Study of Spatial Organization of Chicken α-Globin Gene Domain by 3C Technique
A. A. Gavrilov1* and S. V. Razin1,2
1Institute of Gene Biology, Russian Academy of Sciences, ul. Vavilova 34/5, 119334 Moscow, Russia; fax: (499) 135-4105; E-mail: aleksey.gavrilov@mail.ru2Biological Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received June 23, 2008; Revision received July 23, 2008
This work deals with 3C (Chromosome Conformation Capture) analysis of the chicken α-globin gene domain in embryonic erythrocytes and lymphoid cells. Ligation products were quantitatively analyzed by real-time PCR with TaqMan probes. It was found that in lymphoid cells, where α-globin gene is not active, the domain has a relatively extended configuration. In embryonic erythrocytes that transcribe αD and αA genes, simultaneous interaction of several domain elements was revealed including the major regulatory element, the erythroid-specific DNase I hypersensitive site at a distance of 9 kb upstream from the α-globin gene cluster (-9 DHS), promoter of the housekeeping gene CGTHBA, the αD-globin gene promoter, and the erythroid-specific enhancer located after the α-globin gene cluster. We suppose that such interaction is necessary to provide for the active transcription status of the chicken α-globin gene domains in erythroid cells.
KEY WORDS: chicken α-globin gene domain, genome spatial organization, 3C, real-time PCR with TaqMan probesDOI: 10.1134/S0006297908110047
Abbreviations: BAC) bacterial artificial chromosome; 3C) chromosome conformation capture; DHS) DNase I hypersensitive sites; MRE) major regulatory element; YAC) yeast artificial chromosome.
For many years, the chicken α-globin gene domain has served as a
model for studying the transcription regulation mechanisms in higher
eukaryotes [1-4]. In addition
to three α-type globin genes (embryonic π gene and
“adult” genes αD and αA),
the domain contains a number of regulatory elements located both
upstream [5-8] and downstream
[9] from the gene cluster. The major regulatory
element (MRE) of the chicken α-globin gene domain, similar to the
locus control region, is located within the housekeeping gene intron at
a distance of about 20 kb upstream from gene π, the first one in the
α-globin gene cluster [5, 8]. It is still not clear how this element works.
Based on investigations of globin gene domains in other vertebrates [10, 11], one could propose that
MRE directly interacts with α-globin gene promoters. It was
supposed in the literature that the same regulatory element is not able
to interact simultaneously with two and more promoters. Then the
interaction should be the short-lived and alternating as it is
postulated in the so-called “flip-flop” model [12, 13]. Different authors have
shown that formation of a complex block of regulatory elements
including promoters of active globin genes and those of adjacent
housekeeping genes is possible [14]. Promoter of
the chicken housekeeping gene is localized at a distance of about 4 kb
upstream from the embryonic globin gene π. Further,
erythroid-specific DNase I hypersensitive sites (DHS) are mapped at a
distance of 9.1, 12.8, 14.9, and 20.5 kb upstream from gene π [5, 15, 16].
Some of them are co-localized with such regulatory elements as MRE [5] and insulators [17]. The
significance of others is still unknown. The erythroid-specific
enhancer is located at a distance of about 1 kb downstream from the
gene αA (the last gene of the chicken α-globin
gene cluster) [9].
To understand how the regulation of the chicken α-globin gene transcription is carried out, it is important to know whether different regulatory elements of the domain interact with each other and with promoters of α-globin genes. To elucidate this problem, we have studied spatial organization of the α-globin gene domain in 10-day-old embryonic erythrocytes using the method of Chromosome Conformation Capture (3C) [10, 18, 19]. The 3C method is intended for in vivo study of spatial organization of genomic loci. The method is based on formaldehyde cross-linking of DNA-protein complexes with following DNA cleavage by restriction enzymes and ligation. The ligation is carried out at a low DNA concentration that results in the preferable ligation of cross-linked rather than random fragments. Thus, the amount of specific ligation products is indicative of the frequency of interactions between appropriate restriction fragments.
