Engineering, Expression, and Purification of “Soluble” Human Cytochrome P45017alpha and Its Functional Characterization
T. A. Pechurskaya, O. P. Lukashevich, A. A. Gilep, and S. A. Usanov*
Institute of Bioorganic Chemistry, National Academy of Sciences of Belarus, 220141 Minsk, Belarus; E-mail: usanov@iboch.bas-net.by* To whom correspondence should be addressed.
Received February 11, 2008
To engineer a “soluble” form of membrane-bound cytochrome P45017alpha (CYP17)--a key enzyme in steroid hormone biosynthesis--in the present work we have built a computer model of the tertiary structure of the hemeprotein, identified the surface hydrophobic amino acid residues, substituted these residues for more hydrophilic ones, and expressed and purified hydrophilized forms of CYP17. We have constructed and purified the following mutant forms of human CYP17: CYP17dH (CYP17 with deleted hydrophobic N-terminal sequence (Delta23)) and CYP17mod (CYP17dH with substituted cluster of hydrophobic amino acid residues in the region of the FG-loop). Removal of the N-terminal sequence responsible for interaction with the membrane does not dramatically change the association of the protein with the membrane. However, CYP17mod containing hydrophilic FG-loop is mostly localized in the cytosolic fraction. Thus, in the present work we for the first time engineered a “soluble” form of the usually membrane-bound human CYP17 that is not bound to membrane. The expression degree of CYP17mod is approximately 900 nmol/liter of culture. The hemeprotein can be purified to apparent homogeneity without using detergents at any purification step. It is shown that replacement of hydrophobic amino acid residues in the FG-loop region does not change the metabolic profile during hydroxylation of steroids that is characteristic for wild type CYP17. Besides, the modification of the hemeprotein does not affect the affinity of CYP17 to steroid substrates. The engineered “soluble” form of human CYP17 is used as a subject for crystallization of the hemeprotein.
KEY WORDS: cytochrome P45017alpha, heterologous expression, CYP17 in bacteria, “soluble” form of cytochrome P45017alphaDOI: 10.1134/S0006297908070092
Abbreviations: DTT) dithiothreitol; IPTG) isopropyl-beta-D-thiogalactopyranoside; OD) optical density; P4) progesterone; P5) pregnenolone; PMSF) phenylmethylsulfonyl fluoride.
Cytochrome P450 represents a huge superfamily of hemeproteins,
components of the monooxygenase systems involved in metabolism of a
wide spectrum of endogenous and exogenous compounds including
biosynthesis and catabolism of steroid hormones, and biotransformation
of drugs and toxins [1, 2].
Cytochrome P45017alpha (17alpha-steroid
hydroxylase/17,20-lyase, CYP17) is the key enzyme in steroid hormone
biosynthesis [3]. This microsomal hemeprotein is
unique in its ability to catalyze two independent reactions at the same
active site. CYP17 catalyzes selective 17alpha-hydroxylation of
pregnenolone (P5) and progesterone (P4) to form corresponding
17alpha-hydroxylated derivatives, which serve as precursors of
glucocorticoid hormones. Besides, CYP17 catalyzes conversion of
17alpha-hydroxyprogesterone (17OH-P4) and
17alpha-hydroxypregnenolone (17OH-P5) via the 17,20-lyase
reaction to form androstenedione and dehydroepiandrosterone,
correspondingly, which are intermediate steroids in sex hormone
biosynthesis. The ratio of 17alpha-hydroxylase and 17,20-lyase
activities of CYP17 is an important physiological factor that
determines the directions of steroid hormone biosynthesis either to
biosynthesis of glucocorticoid or sex hormones [4,
5].
Abnormalities in the function of this enzyme are closely associated with the set of serious disturbances in steroidogenesis and are the main reason for severe hereditary disease in human [6-8]. Besides, it has been shown that human CYP17 plays an essential role in the development of some cancers, which makes the development of selective inhibitors for human CYP17 an important task [9]. At the present time there are no effective inhibitors which might be selective and specific with respect to human CYP17 catalyzed reactions. The development of new selective inhibitors as well as studies of structure-function peculiarities of CYP17 needs information on the structure of the active site and tertiary organization of the molecule.
