Phylogeny of Flowering Plants by the Chloroplast Genome Sequences: in Search of a “Lucky Gene”
M. D. Logacheva1*, A. A. Penin2, T. H. Samigullin3, C. M. Vallejo-Roman3, and A. S. Antonov3
1Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: maria.log@gmail.com2Biological Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received August 22, 2007
One of the most complicated remaining problems of molecular-phylogenetic analysis is choosing an appropriate genome region. In an ideal case, such a region should have two specific properties: (i) results of analysis using this region should be similar to the results of multigene analysis using the maximal number of regions; (ii) this region should be arranged compactly and be significantly shorter than the multigene set. The second condition is necessary to facilitate sequencing and extension of taxons under analysis, the number of which is also crucial for molecular phylogenetic analysis. Such regions have been revealed for some groups of animals and have been designated as “lucky genes”. We have carried out a computational experiment on analysis of 41 complete chloroplast genomes of flowering plants aimed at searching for a “lucky gene” for reconstruction of their phylogeny. It is shown that the phylogenetic tree inferred from a combination of translated nucleotide sequences of genes encoding subunits of plastid RNA polymerase is closest to the tree constructed using all protein coding sites of the chloroplast genome. The only node for which a contradiction is observed is unstable according to the different type analyses. For all the other genes or their combinations, the coincidence is significantly worse. The RNA polymerase genes are compactly arranged in the genome and are fourfold shorter than the total length of protein coding genes used for phylogenetic analysis. The combination of all necessary features makes this group of genes main candidates for the role of “lucky gene” in studying phylogeny of flowering plants.
KEY WORDS: molecular phylogeny, angiosperms, gene sampling, chloroplast genome, plastid RNA polymeraseDOI: 10.1134/S0006297907120061
Abbreviations: TNS) translated nucleotide sequences.
From the beginning of molecular systematics to the present day, the most
popular phylogenetic markers in plants are various regions in the
chloroplast genome. They are used both at the high taxonomic levels
(divisions and classes) and at low levels (genera and species). In most
cases protein-coding genes (and corresponding amino acid sequences) are
used for molecular phylogenetic analysis at the high taxonomic level,
although examples of successful usage in such cases of non-coding
plastome sites are known [1, 2]. The sequence of the large subunit of the ribulose
bisphosphate carboxylase gene (rbcL) was one of first molecular
markers. A work appeared in 1993 reporting the results of analysis of
rbcL sequences in 475 species of seed plants [3]. Many conclusions of these authors were later
confirmed. The set of chloroplast genes for phylogenetic analysis at
the high taxonomic level was subsequently supplemented with genes
matK [4-6], atpB
[7, 8], rpoC1 [9], ndhF [10], rps4
[11-13], and some others.
Nevertheless, different authors have repeatedly pointed out that phylogenetic analysis of a single gene or of a small number of genes could not give reliable and well resolved tree topologies [14-17], which according to many of these authors makes necessary building phylogenetic trees using the sequences of complete chloroplast genomes [18, 19]. However, accumulated data has shown that an insufficient taxon sampling can result in artifacts no less significant than insufficient gene sampling [20, 21]. Combined with the fact that sequencing and analysis of complete chloroplast genomes still remains a resource-consuming problem, this makes difficult their use for investigation of phylogeny.
Thus, an ideal phylogenetic marker should meet the following conditions: 1) a phylogenetic tree constructed using this marker should coincide with the tree constructed using the complete genomes (for the same taxon sampling); 2) it must be significantly shorter, and 3) be arranged compactly. Conditions 2 and 3 are necessary to minimize resources (including computational ones) for sequencing and analysis of complete genomes, which would enable significant increase of taxon sampling analyzed. Hypothetical genes whose phylogenetic analysis gives results identical or even better than obtained for large multigene sets have been designated in the literature as “lucky genes” [22, 23]. Investigations on different groups of organisms suggest that the possibility of “lucky gene” detection and its type depend, first of all, on the group under study.
Thus, analysis of a joined set of 106 genes and sets of individual genes in yeasts of the genus Saccharomyces has shown that a combination of genes allows one to build a completely resolved tree with high (maximal) level of node support, whereas none of the individual genes is able to do the same [16]. A randomly selected 8000 nucleotides (from a set of over 100 kb in length) produced a tree topology identical to that obtained by analysis of the complete set and a high (over 95%) node support. To obtain the same result, it was necessary to combine no less than 20 genes (with mean length of about 1180 bp), and it does not matter which genes, only total length of the set is important. This means that in this case the sequence length but not a specific gene is crucial.
