REVIEW: Mechanisms of Single-Stranded DNA-Binding Protein Functioning in Cellular DNA Metabolism
P. E. Pestryakov and O. I. Lavrik*
Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, pr. Akademika Lavrentieva 8, 630090 Novosibirsk, Russia; E-mail: lavrik@niboch.nsc.ru* To whom correspondence should be addressed.
Received February 7, 2008; Revision received May 29, 2008
This review deals with analysis of mechanisms involved in coordination of DNA replication and repair by SSB proteins; characteristics of eukaryotic, prokaryotic, and archaeal SSB proteins are considered, which made it possible to distinguish general mechanisms specific for functioning of proteins from organisms of different life domains. Mechanisms of SSB protein interactions with DNA during metabolism of the latter are studied; structural organization of the SSB protein complexes with DNA, as well as structural and functional peculiarities of different SSB proteins are analyzed.
KEY WORDS: SSB proteins, replication protein A, protein-DNA complexes, DNA replication, DNA repairDOI: 10.1134/S0006297908130026
Abbreviations: a.a., amino acid residues; dsDNA, double-stranded DNA; ecoSSB, Escherichia coli SSB; hsRPA, Homo sapiens RPA; NER, nucleotide excision repair; nt, nucleotide; OB domain, oligosaccharide/oligonucleotide-binding domain; PCNA, proliferating cell nuclear antigen; pol-prim, DNA polymerase α-primase complex; RFC, replication factor C; RPA, replication protein A; SSB, single-stranded DNA-binding protein; ssDNA, single-stranded DNA.
SSB proteins (single-stranded DNA-binding proteins) are among the most
widespread cellular proteins, this being true of proteins of eukaryotic
organisms as well as of eubacterial and archaeal proteins [1, 2]. SSB proteins are also
encoded by bacteriophage and adenovirus genomes [3,
4]. The main feature of this protein group is their
high nonspecific affinity to ssDNA (single-stranded DNA). Just this
peculiarity of SSB proteins and their participation in the main
cellular processes maintaining genome integrity as well as the
possibility of genetic information transfer define their role in the
life cycle of all living cells.
Enzymes and protein factors interact directly with single-stranded DNA at different stages of DNA replication, repair, and recombination. However, ssDNA molecules tend to form secondary structure that may interfere with the course of these processes. Also, ssDNA molecules can be attacked by intracellular nucleases. After binding to ssDNA, SSB proteins protect and stabilize ssDNA by protecting it from nuclease action and by keeping the DNA in its functionally active single-stranded state [1, 2]. The ssDNA binding by SSB proteins often results in formation of a special protein-DNA complex that is later recognized by protein factors and enzymes involved in DNA metabolism (for example, [5]). The structural organization of such a complex probably defines further events in this process. In this case, SSB proteins are able to stimulate both the activity of DNA polymerases, key enzymes of DNA replication and repair, and that of multiprotein complexes providing for strand exchange upon homologous recombination [1, 2, 6, 7].
This review deals both with the mechanisms used by SSB proteins for coordination of DNA replication and repair, and with characteristics of eukaryotic, prokaryotic, and archaeal SSB proteins, which made it possible to distinguish a number of general mechanisms of protein functioning in organisms of different life domains. In the final part of the review, mechanisms of SSB protein interactions with DNA during its metabolism are discussed, and structural organization of SSB protein-DNA complexes is analyzed along with associated structural peculiarities of different SSB proteins. Functions of replication protein A in DNA replication and repair are considered separately.
INVOLVEMENT OF SSB PROTEINS IN DNA METABOLISM
Transfer of genetic information and maintenance of its stability is a complex multistage mechanism. It is difficult to imagine that all diverse catalytic functions necessary for such processes are realized with involvement of a single multifunctional protein. In reality, they are carried out with participation of complex multifunctional ensembles. At the present time it is assumed that systems of DNA replication, repair, recombination, and transcription are based on interaction of functional protein ensembles. In this case, some enzymes and protein factors may be even involved in several of the above-mentioned processes [8, 9]. For example, the “enzymatic machinery” synthesizing DNA in the S phase of the cell cycle is actively used at the steps of the short region re-syntheses during DNA recombination and repair. The process of DNA repair is parallel to replication and transcription, because DNA structure, involved in these processes, is susceptible to damaging agents. Besides, the correction of a DNA lesion is easier right in the course of these processes. DNA recombination is used not only for exchange of genetic information during crossing-over in meiosis, but also as a repair mechanism in the cell cycle S phase associated with DNA replication. Thus, all processes under consideration closely interact with each other. Enzymes and protein factors participating in each of these steps can be considered as elements joining different steps of DNA processing. Undoubtedly, SSB proteins involved in such fundamental processes as DNA replication, repair, and recombination are candidates for this role.
SSB Proteins Are Key Participants of DNA Repair, Replication, and Recombination
It has been found by now that SSB proteins, like their eukaryotic homologs--replication proteins A (RPA), are necessary components of DNA replication, repair, and recombination systems. Upon binding to ssDNA, these proteins prevent formation of DNA secondary structure that makes difficult its replication, repair, and recombination. SSB proteins are known to increase the mean rate of DNA synthesis by bacterial and archaeal DNA polymerases [1, 6, 7]. Besides, in some cases the accuracy of DNA synthesis catalyzed by DNA polymerases also increases, as in the case of Thermus thermophilus DNA polymerase that in itself does not exhibit correcting activity [5, 10]. It is known that a number of SSB proteins are involved in direct protein-protein interactions with different factors and enzymes. Thus, Escherichia coli SSB (ecoSSB) interacts with appropriate DNA polymerase II, gp32 protein of T4 bacteriophage interacts with polymerase gp43, and gp2.5 of T7 bacteriophage similarly interacts with the corresponding DNA polymerase [3, 4, 6].
Eukaryotic RPA, in turn, also interact with appropriate DNA polymerases. Human replication protein A (hsRPA) is able to influence directly the activity of DNA polymerase δ. However, the mechanism of this interaction is complex, because its realization requires assembly of a multiprotein complex, including nuclear antigen of proliferating cells (PCNA) and replication factor C (RFC) [11, 12]. hsRPA stimulates activity of another replication polymerase, DNA polymerase α, by promoting its movement through “pausing” sites during DNA synthesis [13]. It was shown in in vitro experiments that, along with increased processivity, RPA enhances the accuracy of DNA synthesis [14]. It is supposed that in a pair with DNA polymerase α, hsRPA plays the same role as PCNA with DNA polymerase δ, i.e., these proteins “retain” the enzyme on DNA substrate [14]. However, stimulation may be replaced by inhibition. Thus, in the case of hsRPA excess there emerges competition between this protein and DNA polymerase for binding to DNA substrate, which results in polymerase displacement from DNA [14].
There are also examples of the archaeal SSB protein specific interactions with appropriate DNA polymerases. Thus, polymerase PolBI from Methanosarcina acetivorans, like its functional analog eukaryotic DNA polymerase α, is not highly processive. However, it is known that processivity and the rate of DNA synthesis, catalyzed both by this polymerase and by the analogous polymerase from Pyrococcus furiosus, increase in the presence of appropriate archean analogs of PCNA and RFC, as well as of any one of three homologs of M. acetivorans RPA [15, 16]. An opposite situation is characteristic of other archaeal SSB proteins like those of Methanothermobacter thermoautotrophicus RPA (mthRPA) and Methanopyrus kandleri RPA (mkaRPA). SSB proteins of these organisms in a broad interval of concentrations are able to inhibit activity of corresponding DNA polymerases mthPolBI and mkaPolBI [17, 18]. It is interesting that the inhibition is highly specific and is probably not associated with the DNA substrate blocking by SSB protein, because bacterial ecoSSB is not able to exhibit such effect on mthPolBI and mkaPolBI [17, 18]. It should be noted that direct interactions between mthPolBI and mthRPA can be revealed by immune precipitation and combined centrifugation in density gradient [18].
Working mechanisms of enzymic machineries of DNA replication and repair. At the present time it is possible to consider two different models of structures providing for reading and correction of information incorporated in cellular DNA: a model of formed in advance protein ensembles - “superenzyme” and a model of “dynamic” complexes with DNA (the concept of “DNA substrate transfer” from one protein or protein complex to another) [19].
The first model suggests assembly of large protein complexes that join practically all participants of enzymatic transformations of DNA. According to this model, the assembly is followed by the translocation of this complex along processed DNA (or on the contrary, by DNA movement through this complex) with successive realization of all catalytic stages by a multiprotein “superenzyme”. The “replisome concept”, based on results of studying prokaryotic replication system and transferring them to that of eukaryotes, can be considered in the frames of this model [9, 20, 21].
The second model suggests dynamic binding to DNA and following association of just some protein factors whose activity is required at a given concrete stage of DNA processing (see for example [22, 23]). In this case, during the remaining time protein factors are outside such complex in the cell nucleus. The main distinction between this model and the former one is the smaller size of the multiprotein enzyme complex and a special role of weak interactions that enable dynamic formation and changing the body of process participants. The dynamism specific to this model offers great possibilities for process regulation. It is supposed that this model can be the basis of the mechanism of nucleotide excision repair and homologous recombination [22, 23].
In the frames of both models, special attention is paid to such protein peculiarities as the presence of several binding centers able to interact with different ligands like DNA or protein. These properties have to provide for formation of a diverse set of intricate protein-nucleic acid complexes. The competition between two ligand molecules for the same binding center enables transformation of one type complex to another and substrate transfer from one protein to another (no mater whether it is DNA substrate or preformed protein-nucleic complex) [19].
Multicenter interactions between SSB proteins and ligands. Proteins involved in events associated with DNA processing are usually characterized by module structure. Several domains of such proteins carry out different biochemical functions. These structure-functional regions are joined with each other by mobile linker regions. On one side, each domain is highly specialized for optimal realization of a concrete biochemical function, on the other side, combination of different domains in one protein makes possible mutual coordination of these functions. Modular structure of proteins defines functional specificity of multiprotein complexes, because several contacts are at once formed in them between any pair of participants [20, 22, 23]. However, stages of enzymic transformation are carried out without protein dissociation from the complex, but with replacement of one bond in the complex by others, which is accompanied by structural rearrangement of the complex [22]. Such complexes are characterized by variation of association constant of each individual protein in these complexes in broad intervals up to 1010 M-1 depending on the amount of contacts formed between this protein and its partners [19]. On the other side, the fact that components of the complex interact at once via several binding centers provides for several possible ways for regulation of these interactions.