The most effective approach at the present time to study structure of protein molecules is X-ray analysis, which requires the preparation of crystals of macromolecules. So far the crystal structure of steroidogenic cytochrome P450 is not known. This is explained by the fact that as for majority membrane-bound proteins, eukaryotic cytochrome P450s are characterized by high content of exposed hydrophobic regions serving for anchoring of the protein in the membrane and connected with the high tendency of the protein molecules to aggregate even in the presence of detergents, which are necessary for their purification to homogenous state. Crystallization of such proteins is possible only after their hydrophilization to decrease the content of surface-exposed hydrophobic regions.
It has been shown for some microsomal cytochrome P450 metabolizing drugs that removal or modification of the highly hydrophobic N-terminal sequence (signal sequence responsible for translocation and anchoring of cytochrome P450 in membrane [10]) results in increase of expression level and allows the preparation of much higher quantities of functionally active “soluble” protein not associated with membrane [11-14]. In the case of steroidogenic cytochrome P450, this approach proved to be ineffective. In part, it was shown for bovine CYP17 that removal of the highly hydrophobic N-terminal signal sequence (Delta2-17) does not result in a “soluble” form of the hemeprotein [15].
A characteristic feature of eukaryotic cytochrome P450 is the presence of a cluster of hydrophobic amino acid residues in the region of FG-loop. It is thought that this region together with the N-terminal sequence forms a hydrophobic surface of the protein that participates in interaction with microsomal membrane [16], and also is involved in formation of a hydrophobic channel for substrate penetration into the active site of the hemeprotein, which is extremely important for microsomal cytochrome P450 metabolizing highly hydrophobic steroid substrates [17, 18].
In the present work, with the goal of engineering a “soluble” form of normally membrane-bound human CYP17, we computer modeled the tertiary structure of CYP17 and performed site-directed mutagenesis of hydrophobic regions exposed on the surface of protein globule: modified the N-terminal sequence and replaced the cluster of hydrophobic amino acid residues located in the region of the FG-loop. We compared the characteristics of full-length human CYP17 (CYP17fl), CYP17 with deleted N-terminal hydrophobic amino acid sequence (CYP17dH), and CYP17dH with replacement of the cluster of hydrophobic amino acids in the region of the FG-loop (CYP17mod) and engineered for the first time a “soluble” form of human CYP17mod that is not bound with membrane.
MATERIALS AND METHODS
The following chemicals were used in the present work: progesterone (P4), pregnenolone (P5), sodium cholate, Triton X-100, NADPH, phenylmethylsulfonyl fluoride (PMSF), delta-aminolevulinic acid (ALA), SDS, Coomassie brilliant blue R-250, Hepes, and Tris (Sigma, USA); Ni-NTA-Agarose (Qiagen, USA); Emulgen 913 (Kao Atlas, Japan); agarose, isopropyl-beta-D-thiogalactopyranoside (IPTG), and dithiothreitol (DTT) (Gibco BRL, USA); Bacto-Tryptone, Bacto-Peptone, and Bacto-Yeast extract (Difco Laboratories, USA); Bio-Gel HTP (Bio-Rad, USA); methylene chloride (Ekos-1, Russian Federation). Restriction enzymes and other enzymes for DNA modification were from New England Biolabs (USA) and Promega (USA).
Engineering of expression vector for human CYP17fl, CYP17dH, and CYP17mod. To construct an expression vector for human CYP17fl, cDNA coding human CYP17 was amplified using the following primers: 5´-CAAGGAATTCTCTGGGCGGCC and 3´-ATGTCGACCCTTCAGCCTGGGCTTCCC. The fragment obtained after restriction of the amplicon with EcoRI and SalI was cloned into expression plasmid pCW_bov17_his [19], from which the EcoRI-SalI fragment was preliminarily removed.
To obtain expression plasmid CYP17dH, we replaced the highly hydrophobic N-terminal sequence of CYP17 (Delta2-23 N-terminal amino acid residues) for amino acid sequence MAKKT as reported for CYP2C3 [13], while into the C-terminal sequence we added a 4His-cluster to simplify the purification procedure. To construct expression plasmid CYP17mod and replace the hydrophobic cluster located in the FG-loop, cDNA for human CYP17 with modified N-terminal sequence was amplified using the following primers: direct 5´-ACTCCTGCGCAACGTAATCAA-3´ and reverse 5´-TTGATTACGTTGCGCAGGAGT-3´. The constructs containing the cDNA coding the studied hemeproteins were cloned into expression vector pCW (for CYP17fl) or pTrc99 (CYP17dH and CYP17mod).