The real-time PCR technique with TaqMan probes was used for quantitative analysis of ligation products. The data are indicative of interaction in these cells between MRE, -9 DHS, CGTHBA gene promoters, αD gene promoter, and enhancer localized downstream from the α-globin gene cluster.
MATERIALS AND METHODS
Cell culture. Chicken lymphoid cells of DT40 line (CRL-2111, ATCC) were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) containing 8% fetal calf serum, 2% chicken serum, and 50 µM β-mercaptoethanol in the presence of 5% CO2 in the atmosphere. Embryonic erythrocytes were isolated from 10-day-old chicken embryos.
3C analysis. 3C analysis was carried out as described earlier [20] with insignificant modifications. Cells (2·107) were fixed for 10 min at room temperature with gentle shaking in DMEM medium containing serum and 2% formaldehyde. The reaction was stopped by glycine addition to 0.125 M, and then cell suspension was transferred onto ice. Cells were washed with cold PBS, and then nuclei were isolated by 10 min incubation in cold buffer for lysis (10 mM Tris, pH 8.0, 10 mM NaCl, 0.2% NP-40, and one tablet of protease inhibitors (Complete Mini; Roche, Switzerland)). The nuclei were collected by centrifugation, and aliquots of 107 nuclei were frozen in liquid nitrogen and stored at -70°C.
Nuclei (107) were suspended in 0.25 ml of restriction buffer (buffer 3 from New England Biolabs, USA), 20% SDS solution was added to 0.3% concentration, and the mixture was incubated for 1 h at 37°C with intense mixing (1400 rpm). Then 0.25 ml of 1.2× restriction buffer was added, which was followed by addition of 20% Triton X-100 to final concentration of 1.8%, and the mixture was incubated for 1 h at 37°C with intense mixing (1400 rpm). After incubation, BglII (1000 units) and BamHI (1500 units) or MboI (1000 units) (highly concentrated; New England Biolabs) were added and DNA was hydrolyzed for 16 h at 37°C with intense mixing (1400 rpm). The reaction was stopped by addition of 20% SDS solution to 1.3% with heating for 20 min at 65°C. After restriction, the solution was mixed in a 50 ml plastic centrifuge tube with 7 ml of 1× ligation buffer (50 mM Tris, pH 7.5, 10 mM MgCl2, 10 mM DTT, 1 mM ATP), 20% Triton X-100 was added to 1% concentration, and the mixture was incubated for 1 h at 37°C with mixing (400 rpm). Then 100 units of T4 DNA ligase (Fermentas, Lithuania) were added and the mixture was incubated for 4-5 h with mixing at 16°C for 30 min at room temperature. Solution after ligation was incubated for 16 h at 65°C in the presence of 300 µg of proteinase K, then 300 µg RNase was added, and RNA was hydrolyzed for 45 min at 37°C. The solution was extracted in succession by equal volumes of phenol, phenol-chloroform mixture (1 : 1), and chloroform, then diluted by half with water, 3 M sodium acetate (pH 5.2) was added to final concentration 0.2 M, then two volumes of cold 96% ethanol were added and the mixture was kept for 24 h at -70°C. DNA was precipitated by centrifugation for 1 h at 3200g and 4°C, DNA precipitate was washed by cold 70% ethanol and centrifuged again for 20 min. The precipitate was dissolved in 200 µl of 10 mM Tris, pH 7.5, and stored in aliquots at -70°C.
Obtaining equimolar mixture of ligation products. Five micrograms of the bacterial artificial chromosome (BAC) DNA, carrying the chicken α-globin gene domain with adjacent regions (clone CH261-75C12, CHORI BACPAC), was hydrolyzed by restriction endonucleases BamHI and BglII or Sau3A, DNA was purified by extraction with phenol-chloroform and precipitation by ethanol, religated at the DNA concentration of 100 ng/µl, and purified again. The full-sized chicken genomic DNA was hydrolyzed and religated in a similar way.