The work of a replication multiprotein complex consisting of DNA polymerase δ, RFC, and replication protein A can be considered as an example. In the course of replication, RFC interacts with RPA by three of its five subunits [11]. It is assumed that two RFC binding sites are located on the surface of RPA, and one of these sites is overlapped with a binding site of DNA polymerase δ. During switching the replicating machinery from DNA synthesis with involvement of DNA polymerase α to synthesis by DNA polymerase δ, the latter competes with RFC for binding to one of two sites on the RPA surface [11]. However, the second binding site allows RFC to remain in the multiprotein replication complex and this is extremely important for work of the replicating machinery.
The interaction of RPA with ssDNA is another example. The dissociation constant of the complex of each of two main DNA-binding domains of p70 subunit (p70A and p70B) with DNA is in the micromolar region, which is indicative of not high affinity to ssDNA [24]. However, total affinity of the tandem of p70AB domains to DNA exceeds that of individual protein domains by three orders of magnitude and dissociation constant of the p70AB-ssDNA complex is already at the nanomolar interval [24]. The significant difference between these constants is explained by the so-called “linker effect” caused by mobile joining of two neighboring DNA-binding domains via a linker region of 10 amino acid residues.
Versatility of SSB protein domains. A peculiarity of proteins involved in DNA processing is the versatility of domains constituting these proteins. Within SSB proteins, OB-domains (oligosaccharide/oligonucleotide binding domain) should be considered as potentially universal ones. According to the database of the SCOP protein structural classification [25], OB-domains can be involved in binding both to nucleic acids and oligosaccharide fragments, and recently the set of functions of this type of domains was also supplemented with peptide-binding activity [25]. In addition to the above-mentioned DNA replication, repair, and recombination, OB-domains are directly involved in transcription, translation, cell response to low temperatures, and telomere maintenance [26, 27]. OB-domains appear in such different proteins as nucleases, pyrophosphatases, and ssDNA- and RNA-binding proteins [25].
Functional analysis of the SSB protein OB-domains shows that the latter can be universal even in the simplest eubacterial SSB, where these domains are responsible for protein binding to DNA and tetramer formation [28, 29]. The versatility of OB-domains is especially pronounced in more complex eukaryotic SSB proteins. Four human RPA domains (p70A, p70B, p70C, and p32D) out of six OB-domains are involved in high-affinity DNA binding [1, 30]. At the same time, p70C, p32D, and p14 subunit, also representing OB-domain, are involved in formation of heterotrimeric RPA complex [31]. A number of experimental data [32-34] show that the high-affinity DNA-binding domains p70A and p70B are able to interact with protein partners of RPA. The latter include T antigen of SV40 virus, repair and recombination factors XPA and Rad51, etc. N-Terminal OB-domain p70NTD of p70 subunit is involved exclusively in protein-protein interactions [30]. Such proteins as p53, XPA, and transcription factors Ga14 and VP16 are among its partners [35-37].
It is interesting to note that within eukaryotic RPA not only OB-domains are potentially universal by their functional abilities. The unique RPA domain not belonging to OB-domains, namely, p32CTD is attributed to the wHTH domain family (“helix-turn-helix”). In hsRPA this domain is involved only in protein-protein interactions [30, 38], but related domains of the same family are among transcription factors and helicase RecQ of E. coli, causing interaction of these proteins with dsDNA [38, 39].
Ligand competition for binding to SSB proteins. Eukaryotic RPA is characterized by the presence of overlapping binding sites between its partners in DNA metabolism [11, 38, 40]. The competition between the DNA polymerase α-primase complex (pol-prim), RFC, and DNA polymerase δ for binding sites on RPA can be considered in eukaryotic replication as competitive interactions with involvement of SSB proteins. Evidently, each of these three proteins contains overlapping sites of interaction with RPA, which provides for transfer of RPA-bound DNA substrate upon transition from the stage of DNA priming to the stage of primer elongation [11]. The interaction of one and the same site in C-terminal domain of p32 subunit, p32CTD, with three different proteins of the repair process, uracil-DNA-glycosylase (UNG2) involved in base excision repair; nucleotide excision repair factor XPA, and recombination protein Rad52 can be considered as another example concerning RPA [38]. Competition of these proteins for binding to RPA can be used in transfer of the RPA complex with DNA substrate from one repair protein to another. Such a mechanism may coordinate between processes of base excision repair, nucleotide excision repair, and DNA recombination. Recent studies using nuclear magnetic resonance technique show that XPA and Rad51 proteins bind in the canonical region of one of the main DNA-binding RPA domains--p70A [32, 41]. Besides, these experiments have shown that the presence of ssDNA destroys the complex formed with N-terminal domain of recombination protein Rad51 (Rad51N). This can also be indicative of overlapping of binding regions of the above-mentioned ligands [41]. It is quite possible that in this case competition between DNA and Rad51N for binding to RPA is responsible for DNA transfer from RPA to other participants of initial stages of homologous recombination.
SSB PROTEIN STRUCTURES
Proteins capable of high affinity binding to ssDNA and in this case demonstrating much lower affinity to other DNA forms are usually considered as SSB proteins. It is assumed that at least one of the conditions mentioned below should be also observed for the protein: (i) the protein-encoding gene should be necessary for DNA replication; (ii) the protein should stimulate activity of replicating DNA polymerase; (iii) the protein should directly interact with different replication factors and enzymes; (iv) the protein should bind stoichiometric amounts of DNA [6]. It is not surprising that at such rather broad criteria of selection presently known SSB proteins have quite different structures. They can be divided to three groups in accordance with their subunit structure. The first group should include homotetrameric proteins, in which all subunits (protomers) are encoded by the same gene. The next group is represented by homodimeric proteins. Finally, the third group includes the most complexly organized heterotrimers of three different subunits.
Besides distinctions in subunit structure, different groups of SSB proteins also have different amino acid sequences [26, 42, 43], which makes difficult searching for homologs in already known genomes. On the other hand, intensive investigation of tertiary and quaternary structures by X-ray analysis and nuclear magnetic resonance makes it possible to distinguish common structural and functional features in such a diverse group of proteins. Thus, the structural motif called OB-domain or OB-fold (oligosaccharide/oligonucleotide binding fold) is the structural basis of all presently known SSB proteins [26].
Homotetrameric SSB Proteins
Most protomers of eubacterial SSB contain a single OB-fold, but active forms of these proteins exist as homotetramers. Only three of 250 bacterial genomes do not obey the rule “one ssb gene - one OB-domain” [44-47]. The basis of eubacterial SSB promoters is N-terminal DNA-binding domain, just which contains the OB-fold. The C-terminal region of this SSB protein group has a rather labile structure [47]. Within this region there are conserved negatively charged amino acid residues specific of all eubacterial SSB.
The best-studied member of this class of homotetrameric proteins is ecoSSB. This protein, necessary for cell vital activity, is involved in such important processes as DNA replication, repair, and recombination. The protein monomer has molecular mass approximately 19 kD and consists of 177 amino acid residues, 120 of which fall into the DNA-binding OB-domain [48]. In addition to DNA-binding activity, this domain is involved in formation of homotetrameric structure and interaction with OB-domains of other protomers. The functional form of ecoSSB is a complex of four functionally equivalent protomers. Such a “functionally symmetrical” homotetramer is able to form with DNA several different type complexes characterized by different stability and different size of bound DNA, as well as by cooperativity of protein binding [2].
Mitochondrial SSB proteins structurally resemble eubacterial SSB. It is interesting to note that, for example, the coincidence of amino acid sequences of human mitochondrial SSB protein and ecoSSB makes up only 36%, but X-ray analysis reveals practically identical protomer structure and mechanism of homotetramer formation for these proteins [49, 50]. The proteins have also similar biochemical characteristics [49, 51].
Homodimeric SSB Proteins
The next group of SSB proteins are homodimers. The first subgroup of this SSB class should include three members of eubacterial domain that are an exclusion from homotetrameric eubacterial SSB. These are proteins tthSSB, taqSSB, and draSSB isolated from extreme thermophiles Thermus thermophilus, Thermus aquaticus, and Deinococcus radiodurans, respectively. Protomers of this protein subgroup have large dimensions that are not characteristic of other eubacterial SSB. The length of amino acid sequences is 263, 264, and 301 a.a. for tthSSB, taqSSB, and draSSB, respectively, while molecular mass of the protomer is 29.9, 30.0, and 32.6 kD [52-54]. Analysis of amino acid sequences of these proteins suggests the existence of at least two OB-folds within each protomer. These two OB-domains are joined to each other by a linker having a very conserved secondary structure that suggests β-fold formation [52, 53, 55]. Thus, although SSB under consideration have homodimeric structure, they, like the other eubacterial SSB, include four OB-domains.
It is interesting to note that the DNA-binding domains of each protomer have structurally similar elements of different amino acid sequence. Comparative modeling was used to find quite a number of amino acid residues in C-terminal OB-domain draSSB which correspond to residues from OB-fold of eubacterial ecoSSB involved in DNA binding [54, 55]. Simultaneously, an analogous three-dimensional model of N-terminal domain shows the absence of a number of important bonds specific of protein complexes with DNA. It is quite possible that such domain non-equivalence should influence the character of the complete SSB homodimer interaction with DNA, thus causing its difference from homotetrameric eubacterial SSB.
Another peculiarity of eubacterial SSB proteins under consideration is the presence in the functional homodimeric protein of only two conserved C-terminal unstructured regions characteristic of all eubacterial proteins [55]. Since similar regions in eubacterial SSB are responsible for interaction with protein partners, one can suppose that the character of protein-protein interactions of homodimeric eubacterial SSB also differs from those observed in homotetrameric analogs.
Another variant of homodimeric SSB is found in some organisms of the Euryarchaeota kingdom of domain Archaea. The individual protein monomer is structurally even more different from eubacterial protein monomers. Similarly to the first group, there are two OB-folds within the monomer, but unlike all eubacterial proteins, the protomer C-terminal part contains a structural element “zinc finger” instead of the negatively charged region of amino acid residues [17]. This element is not found in eubacterial SSB proteins but it is an obligatory component of eukaryotic ones. However, it should be noted that the “zinc finger” structure in archaeal proteins is somewhat different from that in eukaryotic SSB.
Bacteriophage SSB proteins such as gp32 of phage T4, gp2.5 of phage T7, and gpV of the fd phage (family 50315 of phage proteins that bind to ssDNA, according to database of the SCOP protein structural classification [25]) have the simplest domain structure among homodimeric SSB proteins. Molecular mass of protein monomer for these proteins is low, i.e. only 35 kD for gp32. As a rule, the bacteriophage SSB protomer is the unique DNA-binding OB-domain that can contain C-terminal region carrying negatively charged amino acid residues [28]. According to the SCOP database [25], there is also N-terminal LAST ((Lys/Arg)3(Ser/Thr)2) domain in the structure of gp32 protein of phage T4, which is responsible for cooperative interaction of several proteins with DNA. As a rule, phage proteins exist in solution in the form of dimers, and the process of further oligomerization is carried out at very high protein concentrations. So, only two OB-domains can be distinguished within functional homodimers of phage proteins.