Expression of CYP17fl, CYP17dH, and CYP17mod. Escherichia coli JM109 competent cells were transformed with plasmids containing CYP17 (CYP17fl, CYP17dH, or CYP17mod). Transformed cells were screened on Petri dishes with LB-agar and ampicillin (100 µg/ml). Separate colonies were used to inoculate 5 ml of LB-media containing ampicillin (100 µg/ml), and incubated at 37°C with continuous shaking at 180 rpm for 16-24 h. Then 0.5 liter of TB-medium containing 100 mM potassium-phosphate buffer, pH 7.2, and ampicillin (100 µg/ml) was inoculated with 3 ml overnight culture, and the mixture was incubated at 37°C under continuous shaking at 180 rpm. After reaching absorbance OD600 ~ 0.4 (about 5 h), protein expression was stimulated by adding of IPTG (0.5 mM), also adding ALA (0.65 mM) and ampicillin (100 µg/ml). After 48 h of incubation at 26°C under continuous shaking at 140 rpm, cells were collected by centrifugation at 3000 rpm for 10 min. The pellet was suspended in 50 mM Tris-HCl buffer, pH 7.4, containing 20% glycerol, 0.3 M NaCl, 0.5 mM PMSF, and 50 µM progesterone (1 volume of cells/4 volumes of buffer). Cells were frozen to -73°C.
Isolation and purification of recombinant proteins. To purify recombinant CYP17fl and CYP17dH, cells were thawed and treated on ice with ultrasound using a UZDN-2 device six times for 30 sec with 2 min intervals. The sonicated cell suspension was centrifuged (3000 rpm, 10 min; Beckman, USA) to remove unbroken cells. Recombinant protein was solubilized from membranes by adding drops of Emulgen 913 to final concentration 1% (3 mg detergent per mg protein) under continuous mixing for 1 h. The suspension was centrifuged 1 h at 100,000 rpm to remove non-solubilized membrane structures.
At the first step of purification of recombinant protein, solubilized proteins in 50 mM Tris-HCl buffer, pH 7.4, containing 20% glycerol, 0.3 M NaCl, and 50 µM progesterone were applied to a column with DEAE-cellulose to remove impurities that are not removed by metal-affinity chromatography. The eluate was applied directly to a column with Ni-NTA-Agarose, equilibrated with buffer A (50 mM Tris-HCl buffer, pH 7.4, containing 20% glycerol, 0.3 M NaCl, and 50 µM progesterone). The column was washed with 2-3 volumes of buffer A containing 0.2% Emulgen 913 and then with 10 volumes of buffer A containing 0.2% Emulgen 913 and 100 mM glycine (buffer B). The proteins were eluted from the column with buffer B containing 50 mM histidine. Fractions containing CYP17 were pooled and applied to a column with hydroxyapatite for subsequent purification. The column was washed with 10 volumes of 50 mM potassium-phosphate buffer, pH 7.2, containing 20% glycerol, 50 µM progesterone, 0.2% Emulgen 913, and 0.1 mM DTT. Proteins were eluted from the column with 0.3 M potassium-phosphate buffer, pH 7.2, containing 20% glycerol, 50 µM progesterone, 0.2% Emulgen 913, and 0.1 mM DTT. Highly purified hemeproteins were stored at -73°C.
Preliminary experiments have shown that recombinant CYP17mod is mostly localized in cytosol, and therefore the step of solubilization of the hemeprotein from membrane in the purification protocol was omitted. CYP17mod was purified according to a procedure similar to that developed for purification of full-length CYP17, except that detergent was not used at any steps of the CYP17mod purification procedure. The purified hemeprotein was dialyzed against 50 mM Tris-HCl buffer, pH 7.4, containing 20% glycerol and 0.3 M NaCl.
Highly purified recombinant rat NADPH-cytochrome P450 reductase and rat cytochrome b5 were purified as previously described [19].