Real-time PCR with TaqMan probes. Primers and TaqMan probes were selected using the Primer Premier 5 Program (PRIMER Biosoft International) based on the AY016020 DNA sequence (GeneBank) [5]. The size of PCR products that could be obtained using the equimolar mixture of BamHI/BglII ligation products or MboI fragments of the chicken genome region under investigation made up 120-230 bp. The annealing temperature was 55-58°C for primers and 67-68°C for TaqMan probes.
The amplification reaction was carried out in 20 µl of PCR buffer (50 mM Tris, pH 8.6, 50 mM KCl, 1.5 mM MgCl2, 0.1% Tween 20) containing 30 or 60 ng of the studied DNA or a mixture of 0.4, 2, 10, 50, 250, or 1250 pg of hydrolyzed and religated bacmid with 30 ng of hydrolyzed and religated full-sized genomic DNA, 0.5 µM of each primer, 0.25 µM of TaqMan probe, 0.2 mM of each dNTP, 0.75 activity unit of the hot-start Taq polymerase (Sibenzyme, Russia) according to the following scheme: 94°C, 5 min, 1 cycle; 94°C, 15 sec; 60°C, 60 sec, fluorescence detection, 58 cycles.
RESULTS
Application of 3C technique for studying spatial organization of the chicken α-globin gene domain. The 3C technique was developed earlier [10, 18, 19]. To “fix” the existing intracellular spatial organization of the genome, living cells are treated with formaldehyde and then they are lysed in SDS-containing buffer. In addition to lysis, the detergent provides for removal of non-cross-linked proteins from DNA. SDS is “neutralized” by Triton X-100, and then DNA is hydrolyzed by a restriction endonuclease. The enzyme is inactivated by addition of SDS and short-time heating at 65°C followed by dilution of the resulting solution with ligase buffer (DNA concentration decreases to several ng/µl), then SDS is “neutralized” again and DNA undergoes ligation. At the next stage formaldehyde cross-links are removed during incubation at 65°C in the presence of proteinase K, then RNA is hydrolyzed and ligation products are purified by phenol-chloroform extraction followed by ethanol precipitation (Scheme 1). The amount of specific ligation products is indicative of ligation frequency of appropriate restriction fragments, which, in turn, is the measure of their interaction frequency.
Main steps of 3C analysis. Cells are treated with formaldehyde and lysed with SDS, and then DNA is hydrolyzed by restriction endonuclease and religated. Ligation products are purified and analyzed by PCR
Scheme 1
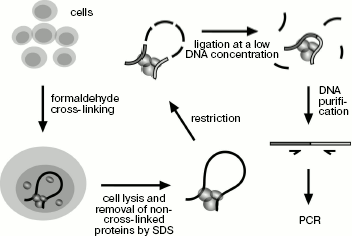
Ligation products are quantitatively analyzed by PCR. Primers for amplification are so selected at the ends of restriction fragments that the 3´-end of the primer should be turned to the fragment end. As a rule, all primers are selected at the same side (left or right) of fragments in order that all of them are directed to the same side. Such primers are used for analysis of ligation products by the type of “head-to-head” or “tail-to-tail”. Unidirectionality of primers excludes the possibility of formation of PCR products on the under-hydrolyzed DNA template. One primer is selected as an “anchor” and is used for PCR in turn with all other primers. TaqMan probes for real-time PCR are selected downstream from the anchor-type primer at the very end of restriction fragment. Thus, the same TaqMan probe can be used for all PCRs with this anchor-type primer.
To determine the amount of DNA template by the PCR signal intensity, it is necessary to keep in mind that the amplification efficiency can be different with different primers. To compare primer efficiencies, one should have a template that contains equal amounts of the DNA matrix for amplification with each pair of primers. DNAs of artificial bacterial or yeast chromosomes (BAC or YAC), carrying the genome region under study, are used to obtain such a template [20]. BAC/YAC is hydrolyzed by a restriction endonuclease, chosen for 3C analysis, and ligation at a high DNA concentration is carried out. The resulting preparation represents an equimolar mixture of all possible ligation products and is used for determination of efficiency of each pair of primers.