Another attempt to find such genes was undertaken with mitochondrial genes of fish [24]. It was shown that analysis of none of the genes most often used in phylogenetic studies gives results identical to those obtained on the complete genomic sequences. In this case, a group of genes with the best congruence of topologies was revealed. The same evaluations for mammals gave different results, i.e., “lucky genes” appeared to be different for two different classes of vertebrates [23].
The question whether it is possible to find a “lucky gene” for analysis of phylogeny of flowering plants is still open. The progress in sequencing complete chloroplast genomes (over 120 and 69 of them in flowering plants) makes possible an experiment on searching for this gene [25].
Complete chloroplast genomes of most higher plants have 120-217 kb in length and about 50% of them belong to the protein-coding genes. The number of genes used for molecular phylogenetic analysis of chloroplasts is 60-61 and total length of their alignment is about 45,000 bp (about 15,000 amino acid residues). The difference between the total number of protein-coding genes and genes used for molecular phylogenetic analysis at a high taxonomical level is due to a certain difference in gene content in different species. Thus, many genes of the NDH group are absent from Phalaenopsis [26], and some legumes do not contain genes rps16 and rpl22 [27]; naturally, when these taxons are included in a set, the genes absent from them are excluded from analysis. The goal of this work was searching for a “lucky gene” for analysis of phylogeny of flowering plants--easily sequenced genes or their combinations whose phylogenetic analysis provides the best approximation to the results obtained using all protein-coding genes--or a proof that this is impossible.
MATERIALS AND METHODS
A set of translated nucleotide sequences (TNS) of 61 chloroplast protein-coding genes in 41 species of flowering plants was used in this work, 40 of which were analyzed in previous works, and data for one species (Fagopyrum esculentum) have been obtained for the first time. Species Pinus thunbergii and Ginkgo biloba belonging to Gymnospermae were chosen as an out group.
Sequences were aligned using the MUSCLE program [28] with subsequent correction. The full length of alignment comprises 14,168 amino acid residues.
Phylogenetic analysis of all data sets, including the complete set, was carried out using the PAUP* 4.08 program [29] with the following settings: optimization criterion, maximum parsimony; search for the most-pasimonious trees, heuristic. A hundred searches with random order of taxon additions were carried out, and gaps were considered as missing data.
Gene suitability (fitness) for phylogenetic analysis was analyzed by comparison of phylogenetic trees obtained for different genes or groups of genes with a phylogenetic tree obtained using all protein-encoding genes. For each resulting tree, the following items were determined.
1. The number of nodes in the tree that coincide by taxon composition with nodes of a tree based on the complete set. In this case, the tree structure inside nodes was not considered. This measure is similar to the value proposed by Simmons and Miya [30] and called the overall success of resolution (OSR), but OSR is the difference between the number of correctly resolved nodes and of those resolved incorrectly.
2. The number of erroneous nodes not coinciding with nodes of the tree constructed on the basis of the complete set. The value of this parameter was determined by subtraction of the number of correct nodes from the total number of nodes in the tree.
To determine the gene efficiency compared to random sequence of the same length, random samples of a predetermined length were analyzed. To do this, a fixed number of positions were randomly chosen from the complete set of 14,168 amino acids, and each obtained subalignment was analyzed by the same method and with the same criteria as the initial alignment. Random samples of 20, 40, 60, 80, 100, 200, 400, 600, 800, 1000, 2000, 4000, 6000, 8000, 10,000, 12,000, and 14,000 amino acids were analyzed. For analysis of same length sequences 10 independent samples were generated, and a phylogenetic tree was constructed on the basis of each of these. Values of the studied parameters were calculated using the Microsoft Office Excel 2003 program.