Heterotrimeric SSB Proteins, Replication Protein A
The third group of SSB proteins includes heterotrimeric complexes consisting of three different subunits. Such proteins appear in all eukaryotic cells - from yeasts to humans - and are the main eukaryotic SSB proteins. Members of this group of SSB proteins are designated by the name of the first discovered member as replication proteins A or RPA [1]. All RPA homologs consist of three different polypeptides whose relationship with each other and with other SSB proteins follows from the fact that each subunit carries at least one OB-domain.
Human replication protein A is the best-studied member of the eukaryotic protein subgroup [1]. Like prokaryotic SSB, RPA plays the key role in DNA replication, repair, and recombination. Structurally, hsRPA is a stable heterotrimer consisting of three subunits with molecular mass values of 70, 32, and 14 kD. Seven domains are distinguished within RPA structure and six of them are represented by OB-folds. Unlike eubacterial proteins, all OB-domains of hsRPA differ from each other both structurally and functionally [30].
The RPA large subunit p70 contains four OB-domains [1]. The N-terminal domain (p70NTD) is separated from the rest of the subunit by a rather extended linker and is considered as one of two RPA domains responsible for protein-protein interactions. The tandem of two DNA-binding domains p70A and p70B, characterized by high DNA-binding activity, is located in the central fragment of the same subunit [56]. Another DNA-binding domain p70C is located in the C-terminal part, and apart from everything else, it is involved in formation of RPA heterotrimer [31]. The “zinc finger” or “zinc ribbon” fragment can be also distinguished in the same domain structure. As already mentioned, this fragment is conserved for all RPA homologs and is also specific of some homodimeric SSB of Euryarchaeota [57, 58]. The intermediate RPA subunit p32 carries an additional DNA-binding domain that is an OB-fold (p32D) [59]. Besides, the C-terminal part of the subunit contains the p32CTD domain responsible for protein-protein interactions and belonging to the family of wHTH domains (winged helix-turn-helix) [38]. Non-structured N-terminal site p32N of the p32 subunit serves as a target for phosphorylation by cellular kinases responsible for the cell cycle and involved in the cell reaction to DNA lesions [7]. The third protein subunit p14 has a single domain, and what is more, it is based on the OB-fold [60].
Although six OB-domains can be counted in the eukaryotic RPA structures, which distinguish these RPA from the earlier described homodimeric and homotetrameric SSB, most researchers still assume that only four of them (p70A, p70B, p70C, and p32D) can be directly involved in DNA binding.
SSB Proteins of Different Subunit Structure
Some presently known SSB proteins do not fit the above-mentioned classification. Thus, proteins from Methanococcus jannaschii and Methanobacterium thermoautotrophicus contain, respectively, four and five OB-domains and a “zinc finger” domain, which distinguishes them from all other SSB proteins [42, 61]. The archaean organism Methanopyrus kandleri differs by the presence of only a single gene encoding a DNA-binding protein, but the protein itself is neither a homotetramer nor a homodimer like in the case of SSB, but it is a homotrimer [17]. The considered members very much resemble eukaryotic or other euryarchaean proteins by the presence of “zinc finger”, but their monomer structures are different.
On the contrary, SSB protein of Sulfolobus solfataricus (Archaea domain, Crenarchaeota kingdom) could be rather attributed by its domain structure to eubacterial homotetrameric proteins, since its OB-domain is flanked by a C-terminal negatively charged region with conserved aspartate residues [62]. However, biochemical investigations show that this protein does not form a multisubunit complex but functions as a monomer [63].
PECULIARITIES OF SSB PROTEIN DOMAIN STRUCTURES AND MECHANISM OF
THEIR INTERACTION WITH DNA
OB-Domain Is Basis of SSB Protein Structural Organization
Results of investigation of the structure formed by OB-domains with DNA and RNA within different protein-nucleic acid complexes indicate that despite the low level of similarity between amino acid sequences of different domains of this family, a number of topological structural peculiarities common for all OB-folds can be distinguished [27].
Individual OB-domains are not big and fit in the interval from 70 to 150 amino acid residues [26]. Dimensional distinctions between OB-domains are first of all caused by the size of variable loops located between rather conserved elements of β-sheets. Topologically, the polypeptide chain of OB-fold (Fig. 1) forms two antiparallel β-sheets with three polypeptide chain regions in each, and one of these β-chains (number 1) is common for both sheets [26]. Resulting β-sheets are located orthogonally to each other and form β-cylinder with chain arrangement topology as β1-β2-β3-β5-β4-β1. Usually the canonical surface of the OB-fold interaction with ligands is located in the region of chains β2 and β3 (Fig. 2). Loops between β1 and β2 (loop L12), β3 and α (loop L3a), α and β4 (loop La4), and finally, between β4 and β5 (loop L45) can be additionally involved in interaction. These loops form a groove over the domain surface that is perpendicular to the axis of topological β-cylinder. Most of oligonucleotide ligands bind just in this groove at right angle to β-sheets. In this case, “polarity”, i.e. the ligand binding orientation, can be different. In the case of oligonucleotide binding from 5′ end towards 3′ end by the polypeptide chain sites β4 and β5 and further towards β2, it is considered as the “direct polarity” of ligand binding.
Fig. 1. OB-domain canonical structure is shown on the example of tertiary structure of N-terminal domain (8-109 a. a.) of p70 subunit of human replication protein A (registered under 1EWI in the RCSB Protein Data Bank database). Elements of the domain secondary structure are also designated in the figure: β chains β1, β2, β3, β3′, and β1, β4, β5 that form, respectively, two β-sheets. Spatial position of the polypeptide chain loops L12, L3a, La4, and L45 is shown.
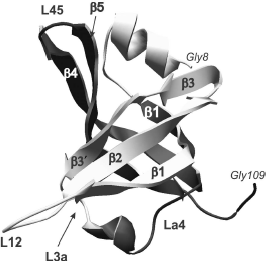
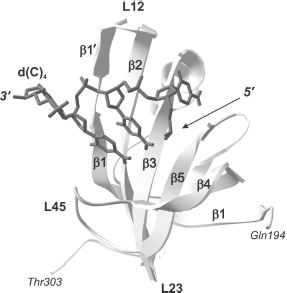
As a whole, according to the data of X-ray and NMR analyses, the above-mentioned loops L12, L3a, La4, and L45 represent a very efficient surface for recognition and binding of single-stranded nucleic acids [27]. Amino acid residues forming these sites in different proteins are also involved both in aromatic “stacking”-interactions, in hydrogen bonding with nucleic acid molecules, and in polar interactions [56].Fig. 2. “Direct polarity” of nucleic acid binding on the example of tertiary structure of a complex of the p70A OB-domain (194-303 a.a.) of hsRPA with oligonucleotide residue d(C)4 (shown in dark gray) [64]. Elements of the domain secondary structure are also designated in the figure: β-chains β1, β1′, β2, β3, β4, and β5, the polypeptide chain loops L12, L23, and L45.
Versatility of OB-domain functions. As follows from the SCOP database [25], proteins having domains formed with involvement of OB-folds are present in 10 superfamilies carrying out appropriate functions. Proteins that bind nucleic acids comprise the main part of proteins incorporating OB-domains. An additional 12 families differing from each other by the domain type can be also distinguished within this superfamily [25]. These proteins can be divided to three groups according to their functions: (i) nucleotide-binding proteins without pronounced specificity towards base sequence (this group includes hsRPA and ecoSSB that are interesting for us); (ii) proteins recognizing specific sites in single-stranded DNA (transcription terminator Rho of E. coli, Cdc13 of Saccharomyces cerevisiae, the telomere end binding protein TEBP, and aspartyl-tRNA synthase); (iii) proteins binding to non-helical structured nucleic acids (ribosomal proteins S12, S17, and initiation factor IF1 of T. thermophilus).
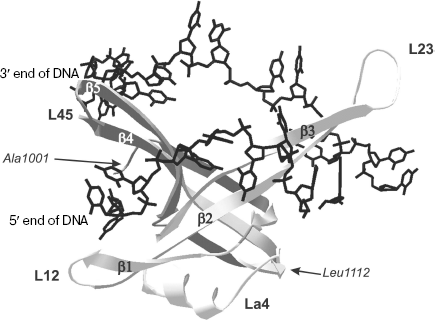
Structural peculiarities of OB-domains involved in nucleic acid binding. The three-dimensional structure of the N-terminal 135 a.a. ecoSSB fragment is now known both in free state and in complex with ssDNA of 35 nt [65, 66]. It should be remembered that just this fragment carries in its structure the DNA-binding OB-domain. In DNA complex with homotetramer ecoSSB, each monomeric subunit forms multiple contacts with DNA [65]. The vast surface of interaction in complexes differs from the “canonical” one observed in other OB-containing proteins and includes both sides of extended β-sheet in the L23 region (Fig. 3). Data of X-ray analysis also indicate that the ligand “binding polarity” for ecoSSB monomeric subunits differs from other proteins and is called “reverse polarity”.
The family of nucleotide-binding OB-domains is characterized by the absence of pronounced similarity of amino acid sequences. At the same time, the above-mentioned domains are characterized by a high extent of structural homology. Structural analysis of 14 OB-folds [27] has revealed structural determinants of nucleotide-binding OB-folds. It was shown that about 30 amino acid residues occupy structurally conserved positions, and in this case the level of the root-mean-square deviation (RMSD) of atom displacements does not exceed 2.1 Å for all structures. Nevertheless, only 12% identity of amino acid sequences was determined in such canonical elements of the OB-fold secondary structures as β-chains and α-helix [27].Fig. 3. Tertiary structure of OB-domain (1001-1112 a.a.) of ecoSSB protomer bound to 23-base oligonucleotide (shown in black) [66]. Secondary structure elements of the domain are also designated in the figure: β-chains β1, β2, β3, β4, and β5; polypeptide chain loops L12, L23, La4, and L45.
Mechanism of OB-domain interactions with DNA. Analysis of three-dimensional structures obtained for a number of OB-containing protein complexes with DNA and RNA shows that most often the protein interacts with the nucleic acid nitrogen bases, whereas its phosphate groups are unfolded in direction “from the protein” [64, 67]. Probably such interactions as a whole are specific of proteins, binding to single-stranded RNA/DNA or to complex nucleic acid structures, formed with emergence of loops. An important role in contacts with protein belongs to stacking interactions between nucleotide residues and aromatic amino acid residues of the protein, as well as to non-polar interactions of ribose or nitrogen base rings with hydrophobic side radicals of amino acid residues, and in some cases with the aliphatic part of the lysine and arginine side radicals [27, 64, 67]. In some OB-containing proteins, like the telomere end-binding protein TEBP and initiation factor IF1, such unusual interactions are revealed as stacking-interactions between the nitrogen base π-orbitals and the charged side radical of arginines. When binding should be carried out with certain nucleic acid sequences, the recognition can also involve polar side radicals capable of hydrogen bonding only with definite nitrogen bases.