An additional important control concerns the cases when it is necessary to compare results of 3C analysis for different cell types. The thing is that the quality and amount of DNA prepared for 3C analysis from the initially equal amount of cells of different types may be different. This control consists in 3C analysis of the genome region that supposedly has an identical spatial organization in all cell types used in the work. Usually housekeeping genes, whose transcription status is the same in all cell types, are chosen. The resulting data are used to normalize data of 3C analysis of the genome site under study [20].
In the first series of experiments on 3C analysis of the chicken α-globin gene domain, we used the combination of restriction endonucleases BglII and BamHI. These enzymes recognize different sequences (AGATCT and GGATCC) but produce compatible DNA ends capable of ligation with each other. In the second series of experiments, we used restriction endonuclease MboI (recognition site GATC) cleaving the domain into smaller fragments. The scheme of the chicken α-globin gene domain showing positions of α-globin genes and important regulatory elements as well as BamHI/BglII- and MboI-restriction maps of the domain are given in figure (a-c, respectively). It is seen that such elements as MRE, promoter of the housekeeping gene CGTHBA, α-globin gene promoter, and the erythroid-specific enhancer are localized in different restriction fragments. Positions of primers and TaqMan probes used for analysis of ligation products are shown over the restriction maps.
The primer and TaqMan probe sequences are shown in Tables 1 and 2. Each pair of primers was used for amplification of DNA of experimental 3C samples as well as of DNA of six successive fivefold dilutions of the ligation product equimolar mixture prepared on the basis of the bacmid. DNA concentration in bacmid dilutions was brought to that in 3C samples using the hydrolyzed and re-ligated chicken genomic DNA. Data on bacmid dilutions were used to plot the calibration curve applied in quantitative determination of ligation products in 3C samples. All components of amplification reaction, except Taq-polymerase, were divided into portions, stored frozen, and were taken right before reaction. Each PCR reaction was carried out in quadruplicate, and corresponding results were averaged. To check reproducibility of the results, all experiments, beginning from live cells, were repeated at least twice.3C analysis of the chicken α-globin gene domain. a) Scheme of the chicken α-globin gene domain showing positions of genes and important regulatory domain elements. b, c) BamHI/BglII and MboI restriction maps of the chicken α-globin gene domain. Short hatches over the scale designate BglII and MboI sites, long hatches correspond to BamHI sites. Point “0” on the scales corresponds to the beginning of AY016020 DNA sequence (GeneBank) [5], scale dimensions are given in kb. Positions of primers and TaqMan probes used for 3C analysis are indicated by ticks and rectangles over maps. d-i) Relative cross-linking frequencies of the anchor-type restriction fragment carrying MRE (d), -9 DHS (e), CpG islet (f), αD gene promoter (g, i), gene αA (i) with other domain fragments (3C analysis was carried out using BamHI and BglII (d-h) or MboI (i)). The X-axis indicates positions of restriction fragments in accordance with the scale of restriction maps. The domain scheme is shown in the upper part of each plot (symbols the same as in the scheme of section (a)). 