as well as their gene-enriched and actively transcribed regions are more often localized closer to the central part of the nucleus, though the position of the latter is to a lesser extent determined in the nucleoplasm [99, 109-113]. Such arrangement of chromosomes is established at an early stage of the cell cycle and is maintained during the whole cycle [99, 111]. In this case, there appears specific alteration of spatial position of chimeric chromosome territories formed after joining of different chromosome regions [105, 111, 114-118]. There are data showing that in some tissues, in particular, in human and other primate fibroblasts, whose nuclei have the shape of 3D ellipsoids, such arrangement of chromosome territories, ordered from the nucleus periphery to its center, more correlates with the chromosome size than with the gene content in it [113, 119, 120].
In 1885, C. Rabl found in cells of salamanders a polar arrangement of centromeres and telomeres in anaphase of mitosis near the cell nucleus surface (Rabl configuration) and supposed that the same order of arrangement of chromosome regions is also retained in interphase nuclei (cited by [121]). This has actually been confirmed in plants and some animals (like drosophila) [122, 123]. However, radial arrangement of chromosomes with preferable location of some chromosome territories at the nuclear periphery and others closer to the center is more characteristic of most animals [121].
However, another type of determination of the chromosome territories arrangement in interphase nuclei is their nonrandom relative position. This model of chromosome arrangement was developed with account of found specific patterns (mitotic “rosettes”) of human metaphase chromosomes on metaphase plates in which the chromosome order (arrangement relative each other) is well reproduced [124, 125]. However, there are also opposing data concerning this problem [126].
The abovementioned evolutionarily conserved features of chromatin architectonics in interphase nuclei point to their functional significance for biological species. Most such kind data do not seem surprising and this is explained by the necessity for coordinated regulation of eukaryotic gene expression by their interaction with each other as well as by our model (see below).
Actively transcribed genes not always are concentrated in external parts of chromosome territories but are disseminated in them. This means that chromosome territories are permeable for substrates, regulatory proteins and their transcription products, and also for DNA-damaging chemical agents [93, 122, 127]. In some cases the expressed genes are characterized by moving away within chromatin loops from the chromosome territory to which they belong [128, 129].
Rapid chromatin movements for distances comparable with the size of chromosome territories are registered only in the first third of G1 phase of the cell cycle [130, 131]. In other cell cycle phases, movements are restricted to small nuclear subdomains whose size depends on the cell type and the extent of their differentiation.
Although as a whole, intranuclear arrangement of chromosome territories and their parts is dynamic, their general position may be genetically determined. In fact, it is difficult to imagine on the basis of general considerations that in the course of metaphase chromosome decondensation during the cell cycle, interphase links, that existed between adjacent chromatin regions in a condensed chromosome, are completely lost, and vice versa. With account of this, specific staining patterns of chromosomes (banding) can be considered as a reflection of ordered and genetically determined spatial structure of chromosome territories [132-134].
Structural analogies with protein organization. The considered hierarchy of the chromatin spatial packing suggests an analogy between structural peculiarities of chromatin and known levels of structural organization of proteins [57]. From this point of view, the chromatin primary structure is formed in a particular chromosome of the nucleosome NS in a nucleosome fiber of 10-nm diameter. Its secondary structure is represented by a more compact fiber of 30-nm diameter. The latter, in turn, forms chromatin domains in the form of loops, rosettes, and highly condensed chromatin, which can be considered as the prototype of its tertiary structure. So organized chromosomes are represented inside the interphase nucleus by chromosome territories interacting with each other, which, taken together, form the quaternary structure of eukaryotic nuclei. In this case, chromatin rearrangements, caused by regulatory factors, resemble mechanisms of allosteric regulation of enzyme activity. Nevertheless, the high dynamism and a complex, changing in time composition of chromatin macromolecular complexes restrict practical application of this kind of analogies.
Genetic loci in the interphase nucleus. Intranuclear gene positions influence gene activities. Movement of quite a number of activated animal genetic loci from the nuclear periphery to the nuclear centers was noted during investigation of the association between chromatin movements and gene expression [131, 135]. In this case, movements appeared to be associated with nuclear actin and myosin [136]. Such kinds of data support the concept of nuclear periphery as a transcriptionally inactive compartment. Nevertheless, the locus transcription status is not defined just by its position relative to the nuclear periphery, but it is rather formed due to other more specific regulatory effects (see references in reviews [131, 137]). The broad use of this strategy of the chromatin radial movements for total transcription regulation (transcriptome) in large animal and plant nuclei is doubtful. Apparently, in these organisms more significant is the locus position relative to extended compartments of constitutive heterochromatin or regulatory NS, often remote by significant distances from genes in a DNA molecule.
Genes are often turned to the repressed state (a mechanism of the gene silencing) due to transfer to the neighborhood of heterochromatized regions of the interphase nucleus [55, 71, 75, 138-142]. The importance of dynamic interchromosomal interactions has been also recently shown for initiation of X-chromosome inactivation in mice [143, 144].
Both the eukaryotic gene position relative to heterochromatin and to each other are important for their regulated expression in the cell cycle interphase [145]. The ordered spatial drawing together of genetic loci located on different chromosomes and their mutual effect on transcription is well known [146, 147]. Spatial approach of in cis- and in trans-activated genes to remote nuclear compartments enriched with RNA polymerase II and necessary transcription factors is probably often used for eukaryotic gene activation [131, 148-150]. This recent concept has been dubbed the active “chromatin hub” or “transcription factory”. It is supposed that this kind of organization of eukaryotic transcription is especially characteristic of ubiquitously expressed “housekeeping” genes.
Epigenetic regulation of spatial arrangement of genetic loci. The abovementioned gene convergence based on looping out of extended NS, containing regulated genes, including those from chromosome areas, can be controlled by epigenetic mechanisms. The epigenetic mechanisms of gene activity regulation are called the inheritable alterations in the patterns of gene expression caused by potentially reversible modifications of chromatin structures and not directly defined by the primary structure of DNA. The abovementioned gene convergence based on looping out of extended NS, containing regulated genes, including those from chromosome areas, can be controlled by epigenetic mechanisms. The events resulting in formation of an epigenotype preserved during the whole life of a eukaryotic organism play the key role in global regulation of its gene transcription (and expression) [151-154]. Among such mechanisms there are, in particular, already mentioned ubiquitous covalent modifications of histones of nucleosome core particles, incorporation into chromatin of rare histone variants and some non-histone proteins, as well as DNA methylation [83, 155-157]. Numerous data of this kind point to the important role of epigenetic mechanisms in establishment of ordered intranuclear structure of individual genetic loci and regions of interphase chromosomes necessary for their correct functioning.
Interphase nucleus as a self-organized system. Analysis of information from the previous section suggests that the ordered spatial arrangement of intranuclear subcompartments, established in interphase nuclei after cell division, is in general evolutionarily conserved and genetically determined. However, the detectable order inside the nucleus is probably not the result of events associated with copying the preexisting template or the effect of organizing framework, in accordance with which intranuclear position of functional elements would be predetermined in advance. On the contrary, intranuclear arrangement of expressed, replicating, or repaired NS of DNA is able to define sites of assembly of supramolecular complexes whose functioning provides for these processes. This may define positions of nucleoli, numerous intranuclear bodies and speckles, and the chromosome territories proper, whose jointly used genetic loci spontaneously find each other.
A typical example of self-assembly of nuclear structure is the emergence of nuclear membrane in mitosis, which begins in anaphase and is finished in early G1 phase of the cell cycle [158]. In this case, the membrane proteins are specifically delivered to certain marker regions of still condensed chromosomes. The position of the latter is defined by arrangement of the cell mitotic apparatus components preserved at the last stage of mitosis. Nuclear membrane locking and the start of nuclear pore complex functioning accompanied by nucleocytoplasmic transport of molecules results in rapid decondensation of the metaphase chromosome chromatin and assembly of the nucleolus. In this case, formation of nuclear lamina is necessary to increase the size of the nucleus and to cause the emergence of the functional structure of the chromatin.
Step-by-step realization of numerous cooperative and specific interactions of macromolecules, probably defined by the order of their appearance in the nucleus, results in establishment of the interphase nucleus spatial structure capable of carrying out genetic functions. As follows from the model shown below, one such function of non-coding NS is protection of the coding NS genes against chemical mutagens, which is realized in intranuclear genome compartments whose structure and spatial arrangement are in general genetically determined.
THE DYNAMIC GENOME
The structure-function state of the genome is highly dynamic. Individual development of organisms and their following existence in changing environmental and internal conditions requires constant and ordered switching the expression status of a gene groups, often associated with alterations in the chromatin spatial structure [159] and sometimes with genomic DNA rearrangements as well. Besides, genomic DNA is in a state of constant change caused by mutations, traces of which are clearly seen during analysis of primary structures of individual eukaryotic chromosomes and total genomes [160]. The capability for global control of mutagenesis rate and genome protection against emergence of harmful mutations is undoubtedly one of the main selectable phenotypic traits of any living organism.
Programmed Alterations of Genome Structure
Gene amplification in eukaryotic somatic cells. A large increase in the copy number (amplification) of some genes takes place in the somatic cell genome during development of some eukaryotic organisms [161]. This genetically determined process is aimed first of all at the increase in the expression level of corresponding genes by increasing the number of templates for transcription. The selective somatic amplification of genetic loci is based on repeated initiation of replication (endoreplication) of appropriate replicons during a single cell cycle (endocycle). Examples of endoreplication are amplification of the drosophila egg membrane (chorion) genes in ovarian follicular cells [162], genes of secreted proteins in the fly Sciara salivary gland cells [163], as well as over 1000-fold amplification of rRNA genes in a ciliate protozoon Tetrahymena [164]. The histone gene amplification with involvement of transposons in response to a decrease in their copy number caused by deletions is an adaptive response of yeasts aimed at restoration of expression level of these genes [165]. Amplification of drug resistance genes resulting in emergence of resistance to insecticides in insects [166] or to chemotherapy in cancer cells [167] is the same adaptive reaction to external insults.
Genome endoreduplication resulting in somatic cell polyploidization or polytenization of their chromosomes, in which sister chromatids do not segregate after replication, can be considered as the extreme case of ontogenetic gene amplification. This event is not rare on the whole; rather, it is extremely widespread in plants and frequent in specialized cells or in those characterized by high metabolic activity [168, 169]. In particular, the cell ploidy in Arabidopsis varies from 4C to 32C, and it can reach ~25,000C in the endosperm cells of Arum maculatum. Amplification of individual genetic loci has also been registered in some cell pathologies such as malignancy, when clonal selection of oncogene expressing cells is observed [170, 171].
In all of the abovementioned examples quantitative changes in the content of individual genetic loci in the genome have been described, which take place during development of eukaryotic organisms on a background of constant primary structure of basic genome. However, there are examples of ontogenetic rearrangements and genome modifications in somatic cells resulting in their sequence alterations.
Changes in eukaryotic genome primary structure during ontogenesis. Chromatin diminution. Chromatin diminution is among the most impressive and global genome rearrangements at the level of its primary structure, regularly happening in ontogeny of eukaryotic organisms. Diminutions are called the high-precision genetically determined elimination of extended heterochromatin regions, containing non-coding DNA, during somatic cell differentiation. As a result, the somatic cell genome is sometimes much reduced in size (~10-20-fold in flagellate protozoa) and undergoes other large rearrangements at the level of primary structure [172]. In this case, the genome of the germ cell line remains unchanged. Chromatin diminutions are rare in plants. They are characteristic of protozoa flagellates, nematodes, copepods, some insects, Japanese hagfish, and even of marsupials [173-176]. The significance of such genome transformations for the organisms is still not understood. It is supposed that the change in DNA content can be due to the ontogenetic necessity of changes in the cell size, body size, and/or cell generation time [175]. In accordance with the concept developed by us, a high content of non-coding NS in the germ line cells can be associated with necessity for more pronounced gene protection in these cells against mutagenesis.
Changes in immunoglobulin genes. The variety of antibodies and antigen receptors required for the adaptive immune response is the result of highly specific genetically determined changes in primary structure of lymphocyte genomic DNA [177, 178].
Changes in antigen-encoding genes. Constant change in antigenic determinants localized on the body surface is one of strategies that help parasitic eukaryotes to escape the neutralizing effect of the host immune system [179]. This result is achieved by mutations in corresponding genes during DNA replication or repair, recombination between distinct genes, or by shuffling of gene segments localized in different genome regions.
Low fidelity DNA synthesis in mutated template regions. No less than 19 DNA polymerases are involved in DNA synthesis in eukaryotes [180]. Approximately half of these enzymes carry out high fidelity DNA synthesis, whereas the rest are originally programmed for poor accuracy in nucleotide incorporation with base substitution errors [181-183]. Enzymes of the latter group are able to copy damaged DNA and have a tendency to form base mispairs rather than correct Watson-Crick base pairs. In this case, the mismatched nucleotide incorporation into the growing DNA strand proceeds at the frequency of ~10-1-10-3. Besides, they are characterized by lowered requirements for complementarity between 3′ terminal primer and template nucleotides, which makes possible initiation of DNA synthesis from such aberrant primer ends. The ability of these low fidelity DNA polymerases to carry out translesion DNA synthesis by filling the gaps in DNA by non-complementary nucleotides, distorting the original information, allows a replication fork that stopped during DNA replication to resume genome replication and escape lethal effect of lesions. Evidently, in this case the place of insertion of additional mutations into genomic DNA by DNA polymerases will be defined by the localization of primary damaged bases (like thymine glycol or 8-oxoguanine), which also often emerge in response to endogenous mutagens (see below). Therefore, factors restricting the appearance of primary DNA lesions in genes (see model proposed by us) in this case also should play a rather significant role in survival of eukaryotic organisms.
Endogenous Mutagenesis
Mutations are assumed to be “spontaneous” or “induced” depending on the factors responsible for their emergence. Spontaneous mutations emerge during the whole life of an organism under normal conditions [184]. At the same time, induced mutations are the result of different mutagenic effects of environmental factors or experimental conditions. Endogenous mutagenesis caused by internal factors is the basis for spontaneous mutations [185]. DNA damage or incorporation of a mismatched nucleotide results in appearance of premutation that is transformed to a mutation after replication if the repair system does not cope with the problem [184]. In this case, the number of mutations emerging in the genome is proportional to the number of preexisting premutations, i.e. eventually to the number of nascent lesions in DNA.
Factors responsible for emergence of spontaneous mutations. Three main reasons for emergence of spontaneous mutations have been revealed. First, in vivo DNA molecules are characterized by chemical instability. At neutral pH and normal temperature, hydrolytic depurination of DNA strands occurs with formation of apurinic (AP)-sites and following degradation (break) of phosphodiester bonds as well as deamination of C and 5-mC residues with formation of U and T, respectively [186]. Errors in DNA synthesis should be mentioned as a second factor. Misinsertion of nucleotides, non-complementary to the template DNA occurs at a frequency of 10-7-10-8 during chromosomal DNA replication [187]. Besides, slippage of the 3′ end of a growing DNA strand along the template (misalignment) is sometimes observed during replication, which can result in the frameshift mutations [188]. Finally, the ability of DNA polymerase for template exchange during DNA synthesis without termination of DNA synthesis (template switching) opens the way to simultaneous replacement of a large number of nucleotides [184]. Third, numerous endogenous mutagens are formed during normal vital activity of eukaryotic cells, which, after interaction with DNA, cause DNA lesions, which in some cases can alter coding potential of DNA [185, 189, 190]. Even more frequent is emergence of the endogenous mutagen adducts with free nucleotides of the endogenous pool, which can incorporate into DNA and eventually alter its primary structure [191].
Under conditions of normal vital activity, endogenous chemical mutagens make the main contribution to spontaneous mutagenesis [185]. In many biochemical reactions, reactive metabolites are formed as intermediates and byproducts able to interact with DNA. In particular, in substrate redox reactions with involvement of oxygen, free radicals are always formed, which can damage genomic DNA [192, 193]. In aerobic organisms 4-5% of molecular O2 is transformed during respiration to reactive oxygen species (ROS) exhibiting mutagenic activity: superoxide anion (O2̅•), hydrogen peroxide (H2O2), hydroxyl radicals (HO•), and singlet oxygen (1O2). ROS also often emerge during normal metabolism, including that involving cytochrome P450.
The effect of ROS on DNA is one of the factors responsible for emergence of multiple modified bases, the elimination of which by DNA glycosylases results in formation of AP sites and following single- and double-stranded breaks in DNA. The total daily number of AP sites formed in genomic DNA of a single human cell is estimated as ~10,000 [186], but according to different data it can reach 50,000-200,000 in cells of various human and rodent tissues [194]. Usually in the absence of repair, AP sites lead to nucleotide substitutions (AP site→T) and can be a source of frameshift and other mutations [185]. As mentioned above, formation of AP sites can be the result of spontaneous DNA depurination, but modification of DNA bases by ROS and different free radicals is considered as one of the main reasons for this event [195].