Determination of 17alpha-hydroxylase activity of CYP17. The 17alpha-hydroxylase activity of recombinant CYP17 was determined in a reconstituted system at 37°C in 25 mM Hepes-NaOH buffer, pH 7.2. Aliquots of concentrated proteins (0.5 µM CYP17 and 1 µM rat NADPH-cytochrome P450 reductase) were mixed and incubated at room temperature for 5 min. Progesterone dissolved in ethanol was added to the reaction mixture to final concentration 50 µM and incubated 10 min at 37°C. The reaction was started by adding NADPH to final concentration 0.25 mM. Aliquots (0.5 ml) were taken from the incubation mixture at selected time intervals. The reaction was stopped by adding 5 ml of methylene chloride. The mixture was vigorously shaken, and layers were separated by centrifugation at 3000 rpm for 10 min. The aqueous layer was carefully removed, and the organic layer was dried under a flow of argon. Steroids were solved in 100 µl of methanol and analyzed by HPLC (Shimadzu LC-10AD using µ-Bondapak C18 column, 3.9 × 300 mm) with an SPD-10A UV spectrophotometric detector.
Determination of 17,20-lyase activity of CYP17. The 17,20-lyase activity of CYP17 was determined in a reconstituted system at 37°C in 25 mM Hepes-NaOH buffer, pH 7.2. Aliquots of concentrated proteins (0.5 µM CYP17 and 1 µM rat NADPH-cytochrome P450 reductase) were mixed and incubated at room temperature for 5 min. The 17alpha-hydroxypregnenolone (17OH-P5), dissolved in ethanol, was added to the reaction mixture to final concentration 50 µM, and the mixture incubated 10 min at 37°C. The reaction was started by adding NADPH to final concentration 0.25 mM. Aliquots (0.5 ml) were taken from the incubation mixture at selected time intervals. The reaction was stopped by adding 5 ml of methylene chloride. Steroids were analyzed using HPLC as described above.
Determination of effect of cytochrome b5 on 17,20-lyase reaction catalyzed by CYP17. The effect of cytochrome b5 on the 17,20-lyase reaction was studied in a reconstituted system at 37°C in 25 mM Hepes-NaOH buffer, pH 7.2, containing concentrated proteins in ratio 0.5 µM CYP17/1 µM NADPH-cytochrome P450 reductase/0.5 µM cytochrome b5. An ethanolic solution of 17OH-P5 was added to the reaction mixture to final concentration 50 µM, and the mixture incubated 10 min at 37°C. The reaction was started by adding NADPH to final concentration 0.25 mM. Aliquots (0.5 ml) were withdrawn from the incubation mixture at selected time intervals. The reaction was stopped by adding 5 ml of methylene chloride. Steroids were analyzed using HPLC as described above.
Modeling of the tertiary structure of CYP17. At the first step of homologous modeling, we did multiple alignments of amino acid sequences of cytochrome P450 with known crystal structure. Based on results of multiple alignments, we chosen as protein template cytochrome P4501A2 (chain A, 2hiv4 A), the crystal structure of which has been solved with resolution 1.95 Å. The amino acid sequence homology for the two proteins is 30%. Using the software package Sybyl 7.3, amino acid residues starting from 48 to 468 of the template protein that were not identical to residues of CYP17 were sequentially replaced with corresponding amino acids of the modeled hemeprotein. Hydrogen atoms were added to construct the model, and the geometry of final model was optimized. Energy minimization was done using the method of Powell in force field Tripos using atomic charges from the Kollman library (“Kollman-All”). Heme was introduced in the active site, and its geometry was optimized in force field Tripos using atomic charges calculated according to Gasteiger-Huckel. The procedure does molecular docking with a small number of iterations of energy minimization.
Analytical methods. Cytochrome P450 concentration was determined spectrophotometrically using molar extinction coefficient for absorbance of complex CO-reduced hemeprotein epsilon450-490 = 91 mM-1·cm-1 [20]. The concentrations of cytochrome b5 and NADPH-cytochrome P450 reductase were determined from the absolute absorption spectrum using molar extinction coefficient epsilon413 = 117 mM-1·cm-1 for cytochrome b5 [21] and epsilon456 = 21.4 mM-1·cm-1 for NADPH-cytochrome P450 reductase [21]. Purification degree of proteins was determined electrophoretically using SDS-PAGE.