Murzin [26] carried out the thorough analysis of three-dimensional structures of five complexes and identified the canonical surface of OB-domain interactions with ligands. The ligand is located between chains β2 and β3, just with which main bonds are established; besides, contacts are made with C-terminal sites of β1 and β5 chains. In some cases, in addition to these canonical contacts, those with other domain regions can be established, which significantly expands the surface of interaction with the ligand. The ligand for proteins binding single-stranded nucleic acids is located in the OB-domain left part, in immediate vicinity of loops L23, L12, L3a, and L45, and it appears to be “pressed down” to the surface of a sheet formed by chains β2 and β3 [26]. The β1 chain N-terminal part (to its bend) and N-terminal part of β4 are not involved in binding and, most likely, their role is only keeping the integrity of the whole domain tertiary structure.
As already stressed above, loops L12, L3a (loop L34 in the absence of α-helix), and L45 play a special role in modulation of OB-domain interactions with random ligands. The loop L23 also plays an important role in the OB-domain binding to nucleic acids [27]. Its dimensions significantly vary from 7 a.a. within E. coli Rho to 28 and 18 a.a. in cdc13 and ecoSSB, respectively; these conclusions are based on information on three-dimensional structures 2A8V, 1KXL, and 1EYG obtained from the PDB database [68]. It should be noted that the large size of just this loop significantly enlarges the area of contact between protein and DNA in the case of ecoSSB [66]. This is also true of other loops--their dimensions in various proteins are very different--and such variability may serve as a good basis for formation of quite different surfaces for ligand binding. Owing to this, proteins based on the OB-domain can have very different dimensions of the nucleic acid binding site, and this correlates with the loop size reaching 31 nt for ribosomal protein S17 of T. thermophilus [69]. In most nucleic acid-binding proteins, the size of the OB-domain binding site is within the interval from 2 to 11 nt [27].
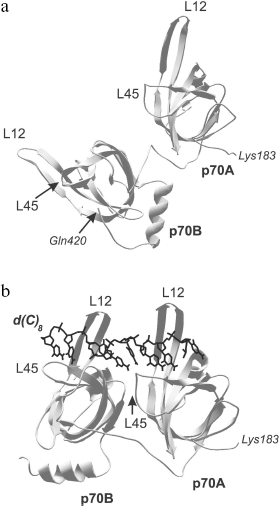
Comparison of three-dimensional structures of several proteins complexed with nucleic acid and in free state shows that, as a rule, OB-domain and/or ligand change their conformations upon binding [56, 65, 66, 70, 71]. The changes first of all touch upon positions of the loops and the OB-fold proper, but conformational rearrangement of other protein domains can also be observed along with the nitrogen base turning inside out and changing spatial structure of the nucleic acid. These alterations can be both separate and combined. They can be of low scale but result in acquirement of specific conformation by the protein or ligand. For example, only slight structural alterations are observed in OB-domains upon binding to DNA of the tandem of DNA-binding domains p70A and p70B of replication protein A (RMSD upon superposition of three-dimensional structures of free and DNA-bound OB-domains does not exceed 1 Å) [56, 65]. These slight alterations practically touch upon just positions of loops L12 and L45. However, reorientation of domains relative each other and their “ordering” on the DNA strand take place within the DNA-protein complex (Fig. 4). The domain tandem forms a channel in which DNA strand is stacked [56]. The tighter DNA clasping by protein and extension both of DNA strand and the tandem of DNA-binding domains along it are observed within such structure.
Presently known three-dimensional structures of OB-domain complexes with nucleic acids show that two different ligand orientations about this domain are possible [26, 27]. As already mentioned above, the variant in which the nucleic acid 5′-end is close to β4 and β5 chains and its 3′-end is near β2 is assumed as “normal orientation”. Most complexes with known three-dimensional structure are characterized by just this orientation. The exception is DNA complexes with ecoSSB proteins and β subunit of the telomere-binding protein TEBP, in which “polarity” of DNA arrangement differs from the above-mentioned one [66, 72].Fig. 4. Three-dimensional structure of the tandem of DNA-binding domains p70A and p70B (183-420 a.a.) of hsRPA in the absence of DNA (a) as well as in the complex with deoxyoctacytidine (shown in black) (b) [70]. Polypeptide chain loops L12 and L45 of each domain participating in interaction with oligonucleotide ligand are designated in the figure.
Other Domains of SSB Proteins
C-Terminal part of eubacterial SSB. As already said above, two parts, N-terminal DNA-binding OB-domain and C-terminal part, are distinguished in the eubacterial SSB protomer structures. Unlike OB-domain, the C-terminal part is considerably less structured, and attempts to resolve its three-dimensional structure by X-ray and NMR analyses are still not successful [73]. However, this region is important for protein functions and carries 10 negatively charged amino acid residues (conserved for eubacterial SSB) including four aspartates. Although the C-terminal region is necessary for cell survival in vivo, it is not involved either in DNA binding or in the homotetrameric structure formation [28] and is, probably, an intermediary in SSB interaction with different proteins.
“Zinc finger” of eukaryotic and euryarchaeal RPA. Metal ions as cofactors often promote stabilization of the protein three-dimensional structure and a certain folding of polypeptide chain, thus playing an important structural role [74]. Zinc ions are widely represented in structural elements called “zinc finger” and “zinc ribbon”. The size of such tertiary structure elements varies from 30 to 100 a.a. [75]. The functional role of these domains consists in binding nucleic acids, proteins and peptides, or small ligands. The “zinc finger”-containing proteins are involved in DNA metabolism, transcription, translation, and metal metabolism. The occurring “zinc finger” fragments can be divided into at least eight groups in which histidine and cysteine residues are invariant amino acid residues creating the coordination sphere for zinc ions [76]. The ion, coordinated in such a way, stimulates the formation of tertiary structure by the fragment with ββα topology, which is not formed in the absence of metal ions [76]. Filling the ion coordination sphere with side radicals of amino acid residues makes impossible reactions catalyzed with the participation of this ion [77].
“Zinc finger” in SSB proteins is conserved for members of eukaryotes and Euryarchaeota. The fragment structure in these two protein groups is different. In eukaryotic proteins, the conserved “zinc finger” fragment occurs in the C-terminal part of the large protein subunit (p70 in hsRPA, Fig. 5). As far as primary structure is concerned, the eukaryotic fragment belongs to the X3CX2-4CX12-15CX2C family (where C is cysteine residues and X is any other amino acid residue) [78]. This fragment is located inside the C-terminal domain in the region of one of the variable loops of the OB-fold. A fragment with similar primary structure was also found in RPA homologs from M. jannaschii and M. thermoautotrophicus--CX2CX12CX2C and CX2CX11CX2C, although these proteins differ from eukaryotic proteins in subunit organization [18, 42]. The “zinc finger” fragment in Euryarchaeota has somewhat different amino acid sequence. It includes one histidine residue--CX2CX8-14CX2H (where C are cysteine residues, H is histidine, and X means any other amino acid residue) [15]. In proteins having heterotrimeric structure “zinc finger” is located, as in eukaryotic RPA, in the C-terminal part of the large subunit (for example, RPA of P. furiosus [79]). In homodimeric proteins carrying two OB-domains in each subunit, “zinc finger” can be found in their C-terminal part. A specific peculiarity of homodimeric proteins of genera Methanosarcinales, Methanopyrales, and Ferroplasmatales is the strict conformity of the “zinc finger” to the CX2CX8CX2H structure, i.e. strictly constant number of random amino acids between cysteine and histidine residues [17].
Although the three-dimensional structure of the “zinc finger” including domain is described only for RPA from human cells [31], it is shown for several euryarchaeal proteins that the amino acid fragment CX2CX8-14CX2H really binds zinc ion. This follows from results of biochemical investigations using mutant proteins [57]. Besides, analysis of circular dichroism spectra of these euryarchaeal RPA has shown that formation of a certain secondary structure takes place in the presence of zinc ion within the protein [57].Fig. 5. Three-dimensional structure of p70C domain (439-616 a.a.) of hsRPA [31]. The “zinc finger” is designated by the dashed line. Positions of cysteine residues, forming the fragment, and zinc ion within the latter (shown in black) are designated. Loops L12 and L45 are also marked in the domain polypeptide chain.
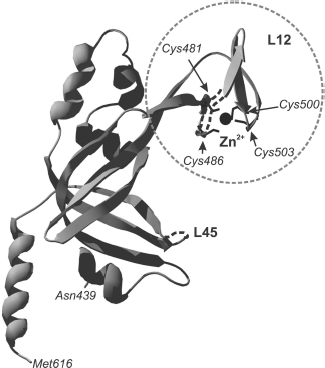
Despite this and the fact that “zinc finger” in eukaryotic proteins is located inside one of the DNA-binding domains, experimental data show that it is not necessary for protein binding to DNA. Some authors only suppose its potential involvement in binding to nucleic acids [78, 80], because the presence of zinc ions in buffer, as well as the presence in solution of reagents, stimulating keeping cysteine residues in the reduced state, slightly increase affinity to DNA. It is interesting that in such euryarchaeal proteins, like mac3RPA from M. acetivorans, “zinc finger” does not play a key role in binding to DNA because dissociation constant of the protein form carrying mutations in this fragment insignificantly differs from that for the wild-type protein [17, 57].
Interaction of SSB Proteins with DNA
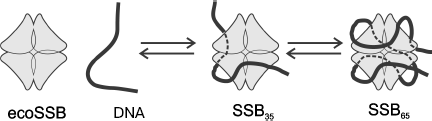
Homotetrameric ecoSSB and heterotrimeric hsRPA are the best-studied objects for investigation of biochemical properties of eubacterial and eukaryotic SSB proteins. Several variants of binding to single-stranded DNA have been shown for both proteins. The most pronounced distinction of these binding types concerns the size of the DNA site interacting with these proteins, which depends on ionic strength of solution, NaCl and divalent cation concentrations, protein concentration, pH, and temperature [30, 81]. Two types of complexes with ssDNA are known and rather well studied for ecoSSB, SSB35 with the binding site of 33-35 nt and SSB65 in which 65 nt are bound. As already mentioned, ecoSSB contains four equivalent DNA-binding OB-domains, one in each subunit. Biochemical investigations and X-ray analysis have shown that the type of SSB35 binding is caused by the contact of two of these DNA-binding domains with DNA (Fig. 6). In complexes formed by the SSB65 type 65 nt of DNA are bound to all four protomers, and it is supposed that in this case a strong alteration of DNA tertiary structure caused by its bending occurs [66, 82]. The C-terminal regions of protomers are not involved in binding, and conserved amino acid residues localized in these regions carry negative charge. Three-dimensional structures of SSB in complex with DNA obtained by X-ray analysis show that the C-terminal protomer regions in such complexes remain non-structured [73].