Among modified bases, the most frequent is 8-hydroxyguanine (8-OH-G) (0.07-145 adducts per 1000 kb of DNA) which pairs with A and causes G→T transversions [185]. In addition, it, like many other modified nucleotides, can incorporate into DNA from intracellular pool of modified dNTP by pairing with C or A and causing not only G→T but A→C transversions as well. Another example of similar numerous reactions is oxidation of pyrimidine bases in DNA accompanied by formation of mutagenic 5-OH-C, 5-OH-U (0.6-53 and 0.7 adduct per 1000 kb of DNA, respectively) [196-198], and uracil glycol which form pairs mainly with A and cause C→T transitions [198]. The last two adducts are dC oxidative deamination products. Overall, the number of nucleotides damaged per day in the human genome caused by ROS is estimated to be about 20,000 [199].
Propano- and etheno-adducts of bases emerging in DNA in response to acrolein, croton aldehyde, and 4-hydroxynonenal and epoxyaldehydes formed from these compounds and appearing in the organism after lipid peroxidation are highly mutagenic [200]. In particular, etheno-DNA base adducts cause mainly base pair substitutions (etheno-dA and -dC are found in DNA as 0.01-0.7 adducts per 1000 kb of DNA, each) [201, 202]. The dG adducts with malonic dialdehyde emerging during lipid peroxidation and biosynthesis of prostaglandins (pyrimido[1,2-α]purin-10-(3H)-on - M1G) are found at the rate of 0.06-0.9 per 1000 kb of DNA [203, 204]. They cause G→T tranversions (40%) and C→T and A→G transitions (60%). It should be kept in mind that pathological states, including inflammatory reactions that constantly emerge during life, can enhance ROS formation manifold accompanied by oxidative stress [192].
Metabolites of endogenous estrogens directly or indirectly stimulate DNA damages by initiation of single-stranded DNA breaks, formation of 8-OH-dG, as well as of numerous adducts of unknown structure. In the process of cyclic reduction-oxidation of catechol estrogens there emerge reactive metabolites forming adducts with bases and following formation of AP sites and emergence of mutations [205].
Endogenous alkylating agents S-adenosylmethionine, betaine, and choline contribute significantly to endogenous mutagenesis. It is supposed that S-adenosylmethionine is able to generate daily in each animal cell formation of ~4000 molecules of 7-methylguanine, 600 molecules of 3-methyladenine, and 10-30 molecules of O6-methylguanine [206]. The first two adducts destabilize glycoside bond, which makes easier formation of mutagenic AP sites. The last adduct is highly mutagenic and causes formation of GC→AT and TA→CG transitions. Other numerous in vivo-formed alkylated derivatives of the DNA bases are also known [185, 189, 190, 207].
Endogenous reactive nitrogen species (RNS) and N-nitrosocompounds, formed from food sources spontaneously and due to the activity of saprophyte microflora and activated macrophages, are also mutagenic [193].
Numerous endogenous DNA damages, including AP sites formed in response to DNA glycosylase activity, block movement of the replication fork along DNA. In this case DNA translesion synthesis through the damaged site is carried out by the abovementioned specialized DNA polymerases that incorporate non-complementary nucleotides during DNA synthetic processes, with formation of premutations [183].
Mobile genetic elements are another endogenous factor able to contribute significantly to endogenous mutagenesis in eukaryotic organisms [208]. Formation of mutations is the last stage of insertional mutagenesis associated with mobilization of transposons after their insertion in the coding genome regions. Thus, up to 10% of all spontaneous mutations in mice are caused by insertions of retroelements [209].
In addition to the abovementioned endogenous biochemical factors, the ecological situation and life habits, including eating habits, are constant sources of mutagens and their precursors. In particular, electromagnetic ionizing radiation is one of the factors responsible for generation of free radicals in the organism, which induce DNA damage. Under anoxic conditions, ionizing radiation generates from water, in addition to •OH, a hydrated electron (e-hyd) and a H atom, whose activities result in formation of highly mutagenic 5,6-dihydrouracil and α-anomer of 2′-deoxyadenosine (αdA) [192]. In this case, 5,6-dihydrouracil generates C→T transitions, whereas α-anomers of 2′-deoxynucleosides generate single-nucleotide deletions. The long-wavelength (UVA, 320-400 nm) and to a lesser extent the middle-wavelength (UVB, 280-320 nm) UV light are also ROS generators [210, 211]. In this case, the most frequent are G→T transversions as the result of oxidation of the DNA bases.
Although much is still not clear in quantitative aspects of endogenous mutagenesis, the available data can be used to approximately evaluate ranges of at least human genomic DNA damage by endogenous mutagens. It seems that (in the steady state) endogenous mutagens cause the presence in the genome of each eukaryotic cell of 104-105 damaged nucleotides. Owing to this, selection should be aimed at fixation of phenotypic features providing for genome protection against chemical, and first of all, against endogenous mutagens. It should also be emphasized that ROS and RNS in vivo play a two-faced role: on one side, they are damaging factors; on the other, they carry out important physiological functions [212, 213]. Therefore, under normal life conditions their level is under strict control.
Systems of genome protection against endogenous mutagenesis. It is known that biological species overcome harmful consequences of DNA damage caused by mutagens using several protection systems that create physical obstacles for mutagen interactions with DNA and provide for anti-mutagenesis and repair of DNA damage [189].
Physical obstacles for mutagens and their precursors are created by intracellular (including nuclear) membranes and by DNA incorporation into chromatin. Macromolecules and low-molecular-weight chemical compounds are exchanged between the cell nucleus and cytoplasm via nuclear pore complex (NPC) [213a, 213b]. NPC are large protein complexes of ~120 MDa consisting of ~30 protein subunits (nucleoporins) that form channels in the bilayer nuclear membrane, joining the cytoplasm and karyoplasms. The NPC content in nuclear envelope is a species characteristic regulated during the cell cycle. Their number gradually increases when mitosis is over after complete formation of nuclear envelope and is maximal in S-phase. Whereas transport of proteins and ribonucleoprotein complexes through nuclear envelope is an active process requiring energy expense, the transmembrane transfer of low-molecular-weight metabolites occurs by passive diffusion. Most endogenous chemical mutagens, the main subject of our discussion in this section of the review, are not hydrophobic molecules. Therefore, it can be supposed that they get into the nucleus mainly as diffusional flow through NPC. Then the total NPC number in nuclear envelope quantitatively defines the flow of chemical mutagens into interphase nuclei and, as a result, the level of spontaneous mutagenesis regulated by this factor.
Chemical or enzymatic inactivation of mutagens and inhibition of promutagen metabolic activation proceed with involvement of antimutagens (enzymes and low-molecular-weight compounds that are traps for free radicals). Examples of low-molecular-weight traps of free radicals are some vitamins (α-tocopherol, ascorbic and lipoic acids, β-carotene), ubiquinones, free cysteine, as well as glutathione that realize their antioxidant properties using different mechanisms like electron donors or direct interaction with radicals with adduct formation [214]. Besides, specialized peptides and proteins of biological fluids play the role of specific chelating agents or receptors for ions of transition metals or metalloproteins, preventing ROS formation in the Fenton reaction (H2O2 + Me2+→ OH- + •OH + Me3+). Superoxide dismutase, peroxidases (such as glutathione peroxidase and peroxiredoxin), and catalase should be mentioned first of all among antioxidant enzymes. In particular, superoxide dismutase converts superoxide anion to H2O2, which is then metabolized by peroxidases to water or by catalase with formation of water and oxygen. Tumor suppressor p53 plays an important role in regulation of the antioxidant system in vivo [215, 216].
At least three DNA repair systems, namely, base excision repair (BER), nucleotide incision repair (NIR), as well as mismatch repair (MMR) systems make the main contribution to repair of DNA damages caused by free radicals of different nature [189, 217-220].
BER in animals can be initiated by at least 11 DNA glycosylases, specific to damaged nucleotides, which remove the latter from DNA, cleaving N-glycoside bonds with formation of AP sites [221, 222]. Then multifunctional AP-endonuclease 1 (APE1), also one of main members of the NIR system, recognizes AP site and cleaves a phosphodiester bond near its 5′ end. Further repair process can be carried out in two ways with involvement of DNA polymerase β. In the case of short-patch BER, there is a single nucleotide substitution in the AP site, whereas in the case of long-patch BER 2-6 nucleotides are substituted in the site proper and in its environment.
In contrast, during NIR system functioning AP-endonuclease introduces a single-stranded break (nick) at the 5′ end of a damaged nucleotide and the formed 3′ DNA end is then used by DNA polymerase for initiation of DNA synthesis with following elimination of the damaged nucleotide [223, 224].
The MMR system removes mismatched DNA nucleotides (including those from the intracellular pool of mutagen-modified nucleotides) that escaped repair activity of DNA polymerase [191, 225]. MMR proteins recognize mismatched DNA nucleotides and interact with them by repair initiation and play the role of sensors of DNA damage by activation, if necessary, of the cell cycle checkpoints, or in the case of significant DNA damage, of apoptosis of the damaged cell.
Neither of the abovementioned systems of genomic DNA protection is absolutely efficient (e.g. [217]). Therefore, damaged DNA nucleotides in daughter strands can be substituted during replication by others, i.e. premutations of the replicating genome are transformed into mutations.
Poly(ADP)-ribosylation is another important biochemical process protecting the integrity of the eukaryotic genome [226-229]. This reaction is considered as a posttranslational protein modification by linear and branched homopolymer chains of ADP-ribose residues, carried out by poly(ADP-ribose) polymerases (PARP) using NAD+ as substrate (a source of monomer). In response to DNA damage by γ-radiation, alkylating agents, or ROS, the level of ADP-ribose in animal cells increases more than 500-fold, which is one of the first cell responses to the genotoxic attack. Simultaneously, PARP complexes with DNA attract the DNA repair enzymes that repair its damages. In accordance with our concept of protective role of non-coding DNA sequences, we suggest that the primary function of poly(ADP-ribose) in the nuclei might be their direct (immediate) antimutagenic effect as traps of chemical genotoxic compounds.
Molecular Mechanisms of Changes in Genome Size
The phenomenon of genome size variability in animals and plants is based on several molecular mechanisms that result in structural rearrangements of the genome. These mechanisms are not alternative and often supplement each other. Thus, polyploidization can result in transposon activation (mobilization), which in turn, is often accompanied by chromosome rearrangements [230]. Some of the most important mechanisms will be considered below.
Polyploidization. Polyploidization (genome size increase relative to its haploid part) along with segment duplication of chromosomes is now considered as the main and usual event in evolution of the genome of higher plants and some vertebrates [231, 232]. In this case, the genome genetic elements do not increase in size, but their total number increases. Polyploidy in flowering plants emerges at a high frequency of 10-5 [233]; 50-70% of such plants underwent polyploidization during evolution, and this process was repeated several times in the phylogeny of many plants [234-236]. Among vertebrates, polyploidy is frequent in fishes and frogs, but it is absent from members of higher taxonomic groups [237]. Moreover, in 10% of cases polyploidy is responsible for spontaneous abortions in humans [238].
The broad spreading of polyploidy in nature suggests that polyploids probably have selective advantages over their diploid ancestors [237]. In particular, they can include continuous heterosis as a result of emergence of new gene combinations, as well as high variability due to initial destabilization of polyploid genomes, which provides for adaptability of a polyploid organism.
Segmental duplications, insertions, and deletions. Along with polyploidization, local changes in NS copy numbers in separate regions (segments) the genome, called segmental duplications or low-copy repeats (no less than two copies, length from 1 to <200 kb, homology 90-100%), are also usual events in genome evolution [239, 240]. In particular, segmental duplications occupy ~5% of the human genome, as well as ~2% and ~3% of mouse and rat genomes, respectively [241, 242]. Unlike other eukaryotic organisms, in plants duplicated loci occupy a significant part of their genomes.
It is assumed that tandem (following one after another) duplications, causing genome size change, are the result of non-allele homologous recombination (unequal crossing-over) and replication errors [243]. According to this, in plants with larger genomes recombinations in meiosis are more frequent [244]. Since genome expansion is a rare event compared to usual homologous recombination, it can be supposed that the higher general frequency in plants with larger genomes may more often result in mismatch recombinations in the form of unequal crossing-over necessary for genome enlargement using this mechanism.
Neither of the abovementioned mechanisms explains the fact that many segmental duplications are non-randomly distributed in animals (in particular, this is observed in primates [245]). However, on the whole both mechanisms not based on NS homology and mechanisms of homologous recombination are involved in NS block expansion in the genome.
Transposon activity. One of the most important factors of genome size expansion is amplification of LTR-containing transposons, whose level and activity spectrum greatly differ in different species. In some cases the number of transposons in the genome can increase by 20-100 copies (0.1-1.0 Mb) in a single generation [246]. In this case, different transposon families can make the main contribution to the genome expansion in different species [247]. In some cases like in the wild rice Oryza australiensis, activity of several families of LTR-containing transposons provided for genome size increase up to the present-day level in a short period of time. In maize, the same but even more impressive (doubling genome size) result appeared after continuous activity of a large number of various transposon families during last several million years [246, 248].
A remarkable consequence of L1 transposon activity is integration (retrotransposition) of NS from the cellular pool of mature mRNA (via cDNA copies) into new genetic loci of the genome [42]. It is believed that just due to the LINE-1-dependent reverse transcription of mRNA and following insertion of formed cDNA into the genome, over 4000 intron-less copies of cellular pseudogenes emerged in mammalian genomes. In this case, about one third of such genes are transcribed [249].
Rates of genomic DNA elimination resulting in genome size reduction also differ greatly in different plant species. Thus, significant interspecies differences in the efficiency of illegitimate recombination and unequal crossing-over mechanisms are described [250, 251]. On the whole, it seems that such processes are able to provide for observed interspecies differences in genome size and structure in the time intervals necessary for phylogeny of the analyzed plant species.
Acquiring of new genes. Gene duplications, though not as vast as the abovementioned ones, are considered as the basic key mechanism of both the emergence of genes with new functions and expansion of the genome coding part [252]. Such doubling of short NS copy number based on meiotic unequal crossing-over can concern the gene region (domain), the whole gene, or a chromosome segment with the gene located in it. After duplication, mutations resulting in rapid divergence of duplicated NS begin to accumulate in one of the gene copies [4]. In this case most mutations are harmful and inactivate the new gene copy, which finally results in the emergence of a pseudogene. However, in rare cases the mutant gene can acquire a new function.
It is supposed that duplication of genes and whole genomes is the basis of enhancement of phenotypic complexity of evolving species [253]. In particular, two duplication rounds of the whole genome of the last common ancestor of a vertebrate resulted in increase in the total number of genes from ~15,000 to ~60,000, which is characteristic of modern animals. This made it possible to formulate the evolutionary rule “one to four”, in accordance with which modern vertebrates in a large number of cases contain four copies of each particular gene [254, 255]. In fishes (one of most numerous and thus evolutionarily successful vertebrate groups), this rule was later changed to “one to eight” [256].
“Return ticket”. Evolutionary transformations of biological species are accompanied by bidirectional changes in genome size, both expansion and compaction [257]. If significant expansion of genome size is easy to explain, in particular, by NS duplications and transposon activity, its reduction is assured by less understandable mechanisms [258]. Analysis of primary structure of the small contemporary genome in Arabidopsis shows that reduction in its dimensions after polyploidization was accompanied by nonrandom elimination of duplicated genes and by highly specific selective expansion of the proteome (the totality of proteins) of this plant [259, 260]. The association of the probability of duplicated gene retention or elimination from a polyploid genome with its function was noted.
On the whole, both expansion and reduction of genome size with involvement of the abovementioned molecular mechanisms are well-proven facts of genetic development of biological species and the subject for intensive investigations. This group of events illustrates one of the global manifestations of mutagenesis in eukaryotes resulting in emergence of large genome rearrangements like genomic, chromosomal, and gene mutations. Much more frequent point mutations, small deletions, and insertions caused by endogenous mutagenesis are not less important in evolution of the eukaryotic genome. Final results of the global mutation process activity are reflected in the genome dimensions of contemporary biological species.