1, 2) Results of 3C analysis for 10-day-old embryonic erythrocytes and for lymphoid DT40 cells. Dark-gray rectangles at the background with the anchor symbol above show position of anchor fragment, light-gray rectangles correspond to test fragments. Borders between neighboring fragments are marked by gray lines. The Y-axis values correspond to frequencies of cross-linking between analyzed fragments normalized relative to frequencies of cross-linking between fragments of control region. Error lines correspond to standard deviation from the mean value in two and more experiments
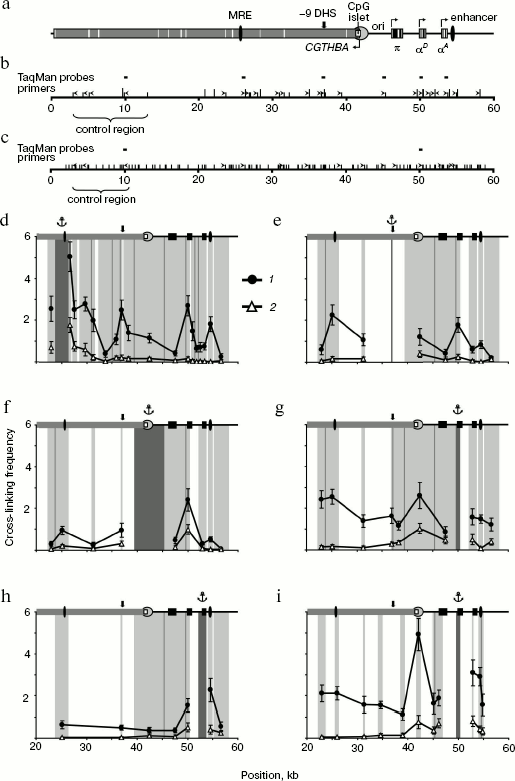
Table 1. Primers used for 3C analysis

Note: Here and in Table 2, the figure in the
primer name corresponds to the primer 5´ end position in
accordance with the AY016020 DNA sequence (GeneBank) [5]. Letters F and R designate forward and reverse
primers (according to the direction of α-globin gene
transcription).
Table 2. TaqMan probes used for 3C
analysis

Note: FAM, fluorescent dye at probe 5´-end; BHQ-1, fluorescence
quencher inside probe at T.
3C analysis of the housekeeping gene CGTHBA site was carried out as the qualitative and quantitative control of DNA in samples obtained from lymphoid and erythroid cells. We have analyzed three pairs of restriction fragments in the case of restriction by BamHI/BglII and two pairs in the case of restriction by MboI (“control region”; figure, panels (b) and (c)). The ligation frequency values, obtained in each case, were averaged and taken by convention as 1.
Comparison of chicken α-globin gene domain spatial organization in erythroid and non-erythroid cells. This work deals with comparison of two types of chicken cells, erythroid cells of 10-day-old embryos, in which gene π is already not active, while genes αD and αA are actively transcribed [21-23], and cultured lymphoid DT40 cells in which α-globin genes are not transcribed.
First, we analyzed the interaction of the major regulatory element (MRE) with the downstream positioned fragments (except very small ones). In this case, the primer complementary to the MRE-containing restriction fragment was used as an anchor. In lymphoid cells the ligation frequency of the MRE-carrying anchor fragment with the rest fragments sharply decreased as the distance between tested fragments increased and then it became rather low (figure, panel (d), 2). This is typical of chromatin regions not organized in loops [10]. In embryonic erythrocytes, on the contrary, the frequency of MRE interaction with most fragments was significantly higher. This especially concerns fragments containing the erythroid-specific enhancer, the αD gene promoter, and the DNase hypersensitive site located 9 kb upstream from α-globin gene cluster (-9 DHS) (figure, panel (d), 1). It is interesting that the MRE did not interact with the π and αA genes. As for the αA gene, this result was unexpected because this gene is actively transcribed in chicken 10-day-old embryonic erythrocytes [21-23]. To confirm this result, we analyzed the ligation frequency of the opposite end of the restriction fragment containing the αA gene. Frequencies of the MRE-containing fragment ligation with both ends of the αA gene-carrying fragment were practically identical.