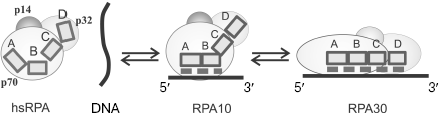
The RPA eukaryotic homolog also forms with ssDNA complexes of various architectures, which differ by the size of the binding site. The best studied are hsRPA complexes with ssDNA in which 8-10 and 30 nt of ssDNA are bound (these complexes are designated as RPA10 and RPA30, respectively). Although no crystals of complete heterotrimeric hsRPA complex have been obtained, three-dimensional structures of all DNA-binding domains in hsRPA have been obtained by X-ray analysis [31, 70], and the structure of the complex consisting of a tandem of p70A and p70B domains with single-stranded 8-nt DNA fragment was resolved [56]. This made it possible to put forward a hypothesis concerning the mechanism of formation of different types of complexes [31].Fig. 6. A hypothetical model of the SSB35 and SSB65 type complex formation upon ecoSSB interaction with ssDNA [2].
It is supposed that several type complexes, successively transformed into each other, are formed in the case of RPA binding to DNA. First, a less stable complex of the RPA10 type is formed in which DNA-binding domains p70A and p70B appear to be bound to 8-10 nt of DNA strand. The possibility of such complex formation was confirmed by X-ray analysis of the tandem of DNA-binding domains p70A and p70B with deoxyoctacytidine [56]. According to the model of the complex three-dimensional structure, 3 nt of ssDNA are in immediate contact with each OB-domain, and another 2 nt are located between protein domains. Thus, 8 nt shielded by the tandem of p70A and p70B domains define the size of the RPA binding site on ssDNA for the RPA10 complexes. If ssDNA is longer than 8-10 nt, then the following alteration of protein conformation, its extension along DNA strand is possible, which results in emergence of a contact of two other DNA-binding domains, p70C and p32D, with ssDNA [83]. It is assumed that this is the way of formation of the RPA17 and RPA30 type complexes with the different size protein-binding site on DNA, 17 and 30 nt, respectively [70, 84]. It was shown by affinity modification and proteolysis that the architecture of protein-DNA interaction in complexes of these types is significantly different [85, 86], which is revealed in their different stability [30]. Complexes whose formation followed the RPA10 binding scheme are the least stable, while those formed using the RPA30 binding scheme are the most stable.
Data on RPA affinity modification in the complex with ssDNA are indicative of specific orientation of eukaryotic SSB protein subunits on single-stranded DNA. The p70 subunit more efficiently interacts with 5′ region of ssDNA, while p32 is more efficient towards the 3′ region. In complexes formed following the RPA30 type, “polar” arrangement of RPA on ssDNA is caused by binding the subunit p32 domain p32D near the 3′ end of ssDNA, while domains p70A, p70B, and p70C of p70 subunit bind preferably near the 5′ end of DNA [21, 83, 84, 87, 88].
As shown by electron microscopy, no protein molecule “entwining” by DNA, like that observed for prokaryotic ecoSSB complexes formed similarly to SSB65, takes place in RPA complexes containing the maximal DNA-binding site (RPA30 type) [89-91]. Thus, mechanisms of formation of eukaryotic and prokaryotic protein complexes have their own peculiarities. There is no main pronounced DNA-binding center in ecoSSB, because the protein is composed of equivalent protomers. Moreover, according to data of X-ray analysis, the tetramer molecule is characterized by space symmetry and interactions between any two ecoSSB protomers, each carrying one DNA-binding domain, are equivalent [65]. In the eukaryotic hsRPA heterotrimer, all subunit pairs make contacts with each other, but analysis of three-dimensional structure of a complex of the trimer-forming subunit regions shows that intersubunit bonds are nonequivalent [31]. Besides, in hsRPA the main DNA-binding activity is located in the large p70 subunit that is able to change its own conformation rather easily. Data in favor of conformational mobility of this RPA subunit were obtained by limited proteolysis [85, 86]. Thus, it can be supposed that the eukaryotic protein has a higher conformational mobility compared to the prokaryotic one. The protein globular conformation changes to the extended one as all DNA-binding domains in hsRPA become bound to DNA [84, 89, 90], i.e. the “extension” of hsRPA molecule along DNA strand takes place (Fig. 7). However, this is accompanied by minimal conformational alterations in the DNA molecule.
The model of one binding type transition to another can be also used to explain the dependence of the formed RPA-DNA complex type on both the size of available ssDNA platform and the ratio of RPA and DNA concentrations [84]. To realize the RPA30 binding type, in which ssDNA is bound by all DNA-binding domains, it is necessary to observe some conditions. First, the size of continuous single-stranded region has to allow successive folding of all DNA-binding protein domains on DNA; second, binding conditions have to promote binding of only a single RPA molecule to this continuous single-stranded region. With allowance made for not high cooperativity upon RPA binding by the type of RPA30 [92], this variant is realized when protein concentration is lower than that of ssDNA. If RPA concentration exceeds the DNA concentration, there dominates the probability that binding will follow the way of successive “loading” of several RPA molecules on ssDNA. In this case, the competitive displacement of low-affinity DNA-binding domains p70C and p32D of one RPA molecule by the high-affinity domains p70A and p70B of another protein molecule is observed [84].Fig. 7. Hypothetical model of RPA10 and RPA30 type complex formation upon interaction of hsRPA with ssDNA. The dashed line shows the region of hsRPA interaction with DNA; participation of DNA-binding RPA domains p70A, p70B, p70C, and p32D in formation of different architecture complexes is demonstrated. Domains are shown schematically as rectangles A, B, C, and D.
For prokaryotic SSB, the affinity of all protomers to DNA is identical and, unlike eukaryotic proteins, for these proteins transition of SSB65 complexes to SSB35 is caused by the difference in the complex topology [2]. It should be remembered that the significant bending of the ssDNA molecule relative the protein molecule is observed in SSB65 complexes. Evidently, the peculiar “entwining” becomes unfavorable in the case of high protein concentrations, and instead, the cooperative binding of the next molecule takes place [2]. The mentioned peculiarity is shown in the difference of the binding cooperativity coefficients for prokaryotic and eukaryotic proteins, which reaches three orders of magnitude [2]. Despite this it is still assumed that the mechanism of ssDNA binding owing to which mutual transition of two binding types is universal for prokaryotic and eukaryotic SSB proteins. In a more stable complex (SSB65 or RPA30) four OB-domains interact with DNA, whereas only two domains interact in a less stable complex (SSB35 or RPA10). The possibility of complex formation with ssDNA not fitting the proposed model has been just recently noted for eukaryotic RPA with the use of photoaffinity modification [87]. The possibility of realization of the RPA-DNA complex architecture in which heterotrimer contacts single-stranded DNA with involvement of p14 subunit has been shown [87]. In the proposed complex, the size of binding site is within 10 nt and interaction with DNA is carried out by p70 and p14 subunits. The gene encoding the RPA small subunit (p14) is absolutely necessary for cell viability [93, 94], which may be indicative of an important role of this type of complexes in the dynamic process of RPA interaction with DNA during DNA replication and repair.
Taking into account common evolutionary roots, one can suppose that archaeal proteins, like eukaryotic and eubacterial ones, are able to form different types of complexes with DNA. However, now the mechanism of the archaeal protein interactions with ssDNA is less studied. Most RPA of the Euryarchaeota kingdom exhibit high affinity to ssDNA (the level of the complex dissociation constant Kd is within 7-80 nM) and in this case the cooperativity of binding to ssDNA for these proteins is not high (values of Hill coefficient n, the cooperativity index, is in the interval of 2-3, which is close to the RPA type characteristics for eukaryotic hsRPA) [17, 92]. The observed dimensions of the ssDNA binding site for euryarchaeal proteins somewhat differ from those known for typical eubacteria and eukaryotes--ecoSSB and hsRPA. It was shown [15] that for the euryarchaeal RPA homologs mac1RPA and mac3RPA from M. acetivorans the size of binding site in the most stable complexes with DNA is 20 nt. On the other side, the ability to bind 10-nt oligonucleotides was shown for these proteins, which is indicative of existence of a second, less stable type of mac1RPA and mac3RPA complexes with DNA [15]. Experimental results have also shown that different mechanisms are involved in formation of these complexes. If in one case the ssDNA molecule is “entwined” around the protein as happens in SSB65 complexes of prokaryotic ecoSSB, the other type of complexes are formed following the way specific of eukaryotic hsRPA, when the DNA molecule does not undergo significant conformational alterations [15].
Although the overwhelming majority of archaeal, eubacterial, and eukaryotic SSB proteins are able to bind DNA in two different ways, in some members just one type of formed complexes is known. Two different RPA that are paralogs to each other are found at once in the archaeal organism Ferroplasma acidarmanus [17]. Only a single type of DNA binding is known for each of them, and the formed complexes have different architecture. Binding to DNA of one of these proteins, fac1RPA, is always accompanied by the DNA strand entwining around the protein, whereas in complexes with another protein, fac2RPA, the DNA molecule retains the extended form [17]. Interestingly, the structural organization of these two proteins is also different, and it evidently defines the described mechanism of interaction with DNA. Structurally, fac1RPA, forming complexes with a DNA topological bend, resembles eubacterial SSB, because only a single domain carrying OB-fold is found in its structure. The fac2RPA protein more resembles euryarchaeal SSB proteins both in the way of its interaction with DNA and by its domain structure, because it contains two OB-folds and one “zinc finger” per protein subunit [17]. This suggests that in the considered case protein paralogs, differing in the ways of interaction with DNA, have different functions in intracellular processes.
The considered variants of SSB protein interactions with DNA concern the single-stranded state of the latter. However, in the key processes of cell viability such as replication, repair, or recombination, DNA is represented by a more complex form. Partial DNA duplex structures with protruding single-stranded regions are better imitated by functionally important DNA forms than by ssDNA [84, 88]. DNA polymerases, the key enzymes of replication processes, fill gaps in double-stranded regions and synthesize the new DNA strand using the 3′ end of a priming nucleotide as primer. Thus, most interesting is the investigation of SSB protein interactions with 3′ end of the partial duplex primer in the transition region from single-stranded DNA to double-stranded (ssDNA-dsDNA). This type of interactions is best studied for eukaryotic SSB replication protein A.