The suitability of individual regions for phylogeny was analyzed both for separate genes and their combinations and for their functional groups as well: genes encoding proteins incorporated in the ATP-synthase enzymatic complex (ATP), ribosomal proteins of large (RPL) and small (RPS) ribosomal subunits, proteins incorporated in plastid RNA polymerase (RPO), proteins of cytochrome f/b6 complex (PET), proteins of photosystem I (PSA) and II (PSB), proteins ycf3 and ycf4 (YCF) supposed to be involved in the assembly of the photosystem I complex, ATP-dependent chloroplast protease (clpP), protein of chloroplast membrane (cemA), a protein involved in cytochrome c biogenesis (ccsA), ribulose bisphosphate carboxylase (rbcL), and maturase K (matK).
Groups of genes and corresponding TNS are designated here and below by capital letters and individual genes by lower-case letters, for example: rpoA, a gene encoding a subunit of plastid RNA polymerase; RPO, a group of genes encoding proteins incorporated in plastid RNA polymerase.
RESULTS
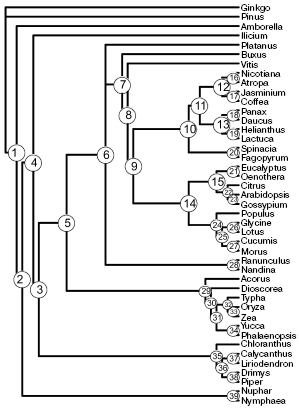
Two trees of minimal length (29,337 steps) with consistency index of 0.544 are the result of phylogenetic analysis of the complete gene set. These trees differ in relative position of Platanus and members of the order Ranunculales (Ranunculus and Nandina). The topology of a strict consensus tree of these two trees (Fig. 1) is similar to those in the most recent works on phylogenetic analysis of complete chloroplast genomes [31-33], and in many respects it is similar to results of analysis of a small number of genes. Most nodes on the tree in Fig. 1 were revealed earlier with high support in the analysis of a large number of genes. Exceptions are in nodes 5, 10, 15, 17, 23, 24, 27, and 35. The tree was used as a reference for comparison with trees derived from individual genes and with trees obtained using random samples.
The number of correctly resolved nodes in trees obtained by analysis of random samples increases in proportion with their length (Fig. 2a). Topologies completely coinciding with the result of the full set analysis appear for the first time at the random sample length of 8000 amino acid residues. However, on average the number of resolved nodes at this length is lower--equal to 37.Fig. 1. A strict consensus tree obtained by analysis of a complete set of protein coding chloroplast sequences.
Analysis of phylogenetic trees of translated nucleotide sequences of individual genes and their functional groups reveals three types.Fig. 2. a) Dependence of the number of correctly resolved nodes on the length of a random sample. Error bars show maximal and minimal values taken as parameters. Genes or groups of genes that were the basis for inferring phylogenetic trees with fewer incorrect nodes compared to the trees based on random sequences of equivalent length (positioned below the curve in Fig. 2b) are shown in bold. b) Dependence of the number of incorrectly resolved nodes on the length of a random sample. Error bars show maximal and minimal values taken as parameters. Genes or groups of genes that were the basis for inferring phylogenetic trees with a larger number of incorrect nodes compared to the trees based on random sequences of equivalent length (positioned above the curve in Fig. 2a) are shown in bold.
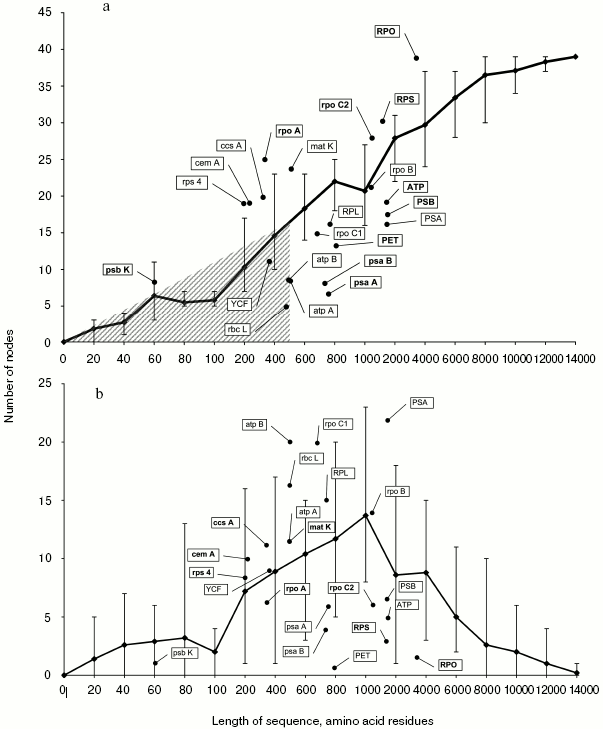
1. The first type includes short genes and gene combinations with product length not exceeding 400-500 amino acid residues. Phylogenetic trees constructed using their TNS contain no more than 15 correctly resolved nodes. The number of correct nodes for these genes is lower than in trees inferred from random samples of the same length. The region of their localization in Fig. 1 is cross-hatched. This type, in particular, includes such widely used phylogenetic markers as rbcL and atpB, as well as the YCF that combines genes ycf3 and ycf4.