EUKARYOTIC GENOME SIZES
Genome size is a reliable taxonomic characteristic of a biological species [261]. In 1948, R. and C. Vendrely formulated a hypothesis concerning the constancy of DNA content in which they emphasized the invariability of its content in all cells of an organism of the same species [262] (cited by [261]). To check this hypothesis, DNA content was soon measured in cells of different tissues of several animals (frog, mouse, and cricket) and plants (spiderwort and maize) and the C-value concept was developed, in accordance with which 1C corresponded to the size of the haploid genome [263, 264]. Gregory in his review [261] emphasizes that in the case of diploid genomes of most animals and, possibly, of the minority of plants the terms “genome size” and “C-value” coincide [261]. The situation with recently emerged polyploid plants is more complicated because in this case 1C includes more than a single genome. These terms have been analyzed in detail in a recent review completely devoted to this problem [1].
The apparent intraspecies invariability of genome size and enormous interspecies differences by this parameter, not correlating with the species phenotypic complexity, resulted in the presently permitted “C-value paradox” formulation (see introductory part of this review). However, the biological sense of this phenomenon and evolutionary forces that control differences in genome size of living organisms are still not understood, which makes this event an intriguing enigma of modern genetics and biology as a whole.
Interspecies Differences in Genome Size of Animals and Plants
The problem of interspecies differences in genome size is now intensely investigated. There is the free access to three independent databases on genome size, which include information about over 10,000 species of plants (www.kew.org/genomesize/homepage.html), animals (www.genomesize.com), and fungi (www.zbi.ee/fungal-genomesize/).
Data on the genome size in living organisms are summarized in Fig. 3. It appeared in general that only a few members of any animal and plant taxon have extremely large genomes [261]. Thus, in plants only certain members of ferns and monocotyledons along with many species of gymnosperms fall into groups with large genomes. Nevertheless, the genome size in other plant taxonomic groups also varies over very broad limits. Even genomes of diploid grasses (although grasses just recently, 50-80 million years ago, separated from a common ancestor, i.e. they are of monophyletic origin) differ in size more than 30-fold [265].
In vertebrates, chondrostean fishes and lungfishes as well of amphibia (especially salamanders) have unusually large genomes. Significantly lower variability in genome size is characteristic of a numerous group of animals that includes mammals, birds, reptiles, and teleost fishes. Large genomes are characteristic of orthopterous insects (crickets) and crustaceans (some shrimp species). Genomes of the largest insect orders (Coleoptera (beetles), Diptera (flies), and Lepidoptera (moths and butterflies)) are compact and characterized by a low size scattering. The genome of mollusks, that are as numerous as crustaceans, is very compact and does not exceed 6 pg in size.Fig. 3. Differences in genome size of members of different taxonomic groups of living organisms [370]. Mean values of genome size in corresponding groups of organisms are shown by dots.
In animals there is ~3300-fold range of interspecies differences in genome size, which is noticeably higher than in terrestrial plants. For the latter, ~1000-fold differences (from 0.11 to 127.4 pg) were registered [11, 261]. However, the size of the smallest algal genome and the largest genome of angiosperms differ more than 8500-fold. In animals, the invertebrate Trichoplax adhaerens (Placozoa), whose organism consists of only four cell types, has the smallest of found genomes (0.04 pg). The marble lungfish Protopterus aethiopicus has the largest known genome (~132 pg) (for comparison, the human genome is ~3.5 pg).
Among chordates, the tunicate Oikopleura dioica has a smallest genome (0.07 pg), while among vertebrates - pufferfishes of the Tetraodontidae family have a genome of 0.4 pg which brings differences in genome size in these taxonomic groups to 1800- and 330-fold, respectively. Differences in genome size among invertebrates approach the latter value (340-fold in flatworms, 240-fold in crustaceans, 190-fold in insects) and far below that in annelids (125-fold), arachnids (70-fold), nematodes (40-fold), mollusks (15-fold), and echinoderms (9-fold).
Leitch et al. [11] analyzed tendencies in the supposed pathways of genome size evolution in terrestrial plants, reconstructed on the basis of analysis of these parameters in over 4500 species, and distinguished the following characteristic features of this event. The size of proposed genome precursors in angiosperms and bryophytes is very small (=1.4 pg), whereas genome precursors in gymnosperms and ferns are of intermediate size (3.5-14.0 pg). In this case, the available data suggest that both enlargement and compaction of genome size can happen during genome evolution, which is valid for most studied terrestrial plant groups [266].
Attempts to understand genome size evolution pathways led Sparrow and Nauman in 1976 to put forward the hypothesis that the present-day genomes of members of all taxons are products of successive doubling of the size of minimal genome precursors [267]. The so-called rule of present-day genome discrete structures was formulated. It is not general, but was confirmed for some groups of organisms, in particular, for several plant genera [268-271] including some algae [272]. The essence of this rule is that in organisms of the same genus not the minimal genome size changes discretely by its successive duplications, but the C value characteristic of the genome with the smallest dimensions. In particular, in 20 studied plant species of genus Tephrosia the genome size changes from 1.3 to 7.4 pg with the pitch of 0.74 pg, which approximately corresponds to half of the minimal genome size in this group [273].
Similar examples of discrete change in genome size are also characteristic of some invertebrate groups [274-277]. The most pronounced are some genome size values found in copepods of the Calanus and Pseudocalanus genera in which size changes with a pitch of ~2 pg from 2.25 to 12.5 pg [276, 278, 279]. As already mentioned, the discussed rule of discreteness is not always observed; this can illustrate one of the particular pathways of eukaryotic genome evolution with still unknown mechanism.
As repeatedly mentioned, large genome size is mainly due to high presence of non-coding NS (table). Coding NS (genes) also contribute to the C-value enigma, though to a lesser extent. The same table shows examples of gene content in some eukaryotic genomes that are indicative of the absence of clear correlation between the content of coding NS in the genome and biological complexity of the corresponding species [252].
Intraspecies Differences in Genome Size
The abovementioned pronounced interspecies distinctions in genome size were for a long time opposed to the invariable genome size of a particular biological species. Actually, high species stability of genome size is probably a general rule. For example, investigations of genome size in onion Allium cepa populations on four continents revealed its remarkable stability [206]. In some cases the appearance of information about intraspecies differences in genome size was finally explained by artifacts due to techniques used for intracellular DNA determination and to the existence of cryptic subspecies in the organisms under study [280-282].
It gradually became clear that the existence of significant intraspecies differences in genome structure as well as of differences between very closely related species were indeed facts. Intraspecies differences in genomic DNA content are especially characteristic of the plant genome that is often considered as unstable, always being in conditions of constant change [283]. In particular, comparison of primary structure of three genome regions of total length of 2.3 Mb in two inbred maize lines revealed the absence of similarity (colinearity) in over 50% of sequenced NS [284]. Intragenome content of mobile genetic elements of some families can significantly vary in biological species and even in their local populations. Thus, copy number of retrotransposon BARE-1 in the genome of certain members of the wild barley Hordeum spontaneum growing in different ecological conditions in Israel differs at least three-fold ((8.3-22.1)⋅103 per haploid genome) [285]. In this case, higher transposon content was noted in plants growing in high and dry areas, i.e. under conditions of intensified stress.
Distinctions in the genomic DNA content in members of different maize lines and varieties are also well documented [286]. In particular, quantitative structural distinctions in maize genome regions are revealed at the cytogenetic level [287]. A part of such distinctions is due to different representation of additional B chromosomes in the genome. Intensive study of genome size in 47 populations of the sand fly Aedes albopictus revealed its 2.5-fold differences [288]. Significant intraspecies differences have been recently detected in populations of eight Drosophila species [289]. Such data introduce certain difficulties into the use of genome size as a taxonomic feature [290]. There are numerous examples of genome size intraspecies variability, and critical analysis of such kind information can be found in recent reviews by T. R. Gregory [261, 291].
Phenotypic Traits Associated with Genome Size
Despite huge distinctions in genome size, the variety of phenotypes associated with genome size and revealed at morphological, physiological, or molecular level is not very striking and in many cases uncertain. Only a few phenotypic features are clearly associated with the genome size. Some more pronounced examples are considered below.
Dimensions of cells and nuclei. Strong positive correlation between the genome and cell dimensions in vertebrates was found more than fifty years ago [12]; its existence has been confirmed for plants and unicellular eukaryotes [9, 275]. The event of cell and whole organism enlargement in plant polyploids along with the increase in genome polyploidy is well known to breeders.
Cell volume influences many physiological parameters of an organism, its suitability for the habitat, and as a result, it is a phenotypic feature susceptible to selection. The cell surface/volume ratio exhibits a significant effect on the cell substance and energy exchange with the environment and their metabolism regulated at genetic level via gene expression [292]. In this aspect, the cell volume is a thoroughly controlled trait, especially in multicellular organisms. Quantitatively the connection between genome size (C value) and cell volume is described by the following formula: V = kCα, where k is constant and α defines slope of the curve describing ratios between V and C in a logarithmic scale and is dependent on the groups of organisms under consideration.
The volume of animal and plant cell nuclei also correlates well with genome size and as a result with volume of cells containing these nuclei. Cavalier-Smith describes the ratio between the nuclear volume and DNA content in it by the following formula: V = aHpsC, where a is a universal constant depending only on measurement units, H is the ratio of total chromatin volume (DNA + proteins) to that of pure DNA, s is the coefficient of chromatin swelling (the ratio of chromatin volume in the cell cycle interphase to that in telophase), p is genome ploidy, and C is its size [9]. The reasons for such correlation are understood as a whole. In particular, efficient cell functioning requires a certain flow of mRNA exported into the cytoplasm through nuclear pore complexes, the number of which may depend on the size of the nuclear surface. Large cells require a larger RNA flow as well as an apparatus able to carry out RNA synthesis and processing, and as a result a larger size of nuclei. On the other side, assuring the functionality of large nuclei requires higher activity of cytoplasmic components and quantitative and qualitative changes in their composition. Owing to this, ratios of the nuclear and cytoplasmic volumes in eukaryotic cells (karyoplasmic ratios) are evolutionarily optimized [9, 293].
Cell cycle duration. In addition to the above-discussed ratios between genome size and cell volumes in various eukaryotic organisms, a clear positive correlation between genome size and cell cycle duration was revealed [10]. Genome size increase does not only result in elongation of the DNA replication time in S phase, but it is accompanied by increased duration of G1 phase. Although ratio between genome size and meiosis duration is quantitatively observed within separate groups of organisms, these ratios are more complicated. As a rule, longer meiosis is characteristic of animals compared to plants with equivalent genome size, while in mammals meiotic cell division takes more time than in amphibia and insects. In this case, the genome of mammals is significantly smaller than that in amphibia, but not in all insects. It is interesting that polyploid plants are often characterized by shorter duration of meiosis than corresponding diploid species.
Growth rate and minimal generation time. Several investigations revealed negative dependence in plants between genome size and relative growth rate as well as positive correlation between genome size and generation time (days before beginning of flowering or fruiting) [294-296]. These facts are in agreement with plant abundance. In fact, the species capable of rapid growth and reproduction during shorter time intervals have more chances for wider spreading in a given ecological niche [297].
Hybrid maize lines with higher genomic DNA content compared to the parental lines are usually characterized by the absence of heterosis [298]. Comparison of different maize lines based on their genome size shows that the content of genomic DNA negatively correlates with the growth and productivity of the line [286]. In accordance with the data mentioned in the preceding paragraph, maize selection for early flowering and, as a result, for more rapid development, is accompanied by reduction in genome size [299]. Similar results were obtained for fodder beans Vicia faba [300]. Increase in the genome size in these plants is accompanied by reduction in the plant size in adults and in green mass during flowering.
Quantitative features at the level of organs and tissues. Genome size also sometimes correlates with the development of other complex traits. Thus, seeds are usually larger in plants with a larger genome size, which is especially characteristic of plants with the largest genomes [295, 301]. This is noted, in particular, for V. faba [300] as well as in the systemic investigation of 1220 plant species [301]. The authors of the last work associate the increase in the seed size with enlargement of cells due to the presence of larger genomes.
The rate of cell division and their size can significantly influence the morphology of plant leaves. One quantitative feature used for characterization of plants is specific leaf area (SLA), which is defined as the leaf area to mass ratio. Comparison of 67 plant species using this feature revealed negative correlation between genome size and SLA, i.e. plants with low SLA values, characteristic of small thick leaves, usually have larger genomes. The association between SLA values and genome size has also been studied in detail elsewhere [18, 295, 301-306].
Morphological complexity and duration of embryonic development in amphibia. The brain morphological organization in vertebrates significantly differs, including nerve tissue types and complexity of relationships between particular cells. It was noted that morphological complexity of separate brain regions in salamanders, whose brain is organized more simply than in frogs, correlates with the size of constituting cells [307]. In this case, frogs with small cells and smaller genome size have a brain arranged in a more complicated manner [307].
The study of complex traits caused by the species genome size in 15 species of lungless salamanders of the Plethodontidae family was continued by comparing this feature with the length of embryonic development [51, 308]. A positive correlation between genome size in these animals and the trait under study was found.
Evolution and ecology. A weak negative relationship between the number of species within a genus and genome size was found [295, 309]. This may be indicative of the fact that plants with large genomes are less variable and have a lower potential for speciation and a higher risk of extinction during evolution. In accordance with this, there are indications that plant species with small genomes exist at all latitudes and in alpine areas, whereas plants with large genomes escape extreme life conditions [295, 310].
It follows from everything written in this section of the review that a great number of quantitative traits correlate with the eukaryotic genome size. At present the mechanisms that are the basis for formation of complex traits are not quite understood. Such quantitative traits are defined by coordinated functioning of a large number of genes located in genome regions known as quantitative trait loci (QTL) [181]. Genetic architecture of complex polygenic traits is defined by the gene number within QTL, their individual contribution to the trait formation, gene positions on the chromosome, as well as by their interaction with each other and with environmental factors [311]. Accounting for all this and also for the fact that functions of most NS of large eukaryotic genomes are unknown, a simplification in interpretation of the revealed direct correlation between genome size and corresponding phenotypic traits can be expected. Such “correlations” can be far from genetic relations.
ON THE WAY TO SOLUTION OF THE “C-VALUE
ENIGMA”
Factors that define size distinctions between eukaryotic genomes are still not clear. Intensive investigations of “C-value enigma” are accompanied by attempts to explain it. Most of presently discussed hypotheses and theories can be divided to two large groups: i) theories considering non-genic DNA as redundant, carrying no definite functions, and ii) theories attributing adaptive functions to non-genic DNA.
Theories of Non-functional Redundant DNA
Theories of non-functional redundant DNA imply that the non-genic DNA fraction in eukaryotic genome changes randomly in response to mutations. Its content in the genome can increase until the excess of non-functional NS begins to exert harmful effect on the cell and organism as a whole.
Junk DNA. Ohno was among the first scientists who put forward a hypothesis concerning the origin of non-coding NS in eukaryotic genome and designated them as debris (fragments) of formerly functioning genes: “Our genome probably carries in itself signs of victories and defeats of former experiments in nature” [312] (cited by [10]). According to this concept, non-coding NS are considered as real debris (junk DNA) that trashes the genome. Although mutation-inactivated genes (pseudogenes) really appear in the genome, they comprise only a small part of the total non-coding DNA and can carry out certain functions (see above). Later the term “junk DNA” was used to designate any non-coding DNA devoid of definite functions, and this term is still used in different contexts (e.g. [283, 308]). Theories of “junk DNA” show that in the absence of constant pressure of selection, aimed at elimination of redundant NS, the eukaryotic genome has the tendency to random increase in size. And this proceeds until cells become unable to bear the load of redundant DNA.
Further development of the group of theories of non-functional redundant DNA was the emergence of the “selfish DNA” concept.
Selfish DNA. Pure theories of useless (junk) DNA suggest the effect of natural selection only at a single stage of eukaryotic genome evolution as a factor limiting random increase in its size above optimal. It is emphasized in the “selfish DNA” theories that selection by phenotype at the level of the whole organism is not a unique evolution strength forming the eukaryotic genome. Moreover, such selection only indirectly influences the structure of genomic DNA. Cells create a habitat for DNA [313]. If mutations increase the probability of preservation of a particular NS in a cell without influencing the phenotype of the whole organism, such NS will inevitably increase their representation in the genome, demonstrating their unique “function”, self-preservation, as well as their parasitic (“selfish”) nature. Independent replicating molecules (replicators), exhibiting the highest replication efficiency, will dominate over their less efficient competitors and increase their representation in the genome [34]. In this case, it is suggested to differentiate between “ignorant” amplification of genomic NS, independent of its primary structure, and “selfish” amplification, dependent on the NS primary structure.