The model describing the RPA arrangement near the region of transition from dsDNA to the ssDNA template protruding in the 5′ direction was based on experimental data on photoaffinity modification and limited proteolysis (Fig. 8) [84, 86, 94, 95]. The similarity in architecture of RPA complexes with partial DNA duplex structures and single-stranded DNA is that DNA-binding domains p70A and p70B of the large subunit define the interaction with single-stranded DNA region, thus providing for necessary protein affinity to the DNA structure [88, 93, 96]. In RPA complexes with extended ssDNA, domains p70C and p32D can also contact with ssDNA, but in the case of interaction with partial DNA duplexes, these domains are located in the region of the single-stranded DNA transition to the double-stranded form (Fig. 8). Data supporting this model of interaction were obtained using partial DNA duplexes with different length protruding template region containing the photoreactive group in the region of the single-stranded DNA transition to double-stranded [84, 95].
The main difference of binding types under consideration is associated with the position of the p32 subunit on the DNA structure. If in the case of RPA complexes with ssDNA, this subunit directly interacts with the single-stranded site; in complexes with partial DNA duplexes it is located in the regions of single-stranded DNA transition to double-stranded [84, 87]. In this case the p70C domain is located on the single-stranded site and p32D directly contacts the 3′ end of the primer [85, 96].Fig. 8. Model of hsRPA oriented interaction with DNA in the region of transition from dsDNA to ssDNA template protruding in the 5′ direction. The dashed line shows the region of hsRPA interaction with the single-stranded template DNA strand. The involvement of DNA-binding domains p70A, p70B, p70C, and p32D and RPA subunit p14 (shown schematically as rectangles) is in formation of a “polar” oriented complex with DNA.
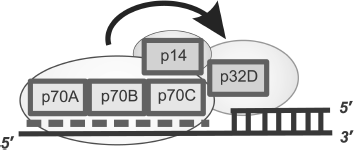
A common feature of all types of binding under consideration is the “polarity” of RPA interaction with DNA. It was shown by photoaffinity labeling using DNA duplexes containing a photoreactive group in the region of double-stranded DNA transition to single-stranded DNA, that in the case of partial DNA duplex binding, the p70 subunit interacts with the 5′ end of the terminating oligonucleotide, whereas p32 interacts with the 3′ end of the priming oligonucleotide, when RPA acquires extended conformation, and p70 interacts with the 3′ end in the case of globular conformation [84, 95]. Thus, orientation of these subunits in RPA complexes with ssDNA coincides with that in complexes with partial DNA duplexes and extended gaps. Moreover, detailed investigation of binding mechanism has shown that the presence of a tandem of main DNA-binding domains p70A and p70B is not obligatory to provide for specific and orientated RPA interaction with partial DNA duplexes [96]. At the same time, the presence of the small p14 subunit in the protein is absolutely necessary. In the absence of this subunit, the ability of RPA for “polar” folding on DNA disappears [97].
Factors Influencing Type of SSB Protein Binding with DNA
As already discussed, the ability of SSB protein to form several types of complexes with DNA can be used in the regulation and coordination mechanisms of DNA repair, replication, transcription, and recombination. The type of protein binding to DNA and protein conformation in the formed complexes depends on the size of available ssDNA site, buffer conditions, and ratio of SSB and DNA concentrations.
The pathway of one or another type of complex formation can regulated in different ways. Thus, binding type for ecoSSB (SSB35 or SSB65) depends on the solution ionic strength, ratio of protein and DNA concentrations, concentration of divalent ions, and the size of ssDNA accessible for binding [81, 91]. For example, complex formation of SSB35 type takes place at NaCl concentration below 10 mM. In the NaCl concentration interval from 200 mM to 5 M, the SSB65 type complexes are formed. At NaCl concentrations between 10 and 200 mM, there are other variants of ecoSSB binding to ssDNA with the binding site from 40 to 56 nt, but these complexes are unstable and intermediate between the two main types [98].
For euryarchaeal and eukaryotic RPA the presence in buffers of oxidizing or reducing agents may be crucial upon formation of one or another type of complexes [78]. The reduced form of cysteine residues is required to maintain structural integrity of “zinc finger” present within these proteins. On one side, reduction conditions prevent formation of disulfide bonds between cysteines, on the other side, oxidative conditions promote the zinc (II) thiolate oxidation and zinc ion release from the complex [80]. The presence of reducing agents like DTT at a concentration up to 1 mM maintains secondary structure of a “zinc finger”. As mentioned earlier, the fragment itself is not directly involved in ssDNA binding, but results of investigations on euryarchaeal mac3RPA from M. acetivorans and eukaryotic human hsRPA stress the effect of the “zinc finger” structural integrity on general protein conformation and its DNA-binding activity [57, 80].
Role of Different Types of SSB Protein Binding to DNA in DNA Replication and Repair
Undoubtedly, the SSB protein conformation and accessibility of its sites to protein-protein interactions define the character of SSB interaction with enzymes and factors of replication, repair, transcription, and recombination. Thus, the assembly of a multiprotein complex or stimulation of enzyme activity, and as a result, the total enzymatic process will depend on the architecture of the SSB protein complex with DNA.
Now several facts are known that are indicative of the role of differences in the SSB protein binding type to DNA during metabolism of the latter [99]. We shall focus on those associated with DNA replication, because just participation in DNA replication serves as a main criterion for the protein inclusion into the class of SSB proteins. Eukaryotic SSB protein RPA is absolutely necessary for all stages of DNA replication [1]. In a model replication system the RPA partner is the key protein of the DNA replication initiation DNA polymerase α-primase (pol-prim), carrying out synthesis of RNA-primer and its elongation at the initial step of replication [1]. It was shown [97] that mutant RPA forms devoid of subunit p32 and/or p14 are not able to provide for synthesis and elongation of RNA-primer catalyzed by pol-prim. Undoubtedly, the absence of small RPA subunits results in the inability of the protein to form the efficient (productive) triple complex pol-prim-DNA substrate-RPA. Along with protein-protein interactions between RPA and pol-prim, the RPA oriented interaction with DNA substrate plays a tremendous role in such complex formation. In the absence of small RPA subunits, a complex incapable of “polar” folding of protein subunits on DNA is formed. Such complex is not able to maintain synthesis and elongation of RNA-primer [97].
Results obtained during investigation of the mechanism of RNA-DNA-primer elongation in replicating SV40 chromatin also revealed different types of RPA binding at different steps of this process [100, 101]. Products of the primer early synthesis make contacts mainly with the RPA subunit p32, which is indicative of “extended” conformation of replication protein A, which corresponds to DNA binding by the RPA30 type. At later stages of synthesis, RPA acquires the compact globular conformation, corresponding to the RPA10 binding type, because only p70 subunit becomes accessible to contacts with the 3′ end of growing chain when the primer undergoes elongation [100, 101]. These data are in full agreement with results of experiments on RPA affinity modification using model DNA duplexes [95]. Transition from one binding type to another can be accompanied by alteration of the replication protein effect on the corresponding DNA polymerase caused by the changed character of protein-protein contacts. Thus, it was found that RPA does not influence DNA synthesis along damaged template by DNA polymerase λ if the template region is of 36 nt [102]. However, the stimulating effect on DNA synthesis was also found in the case of DNA substrate with protruding template site of 16 nt. According to the proposed model of RPA interaction with partial DNA duplexes, RPA within the complex acquires extended conformation in the first case and globular in the second. Thus, the results of investigations show that the ability of the protein to form complexes of necessary architecture is important for RPA function in DNA replication.
As mentioned above, RPA enhances the accuracy of DNA synthesis catalyzed by DNA polymerase α [14]. It has also been found recently that RPA plays a role in providing for the accuracy of DNA synthesis by DNA polymerase λ [103]. It became clear later that this effect is especially pronounced in the case of damaged template. DNA polymerase λ is able to synthesize DNA on template carrying both 8-oxo-guanine and 1,2-dihydro-2-oxo-adenine as a lesion, but the accuracy of synthesis in this case is not high, i.e. the probability of a correct nucleotide incorporation (dCTP and dTTP, respectively) is comparable with the probability of a “wrong” nucleotide incorporation (dATP and dGTP, respectively) [104, 105].
RPA significantly decreases the frequency of the dATP “wrong” incorporation on template carrying 8-oxo-guanine as a lesion. The presence in a mixture, in addition to RPA, of the proliferating cell nuclear antigen (PCNA) stimulates incorporation of dCTP that results in 1200-fold increased probability of the correct nucleotide incorporation by DNA polymerase λ [104]. In the case of synthesis on template carrying the other type of lesion (1,2-dihydro-2-oxo-adenine), the combined addition of PCNA and RPA increases 166-fold the probability of the correct dTTP incorporation [105]. Thus, RPA along with PCNA are important components of the functioning replication complex, which influence both main replication DNA polymerases (α and δ) and specialized polymerases carrying out DNA synthesis in replication fork of complex blocked by a lesion in the template strand.
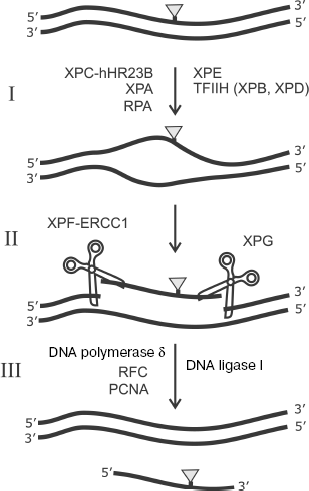
In addition to involvement in DNA replication, RPA is known as an important participant of one of the main pathways of DNA replication, namely, of nucleotide excision repair (NER). A number of the following protein factors are also involved in this process: XPC-hHR23B, TFIIH, XPA, XPE, XPG, and ERCC1-XPF [106]. The scheme of this process is shown in Fig. 9. Bulky lesions emerging in response to UV irradiation or exogenous environmental factors are eliminated from DNA by the NER system.
The NER system exhibits unusually broad substrate specificity. The XPC-hHR23B complex is one of the main candidates for the role of a factor providing for initial recognition of DNA lesions during NER [107-109]. The mechanism of interaction of this protein factor with damaged DNA is under intensive study [106]. XPC-hHR23B attracts the TFIIH complex to the lesion site. This factor with involvement of ATP and two subunits exhibiting helicase activity (XPD and XPB) unwind DNA duplex in the lesion site. Then RPA, XPA, and XPG form a complex that precedes the removal of the lesion-carrying DNA site. At the next stage, endonucleases ERCC1-XPF and XPG remove the 30-nt damaged site of ssDNA by hydrolysis from the 5′ and 3′ ends, respectively. Replication protein A remains bound to ssDNA and facilitates transition to DNA synthesis in order to fill the gap; DNA synthesis at this stage is catalyzed by DNA polymerase δ with involvement of RFC and PCNA. Thus, RPA is first of all important for formation of a multicomponent protein complex with DNA before elimination of a 30-nt lesion-containing DNA site (Fig. 9) [106]. Just at this stage of NER the RPA capability of “polar” binding to the undamaged strand of the DNA duplex is important. The “asymmetry” in arrangement of various DNA-binding RPA domains (p70A, p70B, p70C, and p32D) on the undamaged strand provides the basis for different structural peculiarities at the 5′ and 3′ ends in unwound DNA duplex before removal of the 30-nt damaged region of a complementary strand. This, in turn, defines the precise orientation and activity stimulation of endonucleases XPG (catalyzes cleavage from the 3′ end of the fragment to be removed) and ERCC1-XPF (cleaving DNA from the 5′ end). RPA is also absolutely necessary in this process at the stage of DNA repair in the formed gap, which is catalyzed by DNA polymerase δ supported by RFC and PCNA. Besides, the problem of RPA effect on the lesion recognition by XPC-hHR23B and their further processing is still not unambiguously solved and it is the subject of intensive investigation [110-114].Fig. 9. Scheme of nucleotide excision repair (NER). I) Lesion recognition and formation of an open complex; II) excision of damaged region; III) DNA resynthesis. DNA lesion eliminated by the NER system is shown schematically by the gray triangle. Designations of enzymes and protein factors involved in the process are shown.