In the next series of experiments, the “anchor” was successively transferred onto the fragments containing -9 DHS, the housekeeping gene CGTHBA promoter, the αD gene promoter, and the αA gene. Results of these experiments, shown in figure (e-h), as a whole supported the conclusions drawn from experiments with the “anchor” on MRE. Besides, it was shown that in embryonic erythrocytes the fragment containing the housekeeping gene promoter interacts with all the above-mentioned elements (figure, panel (f), 1). 3C analysis with the anchor primer on fragments containing the housekeeping gene promoter and promoter of the αD gene has shown that these fragments interact both in erythroid and lymphoid cells, although not very intensively (figure, panels (f) and (g)). The interaction of both genes with each other and with the erythroid-specific enhancer was shown in experiments with “anchor” on the αD and αA genes in embryonic erythrocytes (figure, panels (g) and (h)). At the same time, it was shown that gene αA is not adjacent to MRE, -9 DHS, and promoters of the housekeeping and the π genes (figure, panel (h)).
To improve the results, experiments with an anchor on the αD gene promoter were repeated after fixed chromatin cleavage by the fine-cleaving restriction endonuclease MboI. Following such restriction, promoters of the π and αA genes appear within separate fragments, whereas elements like MRE, -9 DHS, and the housekeeping gene promoter appear within smaller fragments. The data support the conclusions drawn from experiments with the BamHI/BglII restriction. Thus, it was shown that the αD gene promoter interacts with MRE, the housekeeping gene CGTHBA promoter, and with the erythroid-specific enhancer. In this case, interaction with the CGTHBA gene promoter was detected both in erythroid and lymphoid cells in full agreement with results of the preceding series of experiments. Besides, the interaction between promoters of the αD and αA genes was also observed (figure, panel (i)). The region including -9 DHS was not analyzed because MboI cleaves this site into very small fragments.
DISCUSSION
To correctly interpret the results of this work, it is necessary to take into consideration that 3C analysis allows one to estimate quantitatively only the probability of interaction between different fragments of the locus under study. Possible alternation of the short-lived configurations of the domain within the same cells should be taken into account. Besides, the domain spatial structure can be different in different cells present in the population. To make the discussion easier, we shall consider only the most interesting spatial configurations of the domain, which are typical of 10-day-old embryonic erythrocytes and cultured lymphoid cells. At the same time, we understand that the most complex configurations can co-exist with others, including the simplest linear configuration.
The most obvious conclusion from this work is that spatial organization of the chicken α-globin gene domain is basically different in erythroid and non-erythroid cells. The domain has a relatively extended configuration in cells not expressing α-globin genes, such as lymphoid cells DT40 (Scheme 2a). The only interaction revealed in these cells was between the αD gene promoter and the CpG islet located at a distance of about 4 kb upstream from the α-globin gene cluster and containing promoter of the housekeeping gene CGTHBA [5, 24] and the site of replication origin [25, 26]. This interaction seems unstable (short-lived) because frequency of interaction between the αD gene promoter and the CpG islet significantly increases in the globin-producing cells. Thus, in non-erythroid cells the linear form of the α-globin gene domain exists in equilibrium with the loop formed upon interaction of the αD gene promoter with the CpG islet. This result contradicts observations concerning the mouse α-globin gene domain, where promoters of α-globin genes interact with CpG islets, surrounding the domain only in cells that transcribe globin genes [14]. Probably, spatial organization of α-globin gene domain in cells that do not express globins is different for chicken and mice. This may be due to differences in the large-scale domain organization [27].
Spatial organization of the chicken α-globin gene domain in erythroid and lymphoid cells: a) DT-40 cells; b) 10-day-old embryonic erythrocytes (large arrows in (b) designate active transcription status of αD and αA genes)
Scheme 2
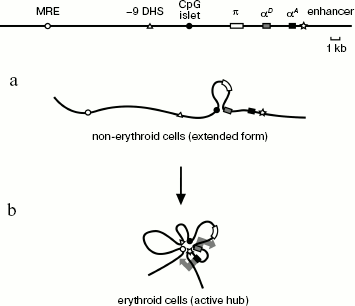
In 10-day-old embryonic erythrocytes actively transcribing adult αD and αA globin genes, interaction between αD gene promoter and CpG islet, containing the housekeeping gene promoter, becomes more frequent (or happens in a larger fraction of cells). Besides, three additional elements are involved in this complex: major regulatory element MRE, erythroid-specific site of hypersensitivity to DNase I localized at a distance of 9 kb upstream from gene π (-9 DHS), and the erythroid-specific enhancer located immediately after the αA gene. As for the DNase I hypersensitivity site, it is only known that this element is co-localized with MAR element [15]. No -9 DHS [28] analog exists in the mouse and human α-globin gene domains. Evidently, this element contains binding sites of some erythroid-specific transcription factors involved in assembly of an active block of regulatory elements (chromatin “hub”), characteristic of globin-expressing erythroid cells (Scheme 2b). We carried out computer analysis of DNA sequence in the -9 DHS region and found four potential binding sites of erythroid-specific transcription factors GATA1 and GATA2.