An evolutionary factor limiting the total genome size is the energy load caused by the necessity for the reproduction of excess DNA. The “selfish” DNA concept suggests that the cleaning selection activity relative excess DNA is weakened for not understood reasons and cannot resist continuous increase in genome size caused by transposons and other genetic mechanisms. The upper limit of genome size is defined by the ability to sustain such additional load for metabolism, which in turn depends on the life style of a biological species and conditions of its interaction with the environment. The increase in the content of useless DNA NS in the genome is compared with propagation of a parasite that does not exhibit a harmful effect on the host organism. From time to time, the redundant parasitic DNA may acquire genetic functions, originally not characteristic of it, including nucleoskeleton functions (see below the nucleoskeleton theory by Cavalier-Smith). Owing to this, the border between terms “selfish” and “parasitic” DNA and “DNA-endosymbiont”, the latter carrying out useful functions, is not clearly outlined.
Variants of the theory of redundant DNA of eukaryotic genome differ mainly in the supposed mechanisms of the origin of the redundancy. In early discussions on the “selfish DNA” concepts, attempts were made to change the term, without significant change in the hypothesis sense, and to call “non-functional” NS “incidental DNA” [314]. In this approach, “redundant” NS are considered as a byproduct of the genome inherent property of variability (mutability), the maintenance of which is provided by natural selection. The appearance of “redundant” repetitive NS is considered as the result of the natural selection effect on maintenance of genome variability as such.
The “selfish DNA” theories suggest the existence of free competition between repetitive NS during their self-reproduction for maximal representation in the genome. Attempts have been made to consider a eukaryotic genome as a peculiar ecosystem in which various transposon families (analogs of biological species) exist and compete for survival [315, 316]. In this aspect, the established ratios between NS of eukaryotic genome are considered as NS symbiosis.
It seems that the inapplicability of such approaches to description of the eukaryotic genome is due to the fact that activity of transposons and expansion of other NS is under strict cell control. This situation excludes the free competition of NS with each other, like that in natural ecosystems or artificial associations of biological species, in which competition is permitted by humans. The situation formed in the eukaryotic nucleus can be to a higher extent compared with a zoological or botanical garden where beasts and their victims are placed in separate cages, while weeds and cultivated plants exist separately due to efforts of agricultural workers. One has only to relax the control for a while, and chaos will come in such artificial ecosystems, which will soon transform this elegant artificial system to natural one based on quite different principles.
One criterion of DNA redundancy is the possibility of its elimination from the genome without visible phenotypic consequences for the organism [317]. In accordance with this, it has been recently shown that the mouse organism “does not notice” the removal of extended non-coding NS [318]. In these experiments the gene knockout technique was used to remove two NS of 1817 kb (chromosome 3) and 983 kb (chromosome 19) which do not contain genes revealed by usual techniques and contain orthologous NS of approximately the same size in the human genome. Comparison of these regions in the mouse and human genomes revealed altogether 1243 conserved NS over 100 bp in length with 70% homology. These data allowed the authors to conclude that the mammalian genome contains really “redundant” DNA. It seems that such a straightforward approach to estimation of NS functional significance is a simplification. The model developed by us suggests a different interpretation of these interesting results.
Theories of Adaptive Non-genic DNA
Unlike the non-functional DNA theories that emphasize unlimited to a certain extent random character of alterations in its content in the genome due to mutations, the theories of non-gene adaptive DNA suggest pressure of natural selection aimed at maintenance of established ratios between coding and non-coding genomic NS. This group of theories implies the existence of a genetic relation between the genome size and functioning of the other systems of a eukaryotic organism. Accordingly, random alterations in non-coding DNA content are fixed in the genome during evolution if they stimulate survival of biological species.
DNA in the role of nucleoskeleton and nucleotype concept. Commoner in his review of 1964 drew a conclusion concerning a dual role of eukaryotic DNA in heredity [319]. According to his concept, on one side the euchromatin DNA provides for exhibition of phenotypic features at the appropriate qualitative level depending on genetic code. On the other side, in accordance with the nucleotide sequestration hypothesis, DNA of heterochromatin regions of the genome, independently of its nucleotide sequence, exerts qualitative effects on cells, namely, on their size, metabolism rate, and generation time. Accordingly, total increase in number of intracellular DNA nucleotides requires the enlargement of the apparatus necessary for cell reproduction, which influences different intracellular processes.
Bennett shows that DNA is able to carry out a skeletal function in the interphase nucleus arrangement, defines its shape and size, and formulates the nucleotype concept [294]. Unlike phenotype, nucleotype is the aggregate of organismal traits that are defined by the genome size as such but not by its primary structure peculiarities, i.e. the incorporated genetic information in the traditional sense of this term. Based on these early achievements and probably inspired by them, Cavalier-Smith, beginning from 1978, has developed the theory of the DNA nucleoskeletal function in eukaryotic organisms, the aim of which is explanation of the “C-value paradox” [9, 320].
The volume of eukaryotic somatic cells defined by the amount of DNA included in their nuclei is regarded as of paramount importance in the present-day variant of the nucleoskeleton theory. In other words, the cell volume is genetically determined by the genome size and is the nucleotypic feature of biological species. The cell volume, like genome size, differs in eukaryotes by ~300,000 times, is adapted to the species life conditions, and is under pressure of selection. The change in cell volume under pressure of selection results in the change in nuclear volume and genome size, which defines its volume. This chain of genetic relations between cell volume and genome size defines the point of natural selection application to the adaptive phenotypic feature--the genome size of any eukaryotic organism.
Other concepts of adaptive non-genic DNA. It is claimed in the hypothesis of the passive buffer role of non-coding DNA that the presence in eukaryotic nuclei of a great amount of DNA contributes to maintenance of intranuclear homeostasis [321]. This allows nuclear enzyme systems to survive easier the fluctuations in ion composition of intercellular liquid and facilitates the existence of the organism under extreme conditions. Besides, according to this hypothesis, the presence of such DNA provides for control over intranuclear content and activity of nuclear proteins interacting with DNA.
Non-gene DNA as a gene expression regulator. There are attempts in many present-day investigations to reveal in non-coding DNA specific genetic functions associated with regulation of gene expression. In the recent review by Shapiro, data on known genetic functions of repetitive NS are summarized and there are numerous examples of the effects of different class repeats on gene activity [322]. Some aspects of the participation of heterochromatin in global regulation of eukaryotic gene expression were discussed above.
Protective role of heterochromatin. Yunis and Yasmineh [323] in 1971 were the first who paid attention to the possible role of heterochromatin in gene protection against chemical mutagens, and their work preceded our model. In accordance with their hypothesis, heterochromatin regions consisting mainly of satellite NS could protect functionally significant intranuclear organelles (kinetochores and regions of nucleolar organizer) against harmful external effects because heterochromatin aggregates were at that time detected mainly in surroundings of these chromosome regions.
Four years later Hsu approved this concept in principle and began to develop the heterochromatin-bodyguard hypothesis [324]. In accordance with this concept, heterochromatin forms a thin layer of a dense substance at the nuclear periphery immediately under the nuclear envelope and in this way it plays the role of a euchromatin gene protector against mutagens, clustogens (substances stimulating chromosome breaks), and even viruses. The variability (“plasticity”) of the composition of repetitive DNA sequences in heterochromatin and morphological heterogeneity of heterochromatin regions in karyotype reveal traces of DNA contacts with mutagens (“bodyguard scars”) and are indicative of their ability for mutational changes that do not exert harmful effect on the organism. Breaks in chromosomes caused by damaging agents, in particular by mitomycin C, are non-randomly distributed and are more frequent in heterochromatin regions.
Investigations of structure-function relationships in eukaryotic genomes just began at the beginning of the 1970s, when the abovementioned hypotheses appeared. There was no information concerning ratios of coding and non-coding NS in different genomes and their spatial arrangement in interphase nuclei. It was not clear just against which mutagens organisms had to be protected first of all, i.e. in what mutagenic conditions eukaryotic genome evolved and continues its evolution. Our model of “altruistic non-coding DNA” answers these and some other questions.
ALTRUISTIC DNA
A quantitative model was developed during our investigations according to which the non-coding NS of eukaryotic genome form a new (third) system for protection of coding genome regions against endogenous mutagens. Endogenous mutagens and their precursors, which penetrate into the nucleus through nuclear pore complexes (NPC), interact with numerous intranuclear macromolecules and low-molecular-weight compounds including DNA nucleotides (in both coding and non-coding genome regions) as well as nucleotides of the intranuclear pool and undergo inactivation. Then, the number of damaged coding and non-coding DNA nucleotides is proportional to their content in the nucleus, i.e. non-coding NS play the role of additional traps for mutagens and eliminate the latter from the intranuclear space. It is assumed that the slow alteration of genome size due to increase of non-coding NS in phylogenesis has little effect on the nuclear membrane permeability for chemical mutagens as a result of maintaining the NPC density in NE constant due to coordinated expression of the appropriate genes. In such a situation, the non-coding DNA of eukaryotic genome behaves quite “altruistically” by putting itself under injuries instead of coding DNA. The main concepts of the proposed model that was already mentioned previously [2, 325-327] are considered in more detail below.
Ratio of Coding and Non-coding Genome Parts May Define the Level of Its Gene Protection
At the present stage of modeling we consider only three processes: (1) the emergence of mutagens within the nucleus, (2) their interaction with DNA resulting in inactivation of mutagen molecules and damaging the reacted nucleotides, and (3) repair of damaged nucleotides.
The model assumes that chemical mutagens can appear in the nucleus only due to penetration through nuclear membrane in the form of (i) reactive chemical compounds (in particular, xenobiotics that underwent metabolic activation) or (ii) low-activity precursors (promutagens) (e.g. endogenous H2O2) with subsequent activation nearby the target nucleotides (contact with transition metal ions - the Fenton reaction). In the last case the site of nucleotide damage in DNA will be defined by a random intranuclear position of the contact between promutagen and activating agent.
It is supposed that chemical mutagens enter the nucleus mainly by diffusion through NPC. It should be taken into account that the changes of coding and non-coding genome region sizes differently influence the flow of mutagens into the nucleus through NPC, since the process of biosynthesis of the NPC protein components is largely associated with the genome coding part (see discussion below).
Inside the nucleus, mutagens can cause two types of primary nucleotide lesions - by forming adducts either directly with DNA nucleosides (bases or deoxyribose residues) of DNA or with both nucleotides of the intranuclear pool and their precursors in the free state [185, 328]. We think that at this step of modeling, the incorporation of modified dNTP into DNA from intranuclear pool happens stochastically as DNA replication proceeds, and the contribution of these two processes to the DNA damage in this model is additive.
The experimentally detected differences of DNA repair rates in different eukaryotic genome regions (including actively transcribed and non-transcribed) [329] are not considered here in the description of repair and anti-mutagenesis system contribution into restoration of DNA from damages and maintenance of its integrity.
It is assumed at this stage of modeling that DNA within chromatin is spread homogenously over the nuclear volume during most of the cell cycle. This supposition is a simplification of the real situation. Due to the limited amount of experimental data describing the genome state in an interphase nucleus as a whole, we do not consider yet the peculiarities of spatial structure of individual intranuclear microcompartments and particular genetic loci, including those distinguished by different level of chromatin condensation.
At the usual mutagen content in the nucleus, the number of events resulting in DNA nucleotide damage by mutagens is high. According to our data (see section devoted to endogenous mutagenesis), the number of nucleotides damaged by endogenous mutagens in the human genome in steady state is on average ~104-105 the total genome size being ~3⋅109 nt. Therefore, the quantities describing the content in the nucleus of mutagens and nucleotides (free and included into DNA) can be considered as continuous variables. For these variables, we introduced a system of differential equations similar to those used in chemical kinetics [326]. Further development of this model gave the following expression for the rate of mutagen flow from cytoplasm into nucleus:
where φ is total mutagen flow into the nucleus, mole/sec; NcT and NncT are total numbers of nucleotides, respectively, in coding and non-coding genome regions (“total” means the sum of damaged and undamaged nucleotides); φ0c and φ0nc are coefficients that describe the effect of coding and non-coding genome regions on the mutagen flow into the real nucleus. This effect is carried out via regulation of the NPC total number on the nuclear surface and/or the NPC permeability for mutagens.
Let β be the fraction of damaged nucleotides in the real genome (ratio of the number of damaged nucleotides to total number of nucleotides in the genome); β0, a similar value for a hypothetical genome containing total coding DNA of real genome and free of its non-coding DNA; NT = NcT + NncT, the total content of all nucleotides in the genome (coding and non-coding, damaged and undamaged). Solution of equations for steady state of the nucleus-mutagen system, which accounts for the processes of mutagen delivery into the nucleus and DNA repair, results in the following dependence (detailed calculations will be published elsewhere):
It follows from Eq. (2) that under the above assumptions the protective effect of non-coding sequences takes place when φ0nc < φ0c, i.e. if the number of NPC and their permeability for mutagens are defined to a higher extent by the amount of coding than non-coding DNA. It should be emphasized that the lower is the dependence of the mutagen flow through the nuclear membrane on the presence of non-coding sequences (i.e. the lower is the coefficient φ0nc), the higher the protective effect of non-coding DNA is. The highest protective effect of non-coding DNA should take place at φ0nc = 0 (equal NPC amount and permeability for mutagens in the presence and absence of non-coding DNA) (see Eq. (1)). In theory, protective effect of non-coding sequences might be even higher if they reduced the total mutagen flow into the nucleus (φ0nc < 0). Finally, protection is absent in the case of equal determinacy of mutagen flows by coding and non-coding sequences (φ0nc = φ0c).
In the case of NcT << NT, Eq. (2) can be approximately written as:
It follows from Eqs. (2) and (3) that as the content of non-coding region in the genome increases, the fraction of mutagen-damaged nucleotides in coding NS decreases and rather greatly at NncT >> NcT. Thus, for the human genome the abovementioned fraction of coding DNA ~3% means that NncT/NcT ~ 0.97/0.03 ~ 32 [326].
Thus, the significant protective effect of non-coding NS found by us points out the existence in eukaryotes of a new, third system of gene protection against chemical mutagens, in addition to the anti-mutagenesis and DNA repair systems.
Possible Reasons for Genome Size Variability in Eukaryotes
In accordance with our model, total frequency of mutations emerging in coding genome regions will depend on coordinated effect of anti-mutagenesis and DNA repair systems as well as on the ratio of coding and non-coding NS in the genome, i.e., ultimately, on the genome size. Taking all the above-said into account, it can be supposed that in close biological species with a low ratio of coding and non-coding NS lengths (larger genome size) a larger amount of chemical mutagens (of endogenous or exogenous origin) is present in the nucleus due to lower activity of anti-mutagenesis system and/or lower activity of DNA repair system. In this case, non-coding NS provide for lowering nucleotide damaging and, as a result, lowering mutation frequency in coding NS to an acceptable level. Investigation of the efficiency of anti-mutagenesis and DNA repair systems depending on the genome size in biological species is one of possible ways for our model to be experimentally verified.
We have found in the literature only indirect data concerning interspecies distinctions in functional activity of anti-mutagenesis and DNA repair systems in organisms the genome sizes of which differ significantly. In particular, the presence of additional homologs for most genes of repair and recombination systems is characteristic of the small genome of Arabidopsis thaliana (~125 Mb), which noticeably distinguishes this plant from other studied eukaryotes, and it is now a specific feature of just this plant [331]. According to this, Arabidopsis can be characterized by an increased rate of DNA repair, which reduces its need for protective non-coding NS. There are indications that in salamanders the activity of photolyase, an enzyme involved in repair of DNA damage caused by UV light, is significantly lower than in toads and frogs having significantly smaller genomes [332].
In accordance with our predictions, the specific activity of O6-methylguanine-DNA-methyl transferase, eliminating in vivo O6-mG from alkylated DNA, in rainbow trout Oncorhynchus mykiss (genome size 2.6 pg) is ~2.4 times lower than in the sword-tailed minnow Xiphophorus maculatus (genome 0.97 pg) (cited by [333]). In this case, regardless of the genome size, there are data showing that low activity of the DNA repair system is specific for fishes on the whole [334]. In diving mammals characterized by rapid transition from hypoxia to reoxygenation and storage in tissues of a great amount of oxygen, the constitutively higher antioxidant status was noted compared with that in terrestrial mammals [335]. Data of this kind suggest that organisms can use different strategies for protection against continuous genotoxic effects.
Although protection of genes against endogenous mutagenesis caused by chemical mutagens is vitally important for biological species, it is not a unique function of non-coding NS and at least a part of them is used for other purposes. In particular, the skeletal function of these NS, mentioned for the first time by Bennett [9], and their associated role in nucleotype formation are doubtless [294, 319, 336]. There are data showing the involvement of repetitive NS in global gene expression at the transcription level via chromatin domain formation, heterochromatization of genetic loci, influencing gene transcription via the transposon regulatory elements, etc. [322]. All this might be the secondary acquiring of new functions after the non-coding NS primary cooptation for genome protection against mutations, and first of all, against endogenous mutagenesis.