RESULTS AND DISCUSSION
Modeling of tertiary structure of CYP17 and analysis of the model. In the present work, in order to construct a “soluble” form of membrane-bound CYP17, we preliminarily built a model of the tertiary structure of the hemeprotein using homology modeling of tertiary structure and performed a search for hydrophobic fragments of CYP17 exposed on the surface of the protein molecule. Since there is still no crystal structure solved for steroidogenic cytochrome P450, soluble bacterial cytochromes P450cam and P450BM-3 usually serve as a model for building the tertiary structure of CYP17 [22-26]. It is necessary to stress that all constructed theoretical models of the tertiary structure do not contain highly hydrophobic N-terminal sequence of human CYP17.
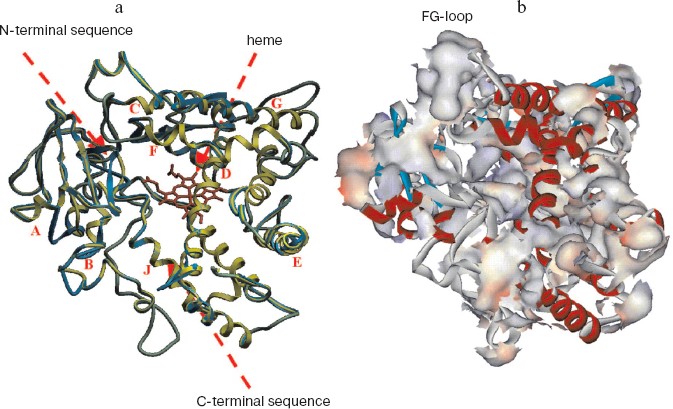
As a template to construct a model for tertiary structure of human CYP17, we chosen microsomal cytochrome P4501A2, the crystal structure of which has been solved recently with resolution 1.95 Å [27], and homology of the amino acid sequences of the two hemeproteins is 30%. The model of the tertiary structure of human CYP17 is shown in Fig. 1 (see color insert). Root mean square deviation of distance between tertiary structure for human CYP17 known from the literature [25] and the constructed model is 7.95 Å. Heme was introduced into the active site. The internal volume of the molecular model of CYP17 is 189 Å3.
Analysis of the surface of the model for tertiary structure of human CYP17 (Fig. 1b) revealed several regions with definitely increased content of hydrophobic residues exposed on the surface of the molecule. Based on the analysis of these data, we chose surface-exposed hydrophobic amino acid residues localized in non-structured elements of the hemeprotein and according with this modified the N-terminal sequence and the cluster of hydrophobic amino acid residues V218T, W220A, L221Q, K222R, I223N, and F224Q located in the region of the FG-loop.Fig. 1. Model of tertiary structure of human CYP17. a) Superposition of the models of tertiary structure of CYP17 constructed in the present work (green) and the model constructed previously (blue) [25]. Latin letters indicate alpha-helixes. b) Distribution of hydrophobic amino acid residues on the surface of human CYP17.
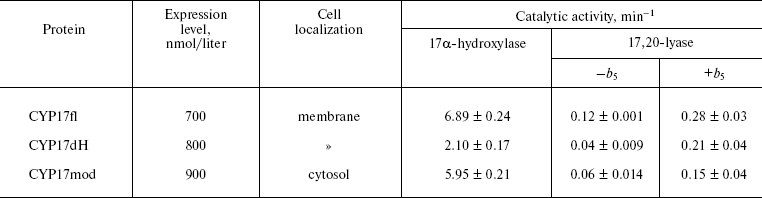
Heterologous expression of CYP17fl, CYP17dH, and CYP17mod. In the present work, we constructed form CYP17dH with shortened N-terminal sequence and CYP17mod with modification of cluster of hydrophobic amino acid residues in the region of the FG-loop (Fig. 2) (see color insert). All abovementioned forms of CYP17 were expressed in E. coli, the expression conditions being optimized to get large quantities of the recombinant hemeprotein (table). The expression level of cytochrome P450 in bacterial cells was estimated from difference spectra of the CO-complex of sodium dithionite reduced hemeprotein using molar extinction coefficient 91 mM-1·cm-1. The 17alpha-hydroxylase activity was determined based on 17alpha-hydroxylation of P4, while 17,20-lyase activity using 17OH-P5 as substrate as described in “Materials and Methods”. In experiments on intracellular localization of expressed proteins, it was shown that localization of the recombinant proteins significantly depends on the degree of modification of the initial form of the full-length hemeprotein. Thus, based on the data on distribution of recombinant protein among subcellular fractions after membrane sedimentation, it was found that the full-length form of CYP17 is predominantly found in membrane fraction (>90%) both at low and high (300 mM NaCl, 20% glycerol) ionic strength. CYP17dH is also mostly associated with membrane, while CYP17mod was found predominantly in cytosol at high ionic strength (300 mM NaCl, 20% glycerol). These data suggest that removal of the N-terminal sequence of CYP17 is not enough to prepare soluble form and indicate the presence of additional contacts of the hemeprotein with the membrane, which appear to be located in the region of the FG-loop.