In both cell models, no interaction of embryonic α-globin gene π with any regulatory element was found. This seems logical because gene π is expressed at the yolk sac stage, whereas it is not active in erythroid cells of 10-day-old embryos [22] as well in lymphoid cells. The observed absence of the αA gene association with the upstream regulatory elements and first of all with MRE was more unexpected. In this connection, it should be remembered that the αA gene is actively transcribed in 10-day-old embryonic erythrocytes [22]. Although the frequency of MRE interaction with the αA gene did not reach the zero level, it was significantly lower than that of MRE interaction with the αD gene promoter (figure, panel (d)). The same is also true of fragments containing -9 DHS (figure, panel (e)) and CpG islet (figure, panel (f)). Keeping in mind the fact that the αA gene promoter is located near the αD gene promoter and erythroid-specific enhancer, which interact on their own with the upstream regulatory elements, it seems unlikely concerning the situation when frequency of αA gene ligation with the upstream regulatory elements reached the same low levels as those in lymphoid cells, even if in reality the αA gene does not interact with these elements. At the same time, we understand that even relatively low ligation frequency is not enough to prove the lack of significance of interaction, because it can be short-term and/or rare, but by no means random. Thus, we cannot exclude the possibility of αA gene promoter interaction with any regulatory element of the domain. Nevertheless, regulation of αD and αA gene activities by different mechanisms seems quite definite. For example, a different situation is observed in the human β-globin gene domain where the locus control region (LCR) alternatively interacts with all active promoters [10, 12, 29].
It seems quite probable that the αA gene in the chicken α-globin gene domain is activated by the erythroid-specific enhancer located in the immediate vicinity of this gene at a distance of about 0.9 kb downstream from it [9]. No similar enhancers exist in the mouse and human α-globin gene domains. However, it is difficult to show reliably by 3C analysis that this enhancer interacts with the αA gene promoter because these elements are too close to each other. In any case, our results suggest such a possibility (figure, panel (h)). On the other hand, it is quite possible that the enhancer, located so closely to the controlled promoter, is able to activate this promoter by a different mechanism, not involving DNA loop formation. Finally, it can be supposed as an additional model of the αA gene transcription activation that interaction of several regulatory elements in the region upstream from the αD gene may provide for activation of the whole “adult” subdomain of α-globin genes, including genes αD and αA.
REFERENCES
1.Higgs, D. R., and Wood, W. C. (2008) Curr. Opin.
Hematol., 15, 176-183.
2.Zhang, H. B., Liu, D. P., and Liang, C. C. (2002)
Int. J. Hematol., 76, 420-426.
3.Recillas-Targa, F., and Razin, S. V. (2001)
Crit. Rev. Eukaryot. Gene Exp., 11, 227-242.
4.Razin, S. V., and Ioudinkova, E. S. (2007)
Biochemistry (Moscow), 72, 467-470.
5.Flint, J., Tufarelli, C., Peden, J., Clark, K.,
Daniels, R. J., Hardison, R., Miller, W., Philipsen, S., Tan-Un, K. C.,
McMorrow, T., Frampton, J., Alter, B. P., Frischauf, A. M., and Higgs,
D. R. (2001) Hum. Mol. Genet., 10, 371-382.
6.Razin, S. V., Ioudinkova, E. S., and Scherrer, K.