Atmospheric Oxygen in Evolution of Biological Species and Their Genomes
According to present-day concepts, life on the Earth originated ~3.5 billion years ago in the absence of oxygen, and originally it was represented by prokaryotes, including methane-producing archaebacteria, using oxidation of hydrogen in the presence of CO2 for energy generation, as well as by other chemotrophs [337]. Purple bacteria, cyanobacteria, and heliobacteria were probably the first representatives of photosynthesizing organisms that used light energy for carbon fixation and water oxidation with formation of O2 [338]. The emergence of these bacteria ~3 billion years ago resulted in gradual increase in O2 content in the Earth's atmosphere from 1% to the present-day level of 21%. Approximately 1.5 billion years later, already in the presence of oxygen, there appeared the first eukaryotes preceded by emergence of aerobic forms of life using O2 for energy generation [339]. Finally, about 600 million years ago the first multicellular organisms appeared.
It can be supposed that the existence of organisms in the presence of oxygen and its active forms required the development of additional factors for protection of genetic information against damaging effects of these compounds. The enlargement of the genome in addition to already existing antioxidant and DNA repair systems could be among the first evolutionary adaptive reactions of organisms to the increase in the free O2 concentration in the atmosphere. Further changes in the genome size followed alterations in functional efficiency of anti-mutagenesis and DNA repair systems in evolving organisms (see below). Finally, the emergence of multicellularity at O2 atmospheric content close to the present-day level could have an appreciable influence on the establishment of larger genomes in eukaryotes, because it required additional decrease of mutation frequency in somatic cells to prevent cell line breakage in ontogeny [2].
Since the emergence of multicellularity, the O2 atmospheric concentration did not remain constant, and these changes correlate with global evolutionary changes in the biosphere [340]. For example, the increase of O2 content about 410 and 300 million years ago was accompanied by development of gigantism in some animal groups. The increase in genome size, correlating with the size of cells, required for creation of additional gene protection against ROS, could be one of basic mechanisms of this phenomenon. In fact, the available experimental data show that exposing of D. melanogaster to high O2 concentrations is accompanied by enlargement of its body, whereas under conditions of hypoxia it diminishes. These phenotypes are retained during several generations after the fly is transferred into the usual atmosphere [341]. In addition to other known mechanisms of cell growth control [341], it can be also due to changes in nucleus and cell volumes related to each other, as mentioned above in the section devoted to phenotypic features associated with genome size. The existence of positive correlation between cell and body size was repeatedly noted in invertebrates [291].
Pathways of Eukaryotic Genome Size Evolution
Based on the above described possible protective role of non-coding NS, the following path of eukaryotic genome evolution can be conceived (Figs. 4 and 5). During the whole life of aerobic organisms, nuclear DNA exists within a continuous flow of endogenous mutagens (Fig. 4). Mutagens, which escaped the neutralizing effect of the anti-mutagenesis systems, damage bases in DNA, while non-coding NS protect genes against such kind of damage and mutation. Most damage is corrected by the DNA repair systems. All this taken together provides for the admissible genetically determined level of spontaneous mutagenesis of the coding NS.
Fig. 4. Supposed control mechanism of non-coding DNA content in eukaryotic genomes. Three levels of eukaryotic genome protection against endogenous chemical mutagens are shown (horizontal bold arrows on the left) which are formed by: 1) anti-mutagenesis system; 2) non-coding NS of genomic DNA; 3) DNA repair system. When all three systems do not provide the necessary gene protection, mutations in hypothetical molecular sensor(s) take place, which is accompanied by transposon mobilization, enlargement of non-coding genome region (without significant alteration of mutagen flow through NPC), and local decrease in spontaneous mutation frequency to the initial optimal level. The coding genome region is shown by the bold dot in the center of the circle, while four fine arrows point to increase in the genome size.
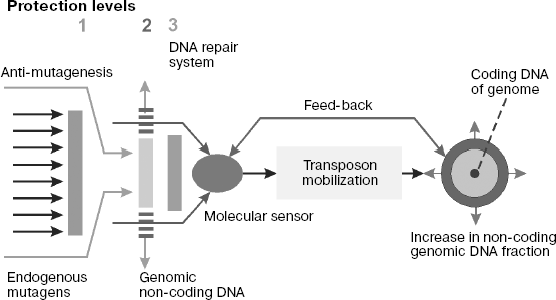
If the intranuclear mutagen concentration increases for any of several reasons, it causes an increase in mutation frequency in coding NS of the genome, among which there are molecular sensor gene(s). Mutational changes in the sensor mobilize retrotransposons that results in local increase of their copy number, genome enlargement (without significant increase in number of NPC and their functional activity, and as a result, without significant change in the endogenous mutagen flow into the nuclei), and, as a result, in lowering the probability of mutations in corresponding coding NS. (See review [343] about the possibility of non-random transposon transfer within a genome, so-called “sectorial mutagenesis”.) As a result, the “genome-endogenous mutagen” system reaches a new steady state in particular genetic loci. A decrease in the background intranuclear mutagen concentration will be accompanied by a slow reduction in genome size due to spontaneous deletions in now redundant (in the aspect of protective function) NS. Actually, investigations on plants and animals have shown that transposons can be efficiently eliminated from eukaryotic genomes [344-347].Fig. 5. Supposed paths of eukaryotic genome size evolution. The initial genome precursor (in the center of the black circle) consists of coding and non-coding NS (light and dark rectangles, respectively). It is characterized by a particular optimal level of gene protection against spontaneous mutagenesis with involvement of endogenous chemical mutagens. This protection level is provided by coordinated action of classical protection systems (anti-mutagenesis and DNA repair) as well as by nucleotides of the non-coding genome region.
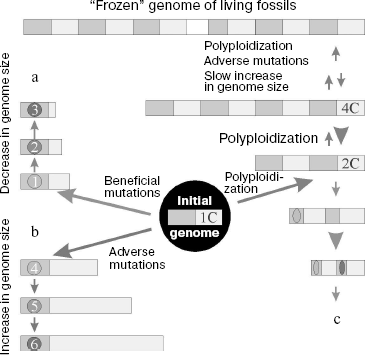
Transposon activity is under strict control, and acts of transposition are rare even in organisms characterized by high transposon activity [26, 31]. Particularly, in humans, who are among such organisms the transpositions are registered at a frequency of ~10-1-10-2 per generation [348]. High transposon activity was registered at certain stages of embryogenesis and in malignant tumor cells. Stress conditions like heat shock, viral infection, or effect of DNA-damaging agents stimulate transposon activation [349, 350]. Mobilization of transposons requires involvement of transposon-encoded proteins and, as a result, transcription of corresponding genes by RNA polymerases II and III [26, 31, 209]. Therefore, mechanisms involved in control of gene transcription by these RNA polymerases can be also used for transposon mobilization control.
At least two molecular mechanisms involved in control of mobile genetic element activity are known: RNA interference with participation of small interfering RNA (siRNA) and methylation of DNA sequences. Since mechanisms of RNA interference are also involved in methylation, these two mechanisms might be interrelated. It has been recently shown that genome instability in human cell cultures induced by radiation is due to change in the character of DNA methylation in NS of retroelements [351]. Taking the above-said into account, C residues the methylation of which is critical for transposon mobilization could be the abovementioned sensor for transposon mobilization. Mutations G→A happening, particularly, in response to ROS could lower the methylation status of corresponding genome regions and activate the adjacent retrotransposons as well as cause local increase in their copy numbers. Changes in spatial structure of corresponding genetic loci, produced by transposon inserted into new genome regions, could be accompanied by alteration of the protection level of these regions against endogenous mutagenesis and would provide for individual protection.
Changes in stationary concentrations of intranuclear mutagens (and increase in frequency of spontaneous mutagenesis) can be caused by different factors. It can be the result of slow alteration of environmental conditions leading to a change in the level of mutagenic effects on organisms living in these conditions. Mutations in enzyme systems of anti-mutagenesis and DNA repair, as well as in metabolic processes generating endogenous mutagens, could result in the same biological consequences (Fig. 5). Besides, mutations in components of the system regulating the number of NPC on NE and their permeability for mutagens can alter mutagen flow inside the nucleus. (For regulation of NPC assembly see reviews [351a, 351b].) For example, the increase in functional efficiency of classical systems of genome protection against mutagens such as anti-mutagenesis and DNA repair should relieve the selection pressure on the genome size that can be now reduced due to spontaneous deletions caused by unequal crossing-over and similar mechanisms (Fig. 5a). On the other side, harmful consequences in the same gene systems, slightly lowering their efficiency, could be compensated by slow genome enlargement to normal size concerning its protective function, including the result of transposon mobilization (Fig. 5b). Transposon activity during periods, comparable with the time of the species existence, might result in significant changes of genome size. In particular, it has been recently shown for the grass Oryza australiensis, in which the retrotransposon activity without polyploidization resulted in doubling of the genome [248].
Genome enlargement due to polyploidization can be a quick solution of the emerging problem of high damaging of coding nucleotides. As mentioned above, polyploid organisms are widespread among plants and are also frequent in fishes and amphibia. According to our model, the saltatory increase in genome size due to polyploidization after getting over the first period of genetic instability could lower damaging of initial coding NS (and as a result, mutation frequency) caused by chemical (including endogenous) mutagens due to reduction of mutagen flow per nuclear volume unit in polyploids comparing to original organisms. Such effect can happen if polyploidization is not accompanied by stepwise increase in NPC number on the polyploid nuclear envelope as a multiple to genome ploidy. In theory the alteration of expression of genes controlling NPC formation in neopolyploids could result in such consequences. The polyploidization effect on expression of such genes can be subjected to experimental investigation.
After successful polyploidization, i.e. overcoming by neopolyploids of an initial period of genetic instability on a background of hypothetical total reduction of the endogenous mutagen flow through NPC per nuclear volume unit, two versions of evolutionary scenario might be realized (Fig. 5c). First, owing to the absence of selective pressure on maintenance of redundant NS in such polyploids, the spontaneous elimination of these NS from the genome due to unequal crossing over and other similar mechanisms will take place. Second, mutations are possible in polyploids that decrease the efficiency of DNA repair and/or anti-mutagenesis systems, or increase the NPC number on nuclear envelope without harmful consequences for the organism, because redundant NS will take upon themselves a part of the protective functions of these systems. In this case, reduction of polyploid genome size will be impossible due to harmful mutagenic consequences for the whole organism.
As mentioned above, in most studied polyploid plants total genome size is diminished and approaches that of the basal genome [352]. Evolutionary reduction of genome size in accordance with this scenario could also take place in diploid mutant organisms having more efficient DNA repair and/or anti-mutagenesis systems as well as the reduced endogenous mutagen flow through NPC compared to the wild-type precursor organism (Fig. 5a). The decrease in spontaneous mutation frequency after polyploidization predicted by our model can explain in a new way species formation inhibition in organisms with large genomes: it is known that the number of biological species in a taxon is inversely proportional to genome size of included species [295]. In this connection, giant genome dimensions characteristic of “living fossils” (mentioned in the introductory part and others) explain evolutionary conservativeness of such species. In accordance with the concept developed by us, inhibition by non-coding NS of spontaneous mutagenesis in coding gene regions could take place in these organisms. Owing to transposon activity, tandem duplications, and polyploidization, the genome size in these species exceeded the admissible threshold level and, according to the picturesque expression of C. Ohno, it became “frozen” and the species proper appeared in evolutional deadlock [253]. In this case, the selective pressure in these particular species can be aimed at the nucleotype, in particular, at their cell size, and this preserves superprotection of genetic loci against endogenous mutants, not required for genome stability, and keeps unchanged the giant genome size of these organisms.
On Differential Gene Protection
Our mathematical model is based on the assumption concerning uneven distribution of DNA nucleotides inside the interphase nucleus, which is a deliberate simplification of the real situation. The highly dynamic state of chromatin in interphase nuclei, fine spatial structure of which changes during the cell cycle, as well as differences in the general chromatin structure in various types of cells and tissues in the organism allow such averaging in our first approximation.
In fact, as shown above in detail, the arrangement of genetic material in interphase nuclei is far from even and is highly ordered. There are vast genetically determined euchromatin and heterochromatin regions in the nucleus, the density of DNA packing in which is different and may change during the cell cycle. Separate chromosomes are arranged as discrete chromosomal territories and the content of non-coding NS is unique for individual chromosomes. Moreover, the ratio between intron and exon lengths in particular genes is a stable characteristic of biological species and in mammals it correlates with the genome size, especially with that of euchromatin region.
Taking into account such facts, main conclusions drawn after analysis of the consequences of endogenous mutagenesis for whole genomes can be also applicable to separate intranuclear microcompartments, including particular genetic loci. In fact, the accessibility of genetic loci to chemical mutagens and DNA repair enzymes also depends on the intranuclear spatial arrangement of the locus, including dependence on the level of chromatin condensation. In addition, the ratio between coding and non-coding NS located in intranuclear compartments should define the genetic effect of mutagen contacts with DNA of particular genetic loci.
Since mutagens enter the nucleus from outside, their concentration should be greatest at the nuclear surface near NPC. In this connection, the Rable's configuration, especially characteristic of plant cells, in which non-coding telomeric and centromeric NS of chromosomes are located on the interphase nucleus periphery oppositely to each other, has been recently confirmed for mouse and human cells. As already mentioned, human chromosomes containing many genes (like chromosome 19) are mainly deeply localized inside the nucleus, while those with low gene content (in particular, chromosome 18) are localized nearer to its envelope [109]. The recently discovered gradient in frequencies of synonymous substitutions in pseudoautosomal regions of human sex chromosomes [353] can also be interpreted with the account of differential protection of genetic loci against endogenous mutagenesis. Besides, the genes, depending on their functional belonging, have tendency to arrangement on chromosomes in clusters, which was shown in particular for the housekeeping genes [354]. All this may point to different spatial availability of appropriate genetic loci for chemical mutagens and, in turn, this suggests differences in mutation rates in these loci.
Some experimental data support such a supposition. In particular, the rate of mutation accumulation in avian microchromosomes significantly exceeds that in macrochromosomes and intermediate size chromosomes [355]. Besides, the variability of separate genetic loci in eukaryotic genome differs significantly. Wolfe et al. [356] were among the first who demonstrated significant differences in the rate of synonymous substitutions in various human and animal genes. In accordance with this, based on the analysis of distribution of synonymous nucleotide substitutions in ~15,000 human genes, the existence of regions with high and low mutation rates has been recently shown for the human genome [357]. The same is confirmed for the mouse genome [358]. In addition, short discrete domains containing linked genes of proteins evolving at different rates were detected in the mouse genome, which correlates with a gene belonging to a certain domain [359]. More than 10-fold distinctions in frequencies of synonymous substitutions in coding genome regions were found in drosophila [360]. Evolutionarily conserved genes have the tendency to increase the content of introns [361], which can be also interpreted from the point of view of fulfillment by introns of protective function relative to coding NS of genes.
Ellegren et al. in their review generalize data obtained during comparative analysis of NS of animal genomes which altogether show that differences in mutation rates depend on the genetic locus belonging to a particular chromosome, position inside the chromosome, as well as on the context of NS containing variable nucleotides [362]. All this suggests that, in accordance with our concept, frequency of spontaneous mutations in separate loci can be genetically determined.
Thus it is not surprising that in established biological species differences in genome size are defined not by even increase of all its parts, but are represented by discrete regions scattered over the euchromatin genome part [246]. Such distinctions are indicative of peculiarities of spatial intranuclear chromatin packing, optimized for its functional activity, including ensuring the necessary stability level of genetic information. Recently detected evolutionarily conserved genome regions composed of non-coding NS [363] could carry out such functions on the chromatin spatial packing.
The described analysis of the problem of eukaryotic genome size variability revealed the existence of a new (third) system of coding NS protection against chemical mutagens. In this case, we tried to emphasize that just endogenous mutagens constantly create conditions favorable for eukaryotic genome evolution. This highly dynamic system functioning in close interaction with two others (anti-mutagenesis and DNA repair) rather significantly contributes to total genome protection. According to calculations, only the existence of non-coding region in human genome provides for ~30-fold protection of coding NS against chemical mutagens of different nature. This contribution is even more significant in organisms with a higher relative content of non-coding DNA. A large genome with an excess of non-coding NS admits lowered efficiency of anti-mutagenesis and repair systems, which opens an additional possibility for their evolution, i.e. accumulation of mutations in genes encoding macromolecular components of these systems. Investigation of efficiency of these systems in organisms with different genome size can serve as an approach to experimental testing of the whole theory.