Perhaps not all SSB proteins have been discovered up to now. It has been rather recently shown that replication protein A is present at the chromosome telomere ends with maximal level of association with telomere ends in the S phase. Perhaps some functions of SSB proteins remain to be discovered. It has been shown relatively recently that RPA plays an important role in telomere processing, making accessible telomere ends of Est1p protein, the most important component of telomerase complex [115, 116]. RPA probably contributes to stabilization of the ssDNA form interacting with the telomerase complex components. A possible mechanism of RPA involvement in telomerase activation is through its ability to destabilize complex DNA structures like guanosine-rich G-quadruplexes [117, 118]. It is interesting that transition between different forms of RPA binding to DNA plays the most important role in unwinding of such DNA structures. The detailed mechanism of the interaction of RPA with telomerase complex is of significant interest.
The involvement of replication protein A in key processes of DNA metabolism has made it the object of thorough investigations. The universal role of SSB proteins in DNA metabolism is supported by the existence of common structural motifs in proteins of organisms that are evolutionarily remote from each other. Further detailed study of the role of separate RPA domains in formation of their complexes with various DNA structures is of obvious interest for understanding the mechanism of protein-nucleic interactions in functional complexes carrying out DNA processing during the life of cells.
The authors are grateful to Dr. S. N. Khodyreva and Prof. G. G. Karpova for discussions during manuscript preparation.
This work was supported by the Russian Foundation for Basic Research (grants 06-04-48612, 08-04-00704-a, and 08-04-91202-YaF), INTAS-SBRAS (grant 06-1000013-9210), ISTC (grant No. 3312) and by Program of the Russian Academy of Sciences on Molecular and Cellular Biology.
REFERENCES
1.Wold, M. S. (1997) Annu. Rev. Biochem.,
66, 61-92.
2.Lohman, T. M., and Ferrari M. E. (1994) Annu.
Rev. Biochem., 63, 527-570.
3.Cha, T. A., and Alberts, B. M. (1989) J. Biol.
Chem., 264, 12220-12225.
4.Kim, Y. T., and Richardson, C. C. (1993) Proc.
Natl. Acad. Sci. USA, 90, 10173-10177.
5.Perales, C., Cava, F., and Meijer, W. J. J. (2003)
Nucleic Acids Res., 31, 6473-6380.
6.Chase, J. W., and Williams, K. R. (1986) Annu.
Rev. Biochem., 55, 103-136.
7.Zou, Y., Liu, Y., Wu, X., and Shell, S. M. (2006)
J. Cell. Physiol., 208, 267-273.
8.Stein, G. S., Zaidi, S. K., Braastad, C. D.,
Montecino, M., van Wijnen, A. J., Choi, J. Y., Stein, J. L., Lian, J.
B., and Javed, A. (2003) Trends Cell. Biol., 13,
584-592.
9.Davey, M. J., and O'Donnell, M. (2000) Curr.
Opin. Chem. Biol., 4, 581-586.
10.Kur, J., Olszewski, M., Dlugolecka, A., and
Filipkowski, P. (2005) Acta Biochim. Pol., 52,
569-574.
11.Yuzhakov, A., Kelman, Z., Hurwitz, J., and
O'Donnell, M. (1999) EMBO J., 18, 6189-6199.
12.Maga, G., Stucki, M., Spadari, S., and Hubscher,
U. (2000) J. Mol. Biol., 295, 791-801.
13.Carty, M. P., Levine, A. S., and Dixon, K. (1992)
Mutat. Res., 274, 29-34.
14.Maga, G., Frouin, I., Spadari, S., and Hubscher,
U. (2001) J. Biol. Chem., 276, 18235-18242.
15.Robbins, J. B., Murphy, M. C., White, B. A.,
Mackie, R. I., Ha, T., and Cann, I. K. (2004) J. Biol. Chem.,
279, 6315-6326.
16.Cann, I. K., Ishino, S., Yuasa, M., Daiyasu, H.,
Toh, H., and Ishino, Y. (2001) J. Bacteriol., 183,
2614-2623.
17.Robbins, J. B., McKinney, M. C., Guzman, C. E.,
Sriratana, B., Fitz-Gibbon, S., Ha, T., and Cann, I. K. (2005) J.
Biol. Chem., 280, 15325-15339.
18.Kelman, Z., Pietrokovski, S., and Hurwitz, J.
(1999) J. Biol. Chem., 274, 28751-28761.
19.Stauffer, M. E., and Chazin, W. J. (2004) J.
Biol. Chem., 279, 30915-30918.
20.Benkovic, S. J., Valentine, A. M., and Salinas,
F. (2001) Annu. Rev. Biochem., 70, 181-208.
21.Khlimankov, D. Yu., Rechkunova, N. A., and
Lavrik, O. I. (2004) Biochemistry (Moscow), 69,
248-261.
22.Rademakers, S., Volker, M., Hoogstraten, D.,
Nigg, A. L., Mone, M. J., van Zeeland, A. A., Hoeijmakers, J. H.,
Houtsmuller, A. B., and Vermeulen, W. (2003) Mol. Cell. Biol.,
23, 5755-5767.
23.Essers, J., Houtsmuller, A. B., van Veelen, L.,
Paulusma, C., Nigg, A. L., Pastink, A., Vermeulen, W., Hoeijmakers, J.
H., and Kanaar, R. (2002) EMBO J., 21, 2030-2037.
24.Arunkumar, A. I., Stauffer, M. E., Bochkareva,
E., Bochkarev, A., and Chazin, W. J. (2003) J. Biol. Chem.,
17, 41077-41082.
25.Murzin, A. G., Brenner, S. E., Hubbard, T., and
Chothia, C. (1995) J. Mol. Biol., 247, 536-540.
26.Murzin, A. G. (1993) EMBO J., 12,
861-867.
27.Theobald, D. L., Mitton-Fry, R. M., and Wuttke,
D. S. (2003) Annu. Rev. Biophys. Biomol. Struct., 32,
115-133.
28.Williams, K. R., Spicer, E. K., LoPresti, M. B.,
Guggenheimer, R. A., and Chase, J. W. (1983) J. Biol. Chem.,
258, 3346-3355.
29.Carlini, L., Curth, U., Kindler, B., Urbanke, C.,
and Porter, R. D. (1998) FEBS Lett., 430, 197-200.
30.Iftode, C., Daniely, Y., and Borowiec, J. A.
(1999) Crit. Rev. Biochem. Mol. Biol., 34, 141-180.
31.Bochkareva, E., Korolev, S., Lees-Miller, S. P.,
and Bochkarev, A. (2002) EMBO J., 21, 1855-1863.
32.Daughdrill, G. W., Buchko, G. W., Botuyan, M. V.,
Arrowsmith, C., Wold, M. S., Kennedy, M. A., and Lowry, D. F. (2003)
Nucleic Acids Res., 31, 4176-4183.
33.Weisshart, K., Taneja, P., and Fanning, E. (1998)
J. Virol., 72, 9771-9781.
34.Golub, E. I., Gupta, R. C., Haaf, T., Wold, M.
S., and Radding, C. M. (1998) Nucleic Acids Res., 26,
5388-5393.
35.Bochkareva, E., Kaustov, L., Ayed, A., Yi, G. S.,
Lu, Y., Pineda-Lucena, A., Liao, J. C., Okorokov, A. L., Milner, J.,
Arrowsmith, C. H., and Bochkarev, A. (2005) Proc. Natl. Acad. Sci.
USA, 102, 15412-15417.
36.Daughdrill, G. W., Ackerman, J., Isern, N. G.,
Botuyan, M. V., Arrowsmith, C., Wold, M. S., and Lowry, D. F. (2001)
Nucleic Acids Res., 29, 3270-3276.
37.He, Z., Brinton, B. T., Greenblatt, J., Hassell,
J. A., and Ingles, C. J. (1993) Cell, 73, 1223-1232.
38.Mer, G., Bochkarev, A., Gupta, R., Bochkareva,
E., Frappier, L., Ingles, C. J., Edwards, A. M., and Chazin, W. J.
(2000) Cell, 103, 449-456.
39.Gajiwala, K. S., and Burley, S. K. (2000)
Curr. Opin. Struct. Biol., 10, 110-116.
40.Braun, K. A., Lao, Y., He, Z., Ingles, C. J., and
Wold, M. S. (1997) Biochemistry, 36, 8443-8454.
41.Stauffer, M. E., and Chazin, W. J. (2004) J.
Biol. Chem., 279, 25638-25645.
42.Kelly, T. J., Simancek, P., and Brush, G. S.
(1998) Proc. Natl. Acad. Sci. USA, 95, 14634-14639.
43.Philipova, D., Mullen, J. R., Maniar, H. S., Lu,
J., Gu, C., and Brill, S. J. (1996) Genes Dev., 10,
2222-2233.
44.De Vries, J., and Wackernagel, W. (1993)
Gene, 127, 39-45.
45.Genschel, J., Litz, L., Thole, H., Roemling, U.,
and Urbanke, C. (1996) Gene, 182, 137-143.
46.Purnapatre, K., and Varshney, U. (1999) Eur.
J. Biochem., 264, 591-598.
47.Sancar, A., Williams, K., Chase, J., and Rupp, W.
(1981) Proc. Natl. Acad. Sci. USA, 78, 4274-4278.
48.Weiner, J. H., Bertsch, L. L., and Kornberg, A.
(1975) J. Biol. Chem., 250, 1972-1980.
49.Webster, G., Genschel, J., Curth, U., Urbanke,
C., Kang, C., and Hilgenfeld, R. (1997) FEBS Lett., 411,
313-316.
50.Yang, C., Curth, U., Urbanke, C., and Kang, C.-H.
(1997) Nat. Struct. Biol., 4, 153-157.