(2000) J. Mol. Biol., 209, 845-852.
7.Razin, S. V., Shen, K., Ioudinkova, E., and
Scherrer, K. (1999) J. Cell. Biochem., 74, 38-49.
8.Vyas, P., Vickers, M. A., Picketts, D. J., and
Higgs, D. R. (1995) Genomics, 29, 679-689.
9.Knezetic, J., and Felsenfeld, G. (1989) Mol.
Cell. Biol., 9, 893-901.
10.Tolhuis, B., Palstra, R. J., Splinter, E.,
Grosveld, F., and de Laat, W. (2002) Mol. Cell, 10,
1453-1465.
11.Vakoc, C. R., Letting, D. L., Gheldof, N.,
Sawado, T., Bender, M. A., Groudine, M., Weiss, M. J., Dekker, J., and
Blobel, G. A. (2005) Mol. Cell, 17, 453-462.
12.Gribnau, J., de Boer, E., Trimborn, T., Wijgerde,
M., Milot, E., Grosveld, F., and Fraser, P. (1998) EMBO J.,
17, 6020-6027.
13.Wijgerde, M., Grosveld, F., and Fraser, P. (1995)
Nature, 377, 209-213.
14.Zhou, G. L., Xin, L., Song, W., Di, L. J., Liu,
G., Wu, X. S., Liu, D. P., and Liang, C. C. (2006) Mol. Cell.
Biol., 26, 5096-5105.
15.De Moura Gallo, C. V., Vassetzky, Y. S., Huesca,
M., and Scherrer, K. (1992) Biochem. Biophys. Res. Commun.,
184, 1181-1189.
16.Razin, S. V., de Moura Gallo, C. V., and
Scherrer, K. (1994) Mol. Gen. Genet., 242, 649-652.
17.Valadez-Graham, V., Razin, S. V., and
Recillas-Targa, F. (2004) Nucleic Acids Res., 32,
1354-1362.
18.Dekker, J., Rippe, K., Dekker, M., and Kleckner,
N. (2002) Science, 295, 1306-1311.
19.Liu, Z., and Garrard, W. T. (2005) Mol. Cell.
Biol., 25, 3220-3231.
20.Splinter, E., Grosveld, F., and de Laat, W.
(2004) Meth. Enzymol., 375, 493-507.
21.Bruns, G. A., and Ingram, V. M. (1973) Dev.
Biol., 30, 455-459.
22.Weintraub, H., Larsen, A., and Groudine, M.
(1981) Cell, 24, 333-344.
23.Singal, R., van Wert, J. M., and Ferdinand, L.,
Jr. (2002) Blood, 100, 4217-4222.
24.Klochkov, D., Rincon-Arano, H., Ioudinkova, E.
S., Valadez-Graham, V., Gavrilov, A., Recillas-Targa, F., and Razin, S.
V. (2006) Mol. Cell. Biol., 26, 1589-1597.
25.Razin, S. V., Kekelidze, M. G., Lukanidin, E. M.,
Scherrer, K., and Georgiev, G. P. (1986) Nucleic Acids Res.,
14, 8189-8207.
26.Verbovaia, L., and Razin, S. V. (1995)
Gene, 166, 255-259.
27.Tufarelli, C., Hardison, R., Miller, W., Hughes,
J., Clark, K., Ventress, N., Frischauf, A. M., and Higgs, D. R. (2004)
Genome Res., 14, 623-630.
28.Hughes, J. R., Cheng, J. F., Ventress, N.,
Prabhakar, S., Clark, K., Anguita, E., de Gobbi, M., de Jong, P.,
Rubin, E., and Higgs, D. R. (2005) Proc. Natl. Acad. Sci. USA,
102, 9830-9835.
29.Palstra, R. J., Tolhuis, B., Splinter, E.,
Nijmeijer, R., Grosveld, F., and de Laat, W. (2003) Nat. Genet.,
35, 190-194.