Our concept does not deny the theories of “selfish” DNA and DNA as nucleoskeleton forming the nucleotype of an organism widely cited in the literature. Each of these describes in its own way different sides of the unique process of eukaryotic genome evolution. In fact, non-coding DNA that could spread in the genome of the first eukaryotes using the “selfish” mechanism appeared to be useful for its protective and nucleoskeletal functions. Accordingly, evolution of systems providing for cell protection against “selfish” DNA and anti-mutagenic protection of genetic information could proceed simultaneously in parallel courses.
Genetic determination of damage (and mutation) frequencies in particular genetic loci, suggested by our concept, in turn allows us to think about the existence of genetic control of phylogenetic trends and reveals a material basis of the possibility for adaptive mutations in eukaryotes [364, 365]. This can give a new impetus to the idea of nomogenesis (phylogenetic development of species on the basis of regularities [366], opposed to the classical Darwinian theory of evolution) [367]. In this aspect, Vavilov's law of homologous series can be considered, in accordance with which similar series of phenotypic variability are observed in related plant species, genera, and even families [368]. The genotype variability, genetically determined by chromatin structure, could provide for formation of homologous series of these phenotypic features, thus enhancing the process of speciation.
The authors are grateful to N. V. Solov'eva and E. V. Zhurba for help in manuscript preparation.
REFERENCES
1.Greilhuber, J., Dolezel, J., Lysa, M. A., and
Bennett, M. D. (2005) Annals Bot., 95, 255-260.
2.Ohno, S. (1973) Genetic Mechanisms of
Progressive Evolution [Russian translation] Mir, Moscow.
3.Eichler, E. E., Nickerson, D. A., Altshuler, D.,
Bowcock, A. M., Brooks, L. D., Carter, N. P., Church, D. M.,
Felsenfeld, A., Lee, C., Lupski, J. R., Mullikin, J. C., Pritchard, J.
K., Sebat, J., Sherry, S. T., Smith, D., and Waterson, R. H. (2007)
Nature, 447, 161-165.
4.Medini, D., Donati, C., Tettelin, H., Masignani,
V., and Rappuoli, R. (2005) Curr. Opin. Genet. Devel.,
15, 589-594.
5.Tettelin, H., Masignani, V., Cieslewicz, M. J.,
Donati, C., Medini, D., et al. (2005) Proc. Natl. Acad. Sci.
USA, 102, 13950-13955.
6.Morgante, M., de Paoli, E., and Radovic, S. (2007)
Curr. Opin. Plant Biol., 10, 149-155.
7.Khesin, R. B. (1980) Mol. Biol. (Moscow),
14, 1205.
8.Khesin, R. B. (1984) Genome Instability [in
Russian], Nauka, Moscow.
9.Cavalier-Smith, T. (2005) Annals Bot.,
95, 147-175.
10.Gregory, T. R. (2001) Biol. Rev.,
76, 65-101.
11.Leitch, I. J., Soltis, D. E., Soltis, P. S., and
Bennett, M. D. (2005) Annals Bot., 95, 207-217.
12.Mirsky, A. E., and Ris, H. (1951) J. Gen.
Physiol., 34, 451-462.
13.Thomas, C. A. (1971) Annu. Rev. Genet.,
5, 237-256.
14.Lander, E. S., Linton, L. M., Birren, B.,
Nusbaum, C., Zody, et al. (2001) Nature, 409,
860-921.
15.Papathanasiou, P., and Goodnow, C. C. (2005)
Annu. Rev. Genet., 39, 241-262.
16.Claverie, J. M. (2001) Science,
291, 1255-1257.
17.Babushok, D. V., Ostertag, E. M., and Kazazian,
H. H. (2007) Cell. Mol. Life Sci., 64, 542-554.
18.Gregory, T. R. (2002) Evolution,
56, 121-130.
19.Britten, R. J., and Davidson, E. H. (1976)
Proc. Natl. Acad. Sci. USA, 73, 415-419.
20.Ugarkovic, D., and Ploh, M. (2002) EMBO
J., 21, 5955-5959.
21.Johnson, D. H., Kroisel, P. M., Klapper, H. J.,
and Rosenkranz, W. (1992) Hum. Mol. Genet., 1,
741-747.
22.Ustinova, J., Achmann, R., Cremer, S., and Mayer,
F. (2006) J. Mol. Evol., 62, 158-167.
23.Takahashi, Y., Mitani, K., Kuwabara, K., Hayashi,
T., Niwa, M., Miyashita, N., Moriwaki, K., and Kominami, R. (1994)
Chromosoma, 103, 450-458.
24.Biemont, C., and Vieira, C. (2006) Nature,
443, 521-524.
25.Kazazian, H. H. (2003) Curr. Opin. Genet.
Devel., 13, 651-658.
26.Kazazian, H. H. (2004) Science,
303, 1626-1632.
27.Kidwell, M. G. (2002) Genetica,
115, 49-63.
28.Berger, S. L. (2002) Curr. Opin. Genet.
Devel., 12, 142-148.
29.Carlson, C. M., and Largaespada, D. A. (2005)
Nature Rev. Genet., 6, 568-580.
30.De Parseval, N., and Heidmann, T. (2005)
Cytogenet. Genome Res., 110, 318-332.
31.Kramerov, D. A., and Vassetzky, N. S. (2005)
Int. Rev. Cytol., 247, 165-221.
32.Ohshima, K., and Okada, N. (2005) Cytogenet.
Genome Res., 110, 475-490.
33.Georgiev, G. P. (1984) Eur. J. Biochem.,
145, 203-220.
34.Orgel, L. E., and Crick, F. H. C. (1980)
Nature, 284, 604-607.
35.Koonin, E. V. (2005) Annu. Rev. Genet.,
39, 309-338.
36.Roy, S. W., and Gilbert, W. (2006) Nature Rev.
Genet., 7, 211-221.
37.Rodriguez-Trelles, F., Tariro, R., and Ayala, F.
J. (2006) Annu. Rev. Genet., 40, 47-76.
38.Jeffares, D. C., Mourier, T., and Penny, D.
(2006) Trends Genet., 22, 16-22.
39.Vinogradov, A. E. (2004) Trends Genet.,
20, 248-253.
40.Gentles, A. J., and Karlin, S. (1999) Trends
Genet., 15, 47-49.
41.Balakirev, E. S., and Ayala, F. J. (2003)
Annu. Rev. Genet., 37, 123-151.
42.Zhang, Z., Carriero, N., and Gerstein, M. (2004)
Trends Genet., 20, 62-67.
43.Eyre-Walker, A., and Hurst, L. D. (2001)
Nature Rev. Genet., 2, 549-555.
44.Costantini, M., Clay, O., Federico, C., Saccone,
S., Auletta, F., and Bernardi, G. (2007) Chromosoma, 116,
29-40.
45.Bernardi, G., Olofsson, B., Filipski, J., Zerial,
M., Salinas, J., Cuny, G., Meunier-Rotival, M., and Rodier, F. (1985)
Science, 228, 953-957.
46.Zoubak, S., Clay, O., and Bernardi, G. (1996)
Gene, 174, 95-102.
47.Federico, C., Saccone, S., and Bernardi, G.
(1998) Cytogenet. Cell. Genet., 80, 83-88.
48.Federico, C., Saccone, S., Andreozzi, L., Motta,
S., Russo, V., Carels, N., and Bernardi, G. (2004) Gene,
343, 245-251.
49.Federico, C., Scavo, C., Cantarella, D. C.,
Motta, S., Saccone, S., and Bernardi, G. (2006) Chromosoma,
115, 123-128.
50.Saccone, S., Federico, C., and Bernardi, G.
(2002) Gene, 300, 169-178.
51.Johnson, J. M., Edwards, S., Shoemaker, D., and
Schadt, E. E. (2005) Trends Genet., 21, 93-102.
52.Kapranov, F., Cawley, S. E., Drenkow, J.,
Bekiranov, S., Strausberg, R. L., Fodor, S. P. A., and Gingeras, T. R.
(2002) Science, 296, 916-919.
53.Bird, C. P., Stranger, B. E., and Dermitzakis, E.
T. (2006) Curr. Opin. Genet. Devel., 16, 559-564.
54.Waterston, R. H., Lindblad-Toh, K., Birney, E.,
Rogers, J., Abril, J. F., et al. (2002) Nature, 420,
520-562.
55.Grewal, S. I. S., and Jia, S. (2007) Nature
Rev. Genet., 8, 35-46.
56.Huisinga, K. L., Brower-Toland, B., and Elgin, S.
C. R. (2006) Chromosoma, 115, 110-122.
57.Rando, O. J. (2006) Trends Genet.,
23, 67-73.
58.Minsky, A. (2004) Annu. Rev. Biophys. Biomol.
Struct., 33, 317-342.
59.Monakhova, M. A. (1990) Uspekhi Sovr.
Biol., 110, 163-178.
60.Spector, D. L. (2003) Annu. Rev. Biochem.,
72, 573-608.
61.Burke, B., and Stewart, C. L. (2006) Annu.
Rev. Genom. Hum. Genet., 7, 369-405.
62.Nickerson, J. A. (2001) J. Cell Sci.,
114, 463-474.
63.Razin, S. V., Gromova, I. I., and Iarovaia, O. V.
(1995) Int. Rev. Cytol., 405-448.
64.Raska, I. (2003) Trends Cell Biol.,
13, 517-525.
65.Raska, I., Shaw, P. J., and Cmarko, D. (2006)
Curr. Opin. Cell Biol., 18, 325-334.
66.Shaw, P. J., and Brown, J. W. S. (2004) Curr.
Opin. Plant Biol., 7, 614-620.
67.Cioce, M., and Lamond, A. I. (2005) Annu. Rev.
Cell Dev. Biol., 21, 105-131.
68.Jackson, D. A. (2003) Chromosome Res.,
11, 387-401.
69.Seeler, J.-S., and Dejean, A. (1999) Curr.
Opin. Gen. Devel., 9, 362-367.
70.Gilbert, N., Boyle, S., Fiegler, H., Woodfine,
K., Carter, N. P., and Bickmore, W. A. (2004) Cell, 118,
555-566.
71.Grigoryev, S. A., Bulynko, Y. A., and Popova, E.
Y. (2006) Chromosome Res., 14, 53-69.
72.Chadwick, B. P., and Willard, H. F. (2004)
Proc. Natl. Acad. Sci. USA, 101, 17450-17455.
73.Lippman, Z., and Martienssen, R. (2004)
Nature, 431, 364-370.
74.Yasuhara, J. C., and Wakimoto, B. T. (2006)
Trends Genet., 22, 330-338.
75.Dernburg, A. F., Broman, K. W., Fung, J. C.,
Marshall, W. F., Philips, J., Agard, D. A., and Sedat, J. W. (1996)
Cell, 85, 745-759.
76.Jia, S., Yamada, T., and Grewal, S. I. (2004)
Cell, 119, 469-480.
77.Razin, S. V., Iarovaia, O. V., Sjakste, N.,
Sjakste, T., Bagdoniene, L., Rynditch, A. V., Eivazova, E. R.,
Lipinski, M., and Vassetzky, Y. S. (2007) J. Mol. Biol.,
doi:10.1016/j.jmb.2007.04.003 in press.
78.Kornberg, R. D. (1974) Science,
184, 868-871.
79.Davey, C. A., Sargent, D. F., Luger, K., Maeder,
A. W., and Richmond, T. J. (2002) J. Mol. Biol., 319,
1097-1113.
80.Rouleau, M., Aubin, R. A., and Poirier, G. G.
(2004) J. Cell Sci., 117, 815-825.
81.Shiio, Y., and Eisenman, R. N. (2003) Proc.
Natl. Acad. Sci. USA, 100, 13225-13230.
82.Chadwick, B. P., and Willard, H. F. (2002) J.
Cell Biol., 157, 1113-1123.
83.Henikoff, S., and Ahmad, K. (2005) Annu. Rev.
Cell Dev. Biol., 21, 133-153.
84.Richmond, T. J. (2005) Nature, 442,
750-752.
85.Segal, E., Fondufe-Mittendorf, Y., Chen, L.,
Thastrom, A., Field, Y., Moore, I. K., Wang, J.-P. Z., Zhang, Z., and
Widom, J. (2005) Nature, 442, 772-778.
86.Kiyama, R., and Trifonov, E. N. (2002) FEBS
Lett., 523, 7-11.
87.Trifonov, E. N. (1995) J. Mol. Evol.,
40, 337-342.
88.Bode, J., Goetze, S., Heng, H., Krawetz, S. A.,
and Benham, C. (2003) Chromosome Res., 11, 435-445.
89.Schlach, T., Duda, S., Sargent, D. F., and
Richmond, T. J. (2005) Nature, 436, 138-141.
90.McBryant, S. J., Adams, V. H., and Hansen, J. C.
(2006) Chromosome Res., 14, 39-51.
91.Fakan, S. (2004) Cell Biol., 122,
83-93.
92.Fakan, S. (2004) Eur. J. Histochem.,
48, 5-14.
93.Visser, A. E., Jaunin, F., Fakan, S., and Aten,
J. A. (2000) J. Cell Sci., 113, 2585-2593.
94.Luger, K. (2006) Chromosome Res.,
14, 5-16.
95.Cremer, T., and Cremer, C. (2006) Eur. J.
Histochem., 50, 161-176.
96.Cremer, T., and Cremer, C. (2006) Eur. J.
Histochem., 50, 223-272.
97.Kramer, J., Zachar, Z., and Bingham, P. M. (1994)
Trends Cell Biol., 4, 35-37.
98.Zirbel, R. M., Mathieu, U. R., Kurz, A., Cremer,
T., and Lichter, P. (1993) Chromosome Res., 1,
92-106.
99.Cremer, T., and Cremer, C. (2001) Nat. Rev.
Genet., 2, 292-301.
100.Walter, J., Schermelleh, L., Cremer, M.,
Tashiro, S., and Cremer, T. (2003) J. Cell Biol., 160,
685-697.
101.Chubb, J. R., and Bickmore, W. A. (2003)
Cell, 112, 403-406.
102.Sachs, R. K., van den Engh, G., Trask, B.,
Yokota, H., and Hearst, J. E. (1995) Proc. Natl. Acad. Sci. USA,
92, 2710-2714.
103.Ma, H., Siegel, A. J., and Berezney, R. J.
(1999) Cell Biol., 146, 531-541.
104.Alexandrova, O., Solovei, I., Cremer, T., and
David, C. N. (2003) Chromosoma, 112, 190-200.
105.Habermann, F. A., Cremer, M., Walter, J.,
Kreth, G., von Hase, J., Bauer, K., Wienberg, J., Cremer, C., Cremer,
T., and Solovei, I. (2001) Chromosome Res., 9,
569-584.
106.Mayr, C., Jasencakova, Z., Meister, A.,
Schubert, I., and Zink, D. (2003) Chromosome Res., 11,
471-484.
107.Postberg, J., Alexandrova, O., Cremer, T., and
Lipps, H. J. (2005) J. Cell Sci., 118, 3973-3983.
108.Federico, C., Cantarella, C. D., Scavo, C.,
Saccone, S., Bed'hom, B., and Bernardi, G. (2005) Chromosome
Res., 13, 785-793.
109.Boyle, S., Gilchrist, S., Bridger, J. M., Mahy,
N. L., Ellis, J. A., and Bickmore, W. A. (2001) Hum. Mol.
Genet., 10, 211-219.
110.Cremer, M., Kupper, K., Wagler, B. L.,
Wizelman, V., Hase, J., Weiland, Y., Kreja, L., Diebold, J., Speicher,
M. R., and Cremer, T. (2003) J. Cell Biol., 162,
809-820.
111.Croft, J. A., Bridger, J. M., Boyle, S., Perry,
P., Teague, P., and Bickmore, W. A. (1999) J. Cell Biol.,
145, 1119-1131.
112.Francastel, C., Schubeler, D., Martin, D. I.,
and Groudine, M. (2000) Nat. Rev. Mol. Cell Biol., 1,
137-143.
113.Neusser, M., Schubel, V., Koch, A., Cremer, T.,
and Muller, S. (2007) Chromosoma, 116, 307-320.
114.Mayer, R., Brero, A., von Hase, J., Schroeder,
T., Cremer, T., and Dietzel, S. (2005) BMC Cell Biol., 6,
44.
115.Mora, L., Sanchez, I., Garcia, M., and Ponsa,
M. (2006) Chromosoma, 115, 367-375.
116.Tanabe, H., Muller, S., Neusser, M., von Hase,
J., Calcagno, E., Cremer, M., Solovei, I., Cremer, C., and Cremer, T.
(2002) Proc. Natl. Acad. Sci. USA, 99, 4424-4429.