51.Curth, U., Urbanke, C., Greipel, J., Gerberding,
H., Tiranti, V., and Zeviani, M. (1994) Eur. J. Biochem.,
221, 435-443.
52.Dabrowski, S., Olszewski, M., Piatek, R.,
Brillowska-Dabrowska, A., Konopa, G., and Kur, J. (2002)
Microbiology, 148, 3307-3315.
53.Eggington, J. M., Haruta, N., Wood, E. A., and
Cox, M. M. (2004) BMC Microbiol., 4, 2.
54.Witte, G., Urbanke, C., and Curth, U. (2005)
Nucleic Acids Res., 33, 1662-1670.
55.Bernstein, D. A., Eggington, J. M., Killoran, M.
P., Misic, A. M., Cox, M. M., and Keck, J. L. (2004) Proc. Natl.
Acad. Sci. USA, 101, 8575-8580.
56.Bochkarev, A., Pfuetzner, R. A., Edwards, A. M.,
and Frappier, L. (1997) Nature, 385, 176-181.
57.Lin, Y., Robbins, J. B., Nyannor, E. K., Chen, Y.
H., and Cann, I. K. (2005) J. Bacteriol., 187,
7881-7889.
58.Lin, Y. L., Shivji, K. K., Chen, C., Kolodner,
R., Wood, R. D., and Dutta, A. (1998) J. Biol. Chem.,
273, 1453-1461.
59.Bochkareva, E., Frappier, L., Edwards, A. M., and
Bochkarev, A. (1998) J. Biol. Chem., 273, 3932-3936.
60.Bochkarev, A., Bochkareva, E., Frappier, L., and
Edwards, A. M. (1999) EMBO J., 18, 4498-4504.
61.Chedin, F., Seitz, E. M., and Kowalczykowski, S.
C. (1998) Trends Biochem. Sci., 23, 273-277.
62.Kerr, I. D., Wadsworth, R. I., Cubeddu, L.,
Blankenfeldt, W., Naismith, J. H., and White, M. F. (2003) EMBO
J., 22, 2561-2570.
63.Wadsworth, R. I., and White, M. F. (2001)
Nucleic Acids Res., 29, 914-920.
64.Bochkarev, A., and Bochkareva, E. (2004) Curr.
Opin. Struct. Biol., 14, 36-42.
65.Raghunathan, S., Ricard, C. S., Lohman, T. M.,
and Waksman, G. (1997) Proc. Natl. Acad. Sci. USA, 94,
6652-6657.
66.Raghunathan, S., Kozlov, A. G., Lohman, T. M.,
and Waksman, G. (2000) Nat. Struct. Biol., 7,
648-652.
67.Antson, A. A. (2000) Curr. Opin. Struct.
Biol., 10, 87-94.
68.Berman, H. M., Westbrook, J., Feng, Z.,
Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., and Bourne,
P. E. (2000) Nucleic Acids Res., 28, 235-242.
69.Carter, A. P., Clemons, W. M., Jr., Brodersen, D.
E., Morgan-Warren, R. J., Hartsch, T., Wimberly, B. T., and
Ramakrishnan, V. (2001) Science, 291, 498-501.
70.Bochkareva, E., Belegu, V., Korolev, S., and
Bochkarev, A. (2001) EMBO J., 20, 612-618.
71.Bogden, C. E., Fass, D., Bergman, N., Nichols, M.
D., and Berger, J. M. (1999) Mol. Cell, 3, 487-493.
72.Peersen, O. B., Ruggles, J. A., and Schultz, S.
C. (2002) Nat. Struct. Biol., 9, 182-187.
73.Savvides, S. N., Raghunathan, S., Futterer, K.,
Kozlov, A. G., Lohman, T. M., and Waksman, G. (2004) Protein
Sci., 13, 1942-1947.
74.Grishin, N. V. (2001) Nucleic Acids Res.,
29, 1703-1714.
75.Matthews, J. M., and Sunde, M. (2002) IUBMB
Life, 54, 351-355.
76.Krishna, S. S., Majumdar, I., and Grishin, N. V.
(2003) Nucleic Acids Res., 31, 532-550.
77.Lachenmann, M. J., Ladbury, J. E., Dong, J.,
Huang, K., Carey, P., and Weiss, M. A. (2004) Biochemistry,
43, 13910-13925.
78.Park, J. S., Wang, M., Park, S. J., and Lee, S.
H. (1999) J. Biol. Chem., 274, 29075-29080.
79.Komori, K., and Ishino, Y. (2001) J. Biol.
Chem., 276, 25654-25660.
80.Bochkareva, E., Korolev, S., and Bochkarev, A.
(2000) J. Biol. Chem., 275, 27332-27338.
81.Bujalowski, W., Overman, L. B., and Lohman, T. M.
(1988) J. Biol. Chem., 163, 4629-4640.
82.Kozlov, A. G., and Lohman, T. M. (2002)
Biochemistry, 41, 11611-11627.
83.Bastin-Shanower, S. A., and Brill, S. J. (2001)
J. Biol. Chem., 276, 36446-36453.
84.Lavrik, O. I., Kolpashchikov, D. M., Weisshart,
K., Nasheuer, H. P., Khodyreva, S. N., and Favre, A. (1999) Nucleic
Acids Res., 27, 4235-4240.
85.Gomes, X. V., Henricksen, L. A., and Wold, M. S.
(1996) Biochemistry, 35, 5586-5595.
86.Pestryakov, P. E., Weisshart, K., Schlott, B.,
Khodyreva, S. N., Kremmer, E., Grosse, F., Lavrik, O. I., and Nasheuer,
H.-P. (2003) J. Biol. Chem., 278, 17515-17524.
87.Pestryakov, P. E., Krasikova, Yu. S., Petruseva,
I. O., Khodyreva, S. N., and Lavrik, O. I. (2007) Dokl. AN SSSR,
412, 118-122.
88.De Laat, W. L., Appeldoorn, E., Sugasawa, K.,
Weterings, E., Jaspers, N. G., and Hoeijmakers, J. H. (1998) Genes
Dev., 12, 2598-2609.
89.Treuner, K., Ramsperger, U., and Knippers, R. J.
(1996) Mol. Biol., 259, 104-112.
90.Blackwell, L. J., Borowiec, J. A., and
Masrangelo, I. A. (1996) Mol. Cell. Biol., 16,
4798-4807.
91.Lohman, T. M., and Overman, L. B. (1985) J.
Biol. Chem., 260, 3594-3603.
92.Kim, C., and Wold, M. S. (1995)
Biochemistry, 34, 2058-2064.
93.Brill, S. J., and Stillman, B. (1991) Genes
Dev., 5, 1589-1600.
94.Heyer, W.-D., Rao, M. R. S., Erdile, L. F.,
Kelly, T. J., and Kolodner, R. D. (1990) EMBO J., 9,
2321-2329.
95.Kolpashchikov, D. M., Khodyreva, S. N.,
Khlimankov, D. Y., Wold, M. S., Favre, A., and Lavrik, O. I. (2001)
Nucleic Acids Res., 29, 373-379.
96.Pestryakov, P. E., Khlimankov, D. Yu.,
Bochkareva, E., Bochkarev, A., and Lavrik, O. I. (2004)
Nucleic Acids Res., 32, 1894-1903.
97.Weisshart, K., Pestryakov, P., Smith, R. W.,
Hartmann, H., Kremmer, E., Lavrik, O., and Nasheuer, H.-P. (2004) J.
Biol. Chem., 279, 35368-35376.
98.Lohman, T. M., and Bujalowski, W. (1994)
Biochemistry, 33, 6167-6176.
99.Fanning, E., Klimovich, V., and Nager, A. R.
(2006) Nucleic Acids Res., 34, 4126-4137.
100.Mass, G., Nethanel, T., and Kaufmann, G. (1998)
Mol. Cell. Biol., 18, 6399-6407.
101.Mass, G., Nethanel, T., Lavrik, O. I., Wold, M.
S., and Kaufmann, G. (2001) Nucleic Acids Res., 29,
3892-3899.
102.Krasikova, Yu. S., Belousova, E. A., Lebedeva,
N. A., Pestryakov, P. E., and Lavrik, O. I. (2008) Biochemistry
(Moscow), accepted for publication.
103.Maga, G., Shevelev, I., Villani, G., Spadari,
S., and Hubscher, U. (2006) Nucleic Acids Res., 34,
1405-1415.
104.Maga, G., Villani, G., Crespan, E., Wimmer, U.,
Ferrari, E., Bertocci, B., and Hubscher, U. (2007) Nature,
447, 606-608.
105.Crespan, E., Hubscher, U., and Maga, G. (2007)
Nucleic Acids Res., 35, 5173-5181.
106.Gillet, L., and Scharer, O. (2006) Chem.
Rev., 106, 253-276.
107.Sugasawa, K., Ng, J. M., Masutani, C., Iwai,
S., van der Spek, P. J., Eker, A. P., Hanaoka, F., Bootsma, D., and
Hoeijmakers, J. H. (1998) Mol. Cell., 2, 223-232.
108.Volker, M., Mone, M. J., Karmakar, P., van
Hoffen, A., Schul, W., Vermeulen, W., Hoeijmakers, J. H., van Driel,
R., van Zeeland, A. A., and Mullenders, L. H. (2001) Mol. Cell.,
8, 213-224.
109.Rechkunova, N. I., Maltseva, E. A., and Lavrik,
O. I. (2008) Mol. Biol. (Moscow), 42, 24-31.
110.Patrick, S. M., and Turchi, J. J. (1999) J.
Biol. Chem., 274, 14972-14978.
111.Missura, M., Buterin, T., Hindges, R.,
Hubscher, U., Kasparkova, J., Brabec, V., and Naegeli, H. (2001)
EMBO J., 20, 3554-3564.
112.Reardon, J. T., and Sancar, A. (2002) Mol.
Cell Biol., 22, 5938-5945.
113.Patrick, S. M., and Turchi, J. J. (1998)
Biochemistry, 37, 8808-8815.
114.Shuck, S. C., Short, E. A., and Turchi, J. J.
(2008) Cell Res., 18, 64-72.
115.Smith, J., Zou, H., and Rothstein, R. (2000)
Biochimie, 82, 71-78.
116.Schramke, V., Luciano, P., Brevet, V., Guillot,
S., Corda, Y., Longhese, M. P., Gilson, E., and Geli, V. (2004) Nat.
Genet., 36, 46-54.
117.Cohen, S., Jacob, E., and Manor, H. (2004)
Biochim. Biophys. Acta, 1679, 129-140.
118.Salas, T. R., Petruseva, I., Lavrik, O.,
Bourdoncle, A., Mergny, J. L., Favre, A., and Saintome, C. (2006)
Nucleic Acids Res., 34, 4857-4865.