Purification of recombinant proteins. The recombinant proteins were purified to apparent homogeneity using metal-affinity chromatography with subsequent chromatography on hydroxyapatite (Fig. 3b). It is necessary to stress that purification of CYP17mod does not require detergent, which is extremely important for the following procedure of protein crystallization. Besides, it was also found that the recombinant hemeprotein does not have a tendency to aggregation in the absence of detergent.Fig. 2. Alignment of amino acid sequences of CYP17fl, CYP17dH, and CYP17mod. The sequence was modified as described in “Materials and Methods”. Model of the tertiary structure of human CYP17. The modified amino acid residues in the region of the FG-loop are indicated in red.
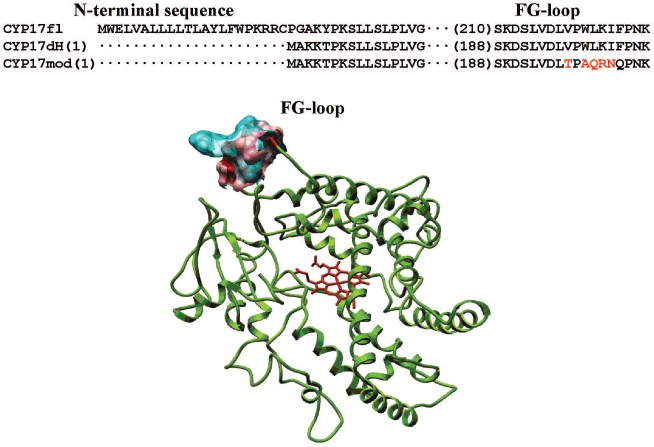
Table. Comparative characteristics of CYP17fl, CYP17dH, and CYP17mod
An absolute absorbance spectrum of highly purified human CYP17mod is shown in the Fig. 3a. The purified hemeprotein is mostly in low-spin state having maximum of absorbance at 414 nm, but addition of substrate results in a shift of the absorption maximum to 393 nm, which is characteristic for high-spin state with substrate bound. Chemical reduction of CYP17mod with sodium dithionite shifts the absorption maximum to 412 nm. Subsequent bubbling of carbon monoxide through the reduced hemeprotein results in the appearance of characteristic carbon monoxide complex with reduced cytochrome P450 with absorbance at 449 nm. Significant prevalence of absorbance at 449 nm over that of at 420 nm indicates that most of the highly purified recombinant CYP17mod is in native form.Fig. 3. a) Absolute absorption spectra of highly purified human cytochrome CYP17mod. Curves: 1-4) oxidized, oxidized in complex with substrate, reduced with sodium dithionite, and carbon monoxide complex of reduced hemeprotein, respectively. b) SDS-PAGE of CYP17mod: 1-3) highly purified protein, extract of E. coli membranes before purification, and standard proteins, respectively. c) Spectral changes induced by adding 25 µM P4 (1), 17OH-P4 (2), 17OH-P5 (3), or P5 (4) to CYP17mod. Each cell contained 2 µM CYP17mod in 50 mM potassium phosphate buffer, pH 7.2.