117.Tanabe, H., Habermann, F. A., Solovei, I.,
Cremer, M., and Cremer, T. (2002) Mutat. Res., 504,
37-45.
118.Taslerova, R., Kozubek, S., Bartova, E.,
Gajduskova, P., Kodet, R., and Kozubek, M. (2006) J. Struct.
Biol., 155, 493-504.
119.Bolzer, A., Kreth, G., Solovei, I., Koehler,
D., Saracoglu, K., Fauth, C., Muller, S., Eils, R., Cremer, C.,
Speicher, M. R., and Cremer, T. (2005) PLoS Biol., 3,
e157.
120.Sun, H. B., Shen, J., and Yokota, H. (2000)
Biophys. J., 79, 184-190.
121.Parada, L. A., and Misteli, T. (2002) Trends
Cell Biol., 12, 425-432.
122.Abranches, R., et al. (1998) J. Cell
Biol., 143, 5-12.
123.Hochstrasser, M., Mathog, D., Gruenbaum, Y.,
Saumweber, H., and Sedat, J. W. (1986) J. Cell Biol.,
102, 112-123.
124.Nagele, R., Freeman, T., McMorrow, L., and Lee,
H.-Y. (1995) Science, 270, 1831-1835.
125.Nagele, R. G., Freeman, T., McMorrow, L.,
Thomson, Z., Kitson-Wind, K., and Lee, H. (1999) J. Cell Sci.,
112, 525-535.
126.Allison, D. C., and Nestor, A. L. (1999) J.
Cell Biol., 145, 1-14.
127.Verschure, P. J., van der Kraan, I., and van
Driel, R. (1999) J. Cell Biol., 147, 13-24.
128.Volpi, E. V., Chevret, E., Jones, T., Vatcheva,
R., Williamson, J., Beck, S., Campbell, R. D., Goldsworthy, M., Powis,
S. H., Ragoussis, J., Trowsdale, J., and Sheer, D. (2000) J. Cell
Sci., 113, 1565-1576.
129.Williams, R. R., Broad, S., Sheer, D., and
Ragoussis, J. (2002) Exp. Cell Res., 272, 163-175.
130.Gasser, S. M. (2002) Science,
296, 1412-1416.
131.Lanctot, C., Cheutin, T., Cremer, M., Cavalli,
G., and Cremer, T. (2007) Nature Rev. Genet., 8,
104-115.
132.Ferreira, J., Paolella, G., Ramos, C., and
Lamond, A. I. (1997) J. Cell Biol., 139, 1597-1610.
133.Sadoni, N., Langer, S., Fauth, C., Bernardi,
G., Cremer, T., Turner, B. M., and Zink, D. (1999) J. Cell
Biol., 146, 1211-1226.
134.Visser, A. E., and Aten, J. A. (1999) J.
Cell Sci., 112, 3353-3360.
135.Tumbar, T., and Belmont, A. S. (2001) Nature
Cell Biol., 3, 134-139.
136.Chuang, C. H., Carpenter, A. E., Fuchsova, B.,
Johnson, T., de Lanerolle, P., and Belmont, A. S. (2006) Curr.
Biol., 16, 825-831.
137.Dillon, N. (2006) Chromosome Res.,
14, 117-126.
138.Brown, K. E., Amoils, S., Horn, J. M., Buckle,
V. J., Higgs, D. R., Merkenschlager, M., and Fisher, A. G. (2001)
Nature Cell Biol., 3, 602-606.
139.Brown, K. E., Baxter, J., Graf, D.,
Merkenschlager, M., and Fisher, A. G. (1999) Mol. Cell,
3, 207-217.
140.Brown, K. E., Guest, S. S., Smale, S. T., Hahm,
K., Merkenschlager, M., and Fisher, A. G. (1997) Cell,
91, 845-854.
141.Csink, A. K., and Henikoff, S. (1996)
Nature, 381, 529-531.
142.Harmon, B., and Sedat, J. (2005) PLoS
Biol., 3, e67.
143.Bacher, C. P., et al. (2006) Nature Cell
Biol., 8, 293-299.
144.Xu, N., Tsai, C. L., and Lee, J. T. (2006)
Science, 311, 1149-1152.
145.Fraser, P., and Bickmore, W. (2007)
Nature, 447, 413-417.
146.Kioussis, D. (2005) Nature, 435,
579-580.
147.Spilianakis, C. G., Lalioti, M. D., Town, T.,
Lee, G. R., and Flavell, R. A. (2005) Nature, 435,
637-645.
148.De Laat, W., and Grosveld, F. (2003)
Chromosome Res., 11, 447-459.
149.Osborne, C. S., Chakalova, L., Brown, K. E.,
Carter, D., Horton, A., Debrand, E., Goyenechea, B., Mitchell, J. A.,
Lopes, S., Reik, W., and Fraser, P. (2004) Nature Genet.,
36, 1065-1071.
150.Pederson, T. (2004) Curr. Opin. Genet.
Devel., 14, 203-209.
151.Bender, J. (2004) Annu. Rev. Plant.
Biol., 55, 41-68.
152.Chan, S. W.-L., Henderson, I. R., and Jacobsen,
S. E. (2005) Nature Rev. Genet., 6, 351-360.
153.Espada, J., and Esteller, M. (2007) Cell.
Mol. Life Sci., 64, 449-457.
154.Freitag, M., and Selker, E. U. (2005) Curr.
Opin. Genet. Devel., 15, 191-199.
155.Jenuwein, T., and Allis, C. D. (2001)
Science, 293, 1074-1080.
156.Lam, A. L., Pazin, D. E., and Sullivan, B. A.
(2005) Chromosoma, 114, 242-251.
157.Robertson, K. D. (2005) Nat. Rev.
Genet., 6, 597-610.
158.Buendia, B., Courvalin, J.-C., and Collas, P.
(2001) Cell. Mol. Life Sci., 58, 1781-1789.
159.Hsieh, T.-F., and Fischer, R. L. (2005)
Annu. Rev. Plant Biol., 56, 327-351.
160.Eichler, E. E., and Sankoff, D. (2003)
Science, 301, 793-797.
161.Tower, J. (2004) Annu. Rev. Genet.,
38, 273-304.
162.Claycomb, J. M., Benasutti, M., Bosco, G.,
Fenger, D. D., and Orr-Weaver, T. L. (2004) Cell, 6,
145-155.
163.Lunyak, V. V., Ezrokhi, M., Smith, H. S., and
Gerbi, S. A. (2002) Mol. Cell Biol., 22, 8426-8437.
164.Kapler, G. M. (1993) Curr. Opin. Genet.
Dev., 3, 730-735.
165.Libuda, D. E., and Winston, F. (2006)
Nature, 443, 1003-1007.
166.Hemingway, J., Field, L., and Vontas, J. (2002)
Science, 298, 96-97.
167.Shimke, R. T. (1986) Cancer, 10,
1912-1917.
168.Nagl, W. (1976) Nature, 261,
614-615.
169.Sugimoto-Shirasu, K., and Roberts, K. (2003)
Curr. Opin. Plant Biol., 6, 544-553.
170.Alitalo, K., and Schwab, M. (1986) Adv.
Cancer Res., 47, 235-281.
171.Garraway, L. A., Widlund, H. R., Rubin, M. A.,
Getz, G., Berger, A. J., Ramaswamy, S., Beroukhim, R., Milner, D. A.,
Granter, S. R., Du, J., Lee, C., Wagner, S. N., Li, C., Golub, T. R.,
Rimm, D. L., Meyerson, M. L., Fisher, D. E., and Sellers, W. R. (2005)
Nature, 436, 117-122.
172.Jahn, C. L., and Klobutcher, L. A. (2002)
Annu. Rev. Microbiol., 56, 489-520.
173.Grishanin, A. K., Brodskii, V. I., and Akif'ev,
A. P. (1994) Dokl. Biol. Sci., 338, 505-506.
174.Kubota, S., Kuro-o, M., Mizuno, S., and Kohno,
S. (1993) Chromosoma, 102, 163-173.
175.Wyngaard, G. A., and Gregory, T. R. (2001)
J. Exp. Zool., 291, 310-316.
176.Yao, M.-C., and Chao, J.-L. (2005) Annu.
Rev. Genet., 39, 537-559.
177.Maizels, N. (2005) Annu. Rev. Genet.,
39, 23-46.
178.Martin, A., and Scharff, M. D. (2004) Nature
Rev. Immunol., 2, 605-614.
179.Antigenic Variation (2003) Elsevier.
180.Hubscher, U., Maga, G., and Spadari, S. (2002)
Annu. Rev. Biochem., 71, 133-163.
181.Goodman, M. F. (2002) Annu. Rev.
Biochem., 71, 17-50.
182.Kamath-Loeb, A. S., Loeb, L. A., Masuda, Y.,
and Hanaoka, F. (2005) DNA Repair, 4, 740-747.
183.Rattray, A. J., and Strathern, J. N. (2003)
Annu. Rev. Genet., 37, 31-66.
184.Maki, H. (2002) Annu. Rev. Genet.,
36, 279-303.
185.De Bont, R., and van Larebeke, N. (2004)
Mutagenesis, 19, 169-185.
186.Lindahl, T. (1993) Nature, 362,
709-715.
187.Kunkel, T. A. (2004) J. Biol. Chem.,
279, 16895-16898.
188.Garcia-Diaz, M., and Kunkel, T. A. (2006)
Trends Biochem. Sci., 31, 206-214.
189.Barnes, D. E., and Lindahl, T. (2004) Annu.
Rev. Genet., 38, 445-476.
190.Klaunig, J. E., and Kamendulis, L. M. (2004)
Annu. Rev. Pharmacol. Toxicol., 44, 239-267.
191.Russo, M. T., de Luca, G., Degan, P., Parlanti,
E., Dogliotti, E., Barnes, D. E., Lindahl, T., Yang, H., Miller, J. H.,
and Bignami, M. (2004) Cancer Res., 64, 4411-4414.
192.Evans, M. D., Dizdaroglu, M., and Cooke, M. S.
(2004) Mutation Res., 567, 1-61.
193.Valko, M., Leibfritz, D., Moncola, J., Cronin,
M. T. D., Mazura, M., and Telser, J. (2007) Int. J. Biochem. Cell
Biol., 39, 44-84.
194.Nakamura, J., and Swenberg, J. A. (1999)
Cancer Res., 59, 2522-2526.
195.Nakamura, J., La, D. K., and Swenberg, J. A.
(2000) J. Biol. Chem., 275, 5323-5328.
196.Lenton, K. J., Therriault, H., Fulop, T.,
Payette, H., and Wagner, J. R. (1999) Carcinogenesis, 20,
607-613.
197.Spencer, J. P., Jenner, A., Aruoma, O. I.,
Cross, C. E., Wu, R., and Halliwell, B. (1996) Biochem. Biophys.
Res. Commun., 224, 17-22.
198.Wagner, J. R., Hu, C. C., and Ames, B. N.
(1992) Proc. Natl. Acad. Sci. USA, 89, 3380-3384.
199.Beckman, K. B., and Ames, B. N. (1997) J.
Biol. Chem., 272, 19633-19636.
200.Bartsch, H., and Nair, J. (2000)
Toxicology, 153, 105-114.
201.Chen, H. J., Chiang, L. C., Tseng, M. C.,
Zhang, L. L., Ni, J., and Chung, F. L. (1999) Chem. Res.
Toxicol., 12, 1119-1126.
202.Kadlubar, F. F., Anderson, K. E., Haussermann,
S., et al. (1998) Mutat. Res., 405, 125-133.
203.Chaudhary, A. K., Nokubo, M., Reddy, G. R.,
Yeola, S. N., Morrow, J. D., Blair, I. A., and Marnett, L. J. (1994)
Science, 265, 1580-1582.
204.Marnett, L. J. (1999) IARC Sci. Publ.,
150, 17-27.
205.Cavalieri, E., Chakravarti, D., Guttenplan, J.,
Hart, E., Ingle, J., Jankowiak, R., Muti, P., Rogan, E., Russo, J.,
Santen, R., and Sutter, T. (2006) Biochim. Biophys. Acta,
1766, 63-78.
206.Bennett, M. D., Johnston, S., Hodnett, G. L.,
and Price, H. J. (2000) Annals Bot., 85, 351-357.
207.Drabløs, F., Feyzi, E., Aas, P. A., Vaagbø,
C. B., Kavli, B., Bratlie, M. S., Peca-Diaz, J., Otterlei, M.,
Slupphaug, G., and Krokan, H. E. (2004) DNA Repair, 3,
1389-1407.
208.Bennetzen, J. L. (2005) Curr. Opin. Genet.
Devel., 15, 621-627.
209.Ostertag, E. M., and Kazazian, H. H. (2001)
Annu. Rev. Genet., 35, 501-538.
210.Besaratinia, A., Synold, T. W., Xi, B., and
Pfeifer, G. P. (2004) Biochemistry, 43, 8169-8177.
211.Pfeifer, G. P., You, Y.-H., and Besaratinia, A.
(2005) Mutation Res., 571, 19-31.
212.Buonocore, G., and Groenendaal, F. (2007)
Semin. Fetal Neonat. Med., doi:10.1016/j.siny.2007.01.020.
213.Droge, W. (2002) Physiol. Rev.,
82, 47-95.
213a.Antonin, W., Ellenberg, J., and Dultz, E. (2008) FEBS Lett.,
582, 2004-2016.
213b.Lim, R. Y. H., Aebi, U., and Fahrenkrog, B. (2008) Histochem.
Cell Biol., 129, 105-116.
214.Sies, H. (1997) Exp. Physiol.,
82, 291-295.
215.Bensaad, K., and Vousden, K. H. (2007)
Trends Cell Biol., 17, 286-291.
216.Sablina, A. A., Budanov, A. V., Ilyinskaya, G.
V., Agapova, L. S., Kravchenko, J. E., and Chumakov, P. M. (2005)
Nature Med., 11, 1306-1313.
217.Chan, K. K. L., Zhang, Q.-M., and Dianov, G. L.
(2006) Mutagenesis, 21, 173-178.
218.Dianov, G. L., and Allinson, S. L. (2005)
Genome Dyn. Stab. DOI 10.1007/7050 007.
219.Kunkel, T. A., and Erie, D. A. (2005) Annu.
Rev. Biochem., 74, 681-710.
220.Mitra, S., Izumi, T., Boldogh, I., Bhakat, K.
K., Hill, J. W., and Hazra, T. K. (2002) Free Radic. Biol. Med.,
33, 15-28.
221.David, S. S., O'Shea, V. L., and Kundu, S.
(2007) Nature, 447, 941-950.
222.Dizdaroglu, M. (2005) Mutat. Res.,
591, 45-59.
223.Daviet, S., Couve-Privat, S., Gros, L.,
Shinozuka, K., Ide, H., Saparbaev, M., and Ishchenko, A. A. (2007)
DNA Repair, 6, 8-18.
224.Ischenko, A. A., and Saparbaev, M. K. (2002)
Nature, 415, 183-187.
225.Colussi, C., Parlanti, E., Degan, P., Aquilina,
G., Barnes, D., Macpherson, P., Karran, M., Crescenzi, P., Dogliotti,
E., and Bignami, M. (2002) Curr. Biol., 12, 912-918.
226.Gagne, J.-P., Hendzel, M. J., Droit, A., and
Poirier, G. G. (2006) Curr. Opin. Cell Biol., 18,
145-151.
227.Huber, A., Bai, P., Menissier, de Murcia, J.,
and de Murcia, G. (2004) DNA Repair, 3, 1103-1108.
228.Muiras, M.-L. (2003) Ageing Res. Rev.,
2, 129-148.
229.Petermann, E., Keil, C., and Oei, S. L. (2005)
Cell. Mol. Life Sci., 62, 731-738.
230.Kashkush, K., Feldman, M., and Levy, A. A.
(2002) Genetics, 160, 1651-1659.
231.Adams, K. L., and Wendel, J. F. (2005) Curr.
Opin. Plant Biol., 8, 135-141.
232.Ware, D., and Stein, L. (2003) Curr. Opin.
Plant Biol., 6, 121-127.
233.Ramsey, J., and Schemske, D. W. (1998) Annu.
Rev. Ecol. Syst., 29, 467-501.
234.Masterson, J. (1994) Science,
264, 421-424.
235.Seoighe, C. (2003) Curr. Opin. Genet.
Dev., 13, 636-643.
236.Wendel, J. F. (2000) Plant Mol. Biol.,
42, 225-249.
237.Comai, L. (2005) Nature Rev. Genet.,
6, 836-846.
238.Eiben, B., Bartels, I., Bahr-Porsch, S.,
Borgmann, S., Gatz, G., Gellert, G., Goebel, R., Hammans, W.,
Hentemaan, M., Osmers, R., Rauskolb, R., and Hansmann, I. (1990) Am.