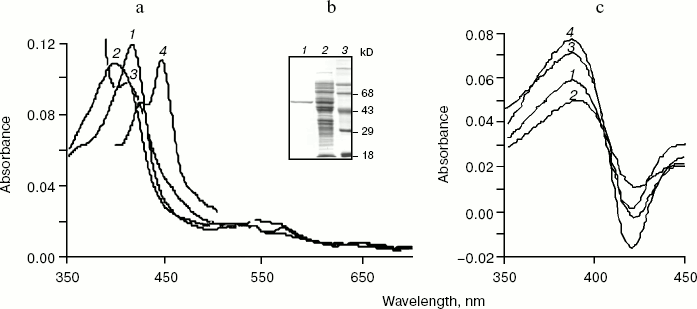
As shown in Fig. 3c, CYP17mod demonstrates characteristic type I spectra on interaction with substrates (P4, P5, 17OH-P4, and 17OH-P5) and has affinity to steroid molecules close to the affinity of CYP17fl.
Kinetic characteristics of highly purified hemeproteins. Catalytic activity of purified hemeprotein was studied as described in “Materials and Methods”. Comparison of hydroxylase activities of the purified hemeproteins showed that as compare to 17alpha-hydroxylase activity characteristic for CYP17fl (6.89 min-1), CYP17dH has 2.4-fold decreased catalytic activity (2.10 min-1) that indicates that the hydrophobic N-terminal amino acid sequence appears to participate in formation of hydrophobic interactions with its redox partner, and that its removal results in decrease of 17alpha-hydroxylase activity of the hemeprotein. However, further modification of CYP17dH does not result in decrease of catalytic activity, and in fact for CYP17mod it is 5.95 min-1 (table).
The removal of the N-terminal sequence is more critical for demonstration of 17,20-lyase activity of human CYP17 (table). The 17,20-lyase activity of CYP17fl is registered at the level 0.12 min-1. In the case of CYP17dH, 17,20-lyase activity is 3-fold decreased to the level 0.04 min-1. CYP17mod demonstrates 2-fold lower 17,20-lyase activity compared to CYP17fl. These data are in accordance with clinical data indicating that mutation of CYP17 result in partial or complete lost of 17,20-lyase activity and much less effect on 17alpha-hydroxylase activity of the mutants [26, 28, 29].
Studies on the effect of the physiological effector, cytochrome b5, on 17,20-lyase reaction catalyzed by CYP17mod have shown that cytochrome b5 causes a common stimulating effect on the reaction as shown earlier [30, 31]. Thus, catalytic activity of CYP17fl increased to 0.28 min-1 in the presence of cytochrome b5, CYP17dH to 0.21 min-1, and CYP17mod to 0.16 min-1 (table). Thus, independent of the degree of modification, the engineered forms of cytochrome P450 are functionally active and retain their ability to catalyze 17alpha-hydroxylase and 17,20-lyase reaction of steroid hydroxylations.
Finally, in the present work we developed an approach and proposed a method for preparation of a water-soluble form of the hydrophobic, membrane-bound microsomal steroidogenic cytochrome CYP17. The removal of N-terminal amino acid sequence of the hemeprotein and modification of hydrophobic amino acid residues near the FG-loop allow to reach very high expression level of functionally active recombinant hemeprotein, which does not have a tendency to form aggregates in the absence of detergent. The cytochrome P450 obtained is catalytically active and retains the profile of activity characteristic for wild type CYP17, which makes our hemeprotein a promising object for further crystallization and resolution of the tertiary structure.
REFERENCES
1.Usanov, S. A., Chashchin, V. L., and Akhrem, A. A.
(1990) in Molecular Mechanisms of Adrenal Steroidogenesis and
Aspects of Regulation and Application, Vol. 3 (Ruckpaul, K., and
Rein, H., eds.) Akademie-Verlag, Berlin, pp. 1-57.
2.Nebert, D. W., and Russel, D. W. (2002)
Lancet, 360, 1155-1162.
3.Lieberman, S., and Warne, P. A. (2001) J.
Steroid Biochem. Mol. Biol., 78, 299-312.
4.Shet, M. S., Fisher, C. W., Tremblay, Y., Belanger,
A., Conley, A. J., Mason, J. I., and Estabrook, R. W. (2007) Drug
Metab. Rev., 39, 289-307.
5.Gilep, A. A., Estabrook, R. W., and Usanov, S. A.
(2004) Biochemistry (Moscow), 69, 364-375.
6.Qin, K. N., and Rosenfield, R. L. (1998) Mol.
Cell. Endocrinol., 145, 111-121.
7.Miller, W. L. (2005) Med. Princ. Pract.,
14, Suppl. 1, 58-68.
8.Auchus, R. J. (2001) Endocrinol. Metab. Clin.
North. Am., 30, 101-119.
9.Ntais, C., Polycarpou, A., and Ioannidis, J. P.
(2003) Cancer Epidemiol. Biomarkers Prev., 12,
120-126.
10.Sakaguchi, M., Mihara, K., and Sato, R.
(1984) Proc. Natl. Acad. Sci. USA, 81,
3361-3364.
11.Neve, E. P., and Ingelman-Sundberg, M.
(1999) FEBS Lett., 460, 309-314.
12.Gillam, E. M., Baba, T., Kim, B. R., Ohmori,
S., and Guengerich, F. P. (1993) Arch. Biochem. Biophys.,
305, 123-131.
13.Von Wachenfeldt, C., Richardson, T. H., Cosme,
J., and Johnson, E. F. (1997) Arch. Biochem.
Biophys., 339, 107-114.
14.Cosme, J., and Johnson, E. F. (2000) J.
Biol. Chem., 275, 2545-2553.
15.Sagara, Y., Barnes, H. J., and Waterman, M. R.
(1993) Arch. Biochem. Biophys., 304, 272-278.
16.De Lemos-Chiarandini, C., Frey, A. B., Sabatini,
D. D., and Kreibich, G. (1987) J. Cell Biol.,
104, 209-219.
17.Graham-Lorence, S., Amarneh, B., White, R. E.,
Peterson, J. A., and Simpson, E. R. (1995) Protein
Sci., 4, 1065-1080.
18.Peterson, J. A., and Graham, S. E. (1998)
Structure, 6, 1079-1085.
19.Gilep, A. A., Guryev, O. L., Usanov, S. A., and
Estabrook, R. W. (2001) Biochem. Biophys. Res. Commun.,
284, 937-941.
20.Omura, T., and Sato, R. (1964) J. Biol.
Chem., 239, 2370-2378.
21.Porter, T. D., Wilson, T. E., and Kasper, C. B.
(1987) Arch. Biochem. Biophys., 254, 353-367.
22.Poulos, T. L., Finzel, B. C., and Howard, A. J.
(1987) J. Mol. Biol., 195, 687-700.
23.Boddupalli, S. S., Hasemann, C. A., Ravichandran,
K. G., Lu, J. Y., Goldsmith, E. J., Deisenhofer, J., and Peterson, J.
A. (1992) Proc. Natl. Acad. Sci. USA, 89,
5567-5571.
24.Lin, D., Zhang, L. H., Chiao, E., and Miller, W.
L. (1994) Mol. Endocrinol., 8, 392-402.
25.Auchus, R. J., and Miller, W. L. (1999)
Mol. Endocrinol., 13, 1169-1182.
26.Qiao, J., Hu, R. M., Peng, Y. D., Song, H.
D., Peng, Y. W., Gao, G. F., Hao, J. H., Hu, N. Y., Xu, M. Y., and
Chen, J. L. (2003) Mol. Cell Endocrinol.,
201, 189-195.
27.Sansen, S., Yano, J. K., Reynald, R. L., Schoch,
G. A., Griffin, K. J., Stout, C. D., and Johnson, E. F. (2007) J.
Biol. Chem., 282, 14348-14355.
28.Hahm, J. R., Kim, D. R., Jeong, D. K., Chung, J.
H., Lee, M. S., Min, Y. K., Kim, K. W., and Lee, M. K. (2003)
Metabolism, 52, 488-492.
29.Gupta, M. K., Geller, D. H., and Auchus, R. J.
(2001) J. Clin. Endocrinol. Metab., 86,
4416-4423.
30.Katagiri, M., Suhara, K., Shiroo, M.,
and Fujimura, Y. (1982) Biochem. Biophys. Res.
Commun., 108, 379-384.
31.Akhtar, M. K., Kelly, S. L., and Kaderbhai, M. A.
(2005) J. Endocrinol., 187, 267-274.