J. Hum. Genet., 47, 656-663.
239.Hancock, J. M. (2005) Trends Genet.,
21, 591-595.
240.Moore, R. C., and Purugganan, M. D. (2005)
Curr. Opin. Plant Biol., 8, 122-128.
241.Bailey, J. A., Church, D. M., Ventura, M.,
Rocchi, M., and Eichler, E. E. (2004) Genome Res., 14,
789-801.
242.Tuzun, E., Bailey, J. A., and Eichler, E. E.
(2004) Genome Res., 14, 493-506.
243.Koszul, R., Caburet, S., Dujon, B., and
Fischer, G. (2004) EMBO J., 23, 234-243.
244.Ross-Ibarra, J. (2007) J. Compilation,
20, 800-806.
245.Bailey, J. A., and Eichler, E. E. (2006)
Nature Rev. Genet., 7, 552-564.
246.Petrov, D. A. (2001) Trends Genet.,
17, 23-28.
247.Vitte, C., and Bennetzen, J. L. (2006) Proc.
Natl. Acad. Sci. USA, 103, 17638-17643.
248.Piegu, B., Guyot, R., Picault, N., Roulin, A.,
Saniyal, A., Kim, H., Collura, K., Brar, D. S., Jackson, S., Wing, R.
A., and Panaud, O. (2006) Genome Res., 16, 1262-1269.
249.Harrison, P. M., Zheng, D., Zhang, Z.,
Carriero, N., and Gerstein, M. (2005) Nucleic Acids Res.,
33, 2374-2383.
250.Bennetzen, J. L., Ma, J., and Devos, K. M.
(2005) Annals Bot., 95, 127-132.
251.Vitte, C., and Panaud, O. (2005) Cytogenet.
Genome Res., 110, 91-107.
252.Betran, E., and Long, M. (2002)
Genetica, 115, 65-80.
253.Monakhova, M. A. (1990) Uspekhi Sovr.
Biol., 110, 163-178.
254.Ohno, S. (1999) Cell. Mol. Life Sci.,
55, 824-830.
255.Spring, J. (1997) FEBS Lett.,
400, 2-8.
256.Meyer, A., and Schartl, M. (1999) Curr.
Opin. Cell Biol., 11, 699-704.
257.Wendel, J. F., Cronn, R. C., Johnston, J. S.,
and Price, H. J. (2002) Genetica, 115, 37-47.
258.Vinogradov, A. E. (2004) Curr. Opin. Genet.
Devel., 14, 620-626.
259.Blanc, G., and Wolfe, K. H. (2004) Plant
Cell, 16, 1679-1691.
260.Seoighe, C., and Gehring, C. (2004) Trends
Genet., 20, 461-464.
261.Gregory, T. R. (2005) Annals Bot.,
95, 133-146.
262.Vendrely, R., and Vendrely, C. (1948)
Experientia, 4, 434-436.
263.Swift, H. (1950) Physiol. Zool.,
23, 169-198.
264.Swift, H. (1950) Proc. Natl. Acad. Sci.
USA, 36, 643-654.
265.Kellogg, E. A., and Bennetzen, J. L. (2004)
Am. J. Bot., 91, 1709-1725.
266.Soltis, D. E., Soltis, P. S., Bennett, M. D.,
and Leitch, I. J. (2003) Am. J. Bot., 90, 1596-1603.
267.Sparrow, A. H., and Nauman, A. F. (1976)
Science, 192, 524-527.
268.Narayan, R. K. J. (1985) J. Genet.,
64, 101-109.
269.Narayan, R. K. J. (1988) Evol. Trends
Plants, 2, 121-130.
270.Narayan, R. K. J. (1998) Annals Bot.,
82 (Suppl. A), 57-66.
271.Sparrow, A. H., and Nauman, A. F. (1973)
Brookhaven Symp. Biol., 25, 367-389.
272.Maszewski, J., and Kolodziejczyk, P. (1991)
Plant Syst. Evol., 175, 23-38.
273.Raina, S. N., Srivastav, P. K., and Rama, R. S.
(1986) Genetica, 69, 27-33.
274.Finston, T. L., Hebert, P. D. N., and Foottit,
R. B. (1995) Insect Biochem. Mol. Biol., 25, 189-196.
275.Gambi, M. C., Ramella, L., Sella, G., Protto,
P., and Aldieri, E. (1997) J. Marine Biol. Assoc. UK, 77,
1045-1057.
276.Gregory, T. R., Hebert, P. D. N., and Kolasa,
J. (2000) Heredity, 84, 201-208.
277.Sella, G., Redi, G. A., Ramella, L., Soldi, R.,
and Premoli, M. C. (1993) Genome, 36, 652-657.
278.McLaren, I. A., Sevigny, J.-M., and Corkett, C.
J. (1988) Hydrobiologia, 167/168, 275-284.
279.McLaren, I. A., Sevigny, J.-M., and Frost, B.
W. (1989) Can. J. Zool., 67, 565-569.
280.MacCulloch, R. D., Upton, D. E., and Murphy, R.
W. (1996) Comp. Biochem. Physiol., 113B, 601-605.
281.Noirot, M., Barre, P., Louarn, J., Duperray,
C., and Hamon, S. (2002) Annals Bot., 89, 385-389.
282.Price, H. J., Hodnett, G., and Johnston, J. S.
(2000) Annals Bot., 86, 929-934.
283.Morgante, M. (2006) Curr. Opin.
Biotechnol., 17, 168-173.
284.Brunner, S., Fengler, K., Morgante, M., Tingey,
S., and Rafalski, A. (2005) Plant Cell, 17, 343-360.
285.Kalendar, R., Tanskanen, J., Immonen, S., Nevo,
E., and Schulman, A. H. (2000) Proc. Natl. Acad. Sci. USA,
97, 6603-6607.
286.Biradar, D. P., Bullock, D. G., and Rayburn, A.
L. (1994) Theor. Appl. Genet., 88, 557-560.
287.Poggio, L., Rosato, M., Chiavarino, A. M., and
Naranjo, C. A. (1998) Annals Bot., 82A, 107-115.
288.Rai, K. S., and Black, W. C. (1999) Adv.
Genet., 41, 1-33.
289.Boulesteix, M., Weiss, M., and Biemont, C.
(2006) Mol. Biol. Evol., 23, 162-167.
290.Murray, B. G. (2005) Annals Bot.,
95, 119-125.
291.Gregory, T. R. (2005) in The Evolution of
the Genome (Gregory, T. R., ed.) Elsevier Inc., pp. 3-87.
292.Waldegger, S., and Lang, F. (1998) J. Membr.
Biol., 162, 95-100.
293.Cavalier-Smith, T. (1980) Nature,
285, 617-618.
294.Bennett, M. D. (1972) Proc. R. Soc. Lond. B
Biol. Sci., 181, 109-135.
295.Knight, C. A., Molinari, N. A., and Petrov, D.
A. (2005) Annals Bot., 95, 177-190.
296.Wakamiya, I. (1993) Am. J. Bot.,
80, 1235-1241.
297.Bennett, M. D., Leitch, I. J., and Hanson, L.
(1998) Annals Bot., 82, 121-134.
298.Biradar, D. P., and Rayburn, A. L. (1993)
Heredity, 71, 300-304.
299.Rayburn, A. L., Dudley, J. W., and Biradar, D.
P. (1994) Plant Breeding, 112, 318-322.
300.Minelli, S., Moscariello, P., Ceccarelli, M.,
and Cionini, P. G. (1996) Heredity, 76, 524-530.
301.Beaulieu, J. M., Moles, A. T., Leitch, I. J.,
Bennett, M. D., Dickie, J. B., and Knight, C. A. (2007) New
Phytologist, 173, 422-437.
302.Charlesworth, D. (2002) Heredity,
88, 94-101.
303.Costich, D. E., Meagher, T. R., and Yurkow, E.
J. (1991) Plant Mol. Biol. Report., 9, 359-370.
304.Delph, L. F., Gehring, J. L., Frey, F. M.,
Arntz, A. M., and Levri, M. (2004) Evolution, 58,
1936-1946.
305.Vinogradov, A. E. (1995) Evolution,
49, 1249-1259.
306.Vinogradov, A. E. (1997) Evolution,
51, 220-225.
307.Roth, G., Blanke, J., and Wake, D. B. (1994)
Proc. Natl. Acad. Sci. USA, 91, 4796-4800.
308.Pagel, M., and Johnstone, R. A. (1992) Proc.
R. Soc. Lond., B 249, 119-124.
309.Vinogradov, A. E. (2003) Trends Genet.,
19, 609-614.
310.Knight, C. A., and Ackerly, D. D. (2002)
Ecol. Lett., 5, 66-76.
311.Mackay, T. F. C. (2001) Annu. Rev.
Genet., 35, 303-339.
312.Ohno, S. (1972) in Evolution of Genetic
Systems (Smith, H. H., ed.) pp. 366-370.
313.Doolittle, W. F., and Sapienza, C. (1980)
Nature, 284, 601-603.
314.Jain, H. K. (1980) Nature, 288,
647-648.
315.Charlesworth, B., Sniegowski, P., and Stephan,
W. (1994) Nature, 371, 215-220.
316.Le Rouzic, A., Dupas, S., and Capy, P. (2007)
Gene, 390, 214-220.
317.Orgel, L. E., Crick, F. H. C., and Sapienza, C.
(1980) Nature, 288, 645-646.
318.Nobrega, M. A., Zhu, Y., Plajzer-Frick, I.,
Afzal, V., and Rubin, E. M. (2004) Nature, 431,
988-993.
319.Commoner, B. (1964) Nature, 202,
960-968.
320.Cavalier-Smith, T. (1978) J. Cell Sci.,
34, 247-278.
321.Vinogradov, A. E. (1998) J. Theor.
Biol., 193, 197-199.
322.Shapiro, J. A., and von Sternberg, R. (2005)
Biol. Rev., 80, 1-24.
323.Yunis, J. J., and Yasmineh, W. G. (1971)
Science, 174, 1200-1209.
324.Hsu, T. C. (1975) Genetics, 79,
137-150.
325.Minkevich, I. G., and Patrushev, L. I. (2007)
Bioorg. Khim., 33, 474-477.
326.Patrushev, L. I., and Minkevich, I. G. (2006)
Bioorg. Khim., 32, 408-413.
327.Patrushev, L. I. (1997) Biochem. Mol. Biol.
Int., 41, 851-860.
328.Cooke, M. S., Evans, M. D., Dizdaroglu, M., and
Lunec, J. (2003) FASEB J., 17, 1195-1214.
329.Laine, J.-P., and Egly, J.-M. (2006) Trends
Genet., 22, 430-436.
330.Jaillon, O., Aury, J.-M., Brunet, F., Petit,
J.-L., Stange-Thomann, N., Mauceli, E., Bouneau, L., Fischer, C.,
Ozouf-Costaz, C., Bernot, A., et al. (2004) Nature, 431,
946-957.
331.Kaul, S., Koo, H. L., Jenkins, J., Rizzo, M.,
Rooney, T., Tallon, L. J., Feldblyum, T., Nierman, W., et al. (2000)
Nature, 408, 796-815.
332.Blaustein, A. R., and Belden, L. K. (2003)
Evol. Devel., 5, 89-97.
333.David, W. M., Mitchell, D. L., and Walter, R.
B. (2004) Comp. Biochem. Physiol. Pt. C, 138,
301-309.
334.Willett, K. L., Lienesch, L. A., and di Giulio,
R. T. (2001) Comp. Biochem. Physiol. Pt. C, Toxicol. Pharmacol.,
128, 349-358.
335.Filho, D. W., Sell, F., Ribeiro, L., Ghislandi,
M., Carrasquedo, F., Fraga, C. G., Wallauer, J. P., Simoes-Lopes, P.
C., and Uhart, M. M. (2002) Comp. Biochem. Physiol. Pt. A,
133, 885-892.
336.Bennett, M. D. (1971) Proc. R. Soc. Lond. B
Biol. Sci., 178, 277-299.
337.Samuilov, V. D. (2005) Biochemistry
(Moscow), 70, 246-250.
338.Blankenship, R. E. (2001) Trends Plant
Sci., 6, 4-6.
339.Rye, R., and Holland, H. D. (1998) Am. J.
Sci., 298, 621-672.
340.Berner, R. A., van den Brooks, J. M., and Ward,
P. D. (2007) Science, 316, 557-558.
341.Frazier, M. R., Woods, H. A., and Harrison, J.
F. (2001) Physiol. Biochem. Zool., 74, 641.
342.Edgar, B. A. (2006) Nature Rev. Genet.,
7, 907-916.
343.Shapiro, J. A. (1999) Ann. N. Y. Acad.
Sci., 870, 23-35.
344.Chantret, N., Salse, J., Sabot, F., Rahman, S.,
Bellec, A., et al. (2005) Plant Cell, 17, 1033-1045.
345.Ma, J., Devos, K. M., and Bennetzen, J. L.
(2004) Genome Res., 14, 860-869.
346.Petrov, D. A., Lozovskaya, E. R., and Hartl, D.
L. (1996) Nature, 384, 346-349.
347.Shirasu, K., Schulman, A. H., Lahaye, T., and
Schulze-Lefert, P. (2000) Genome Res., 10, 908-915.
348.Brouha, B., Schustak, J., Badge, R. M., et al.
(2003) Proc. Natl. Acad. Sci. USA, 100, 5280-5285.
349.Rudin, C. M., and Thompson, C. B. (2001)
Genes Chromosomes Cancer, 30, 64-71.
350.Servomaa, K., and Rytomaa, T. (1990) Int. J.
Radiat. Biol., 57, 331-343.
351.Kaup, S., Grandjean, V., Mukherjee, R., Kapoor,
A., Keyes, E., Seymour, C. B., Mothersill, C. E., and Schofield, P. N.
(2006) Mutat. Res., 597, 87-97.
351a.D'Angelo, M. A., and Hetzer, M. W. (2008) Trends Cell Biol.,
18, 456-466.
351b.Antonin, W., Ellenberg, J., and Dultz, E. (2008) FEBS Lett.,
582, 2004-2016.
352.Leitch, I. J., and Bennett, M. D. (2004)
Biol. J. Linnean Soc., 82, 651-663.
353.Filatov, D. A. (2004) Mol. Biol. Evol.,
2, 1410-1417.
354.Lercher, M. J., Urrutia, A. O., and Hurst, L.
D. (2002) Nature Genet., 31, 180-183.
355.Axelsson, E., Webster, M. T., Smith, N. G. C.,
Burt, D. W., and Ellegren, H. (2005) Genome Res., 15,
120-125.
356.Wolfe, K. H., Sharp, P. M., and Li, W. H.
(1989) Nature, 337, 283-285.
357.Chuang, J. H., and Li, H. (2004) PLoS
Biol., 2, 253-263.
358.Gaffney, D. J., and Keightley, P. D. (2005)
Genome Res., 15, 1086-1094.
359.Williams, E. J. B., and Hurst, L. D. (2000)
Nature, 407, 900-903.
360.Zeng, L., Comeron, J. M., Chen, B., and
Kreitman, M. (1998) Genetica, 102/103, 369-382.
361.Carmel, L., Rogozin, I. B., Wolf, Y. I., and
Koonin, E. V. (2007) Genome Res., 17, 1045-1050.
362.Ellegren, H., Smith, N. G. C., and Webster, M.
T. (2003) Curr. Opin. Genet. Dev., 13, 562-568.
363.Drake, J. A., Bird, C., Nemesh, J., Thomas, D.
J., Newton-Cheh, C., Reymond, A., Excoffier, L., Attar, H.,
Antonarakis, S. E., Dermitzakis, E. T., and Hirschhorn, J. N. (2006)
Nature Genet., 38, 223-227.
364.Chicurel, M. (2001) Science, 292,
1824-1827.
365.Hall, B. G. (1998) Genetica,
102/103, 109-125.
366.Berg, L. S. (1977) Works on Theory of
Evolution [in Russian], Nauka, Moscow.
367.Timofeev-Ressovskii, N. V., Vorontsov, N. N.,
and Yablokov, A. V. (1969) A Short Essay of Evolution Theory [in
Russian], Nauka, Moscow.
368.Vavilov, N. I. (1987) The Law of Homologous
Rows in Hereditary Variability [in Russian], Nauka, Moscow.
369.Biemont, C., and Vieira, C. (2005)
Cytogenet. Genome Res., 110, 25-34.
370.Gregory, T. R. (2005) Nature Rev.
Genet., 6, 699-708.
371.Luger, K. (2003) Curr. Opin. Genet.
Devel., 13, 127-135.