2. Some genes and functional groups for which the number of correctly resolved nodes is lower than on the average for random samples of equivalent length. This type includes many groups of genes, in particular, those that combine sequences of genes PET, PSA, ATP, PSB, and RPL. In most cases the number of correctly resolved nodes is lower than minimally obtained using random samples, and only separate genes or groups of genes (rpoC1, RPL, ATP, rpoB) produce comparable resolution.
3. The third type includes eight genes and two groups, for which the number of correctly resolved nodes exceeds the mean in random samples of equivalent length. One gene of this group, matK, is often used in phylogenetic analysis, the other genes (rpoC2, rps4) being used less frequently or not at all. One gene of this type, psbK, is short (its TNS codes 60 amino acid residues) and conserved (10% variable characters), which makes its consideration as an independent phylogenetic marker inexpedient, but it is possible to use it in multigene sets in combination with rapidly evolving genes.
Along with the number of correctly resolved nodes, a critical parameter for sequence usage in phylogenetic analysis is the number of incorrectly resolved nodes in the resulting tree. The existence of such nodes may result in formulation of false hypotheses.
The number of incorrectly resolved nodes increases in trees inferred from the analysis of random samples before the sequence under analysis reaches the length of 1000 amino acid residues, and the number of such nodes exceeds 60% of the total number of nodes. This is due to increase in the total number of nodes against the background of deficiency of informative characters for their correct resolution. In the case of further extension of the sequence length, the number of informative characters increases, and the number of incorrectly resolved nodes decreases (Fig. 2b).
Each of two earlier distinguished gene types (2 and 3) producing a high number of correctly resolved nodes when used in phylogenetic analysis contains both genes associated with a large numbers of incorrectly resolved nodes and those associated with a small number of such nodes (Fig. 2b and table). This allows us to divide all genes into four classes.
Ratio of the number of correctly and incorrectly resolved nodes for
genes and groups of type 2 and 3 genes. The number of correct/incorrect
nodes and their ratios are shown in parentheses next to each gene
(group)

1. Genes or groups of genes that serve as a basis for phylogenetic trees with a low number of correctly resolved nodes and a high number of incorrect nodes. This class is rather heterogeneous and includes both rapidly evolving genes (rpoB, rpoC1) and highly conserved genes (the group of PSA genes); therefore, the explanations of the observed structures for trees of different genes are probably different. As to the rapidly evolving genes, their high variability may result in numerous reversions and parallel occurrences of features in not directly related groups (homoplasy), and this leads to a high number of false nodes. For highly conserved genes, such structure is evidently caused by the lack of variable characters (which is expressed by a small number of correctly resolved nodes) and evolutionary peculiarities of certain regions that could result in a situation when alterations in a given region contradict alterations in the whole genome.
2. The class including genes and groups of genes that are the basis for phylogenetic trees with a low number of correctly resolved nodes and few mistakes. The main reason for unresolved nodes in this case is lack of information due to a high conservativeness of sequences. Such genes may be perspective markers at a higher taxonomical level, like all seed plants, not only angiosperms. According to our results, the best for such analysis is the group of PET genes.
3. This class includes one of the most popular genetic markers, matK, and genes and groups of genes that are the basis for phylogenetic trees with a high number of both correct and incorrect nodes. All genes of this class are rapidly evolving, and the incorrectly resolved nodes are probably the result of homoplasy. Owing to this, the combination of these genes (matK + cemA + ccsA) gives a tree with characteristics (26/9 = 2.9) not much exceeding that of matK, although they are much longer. It is possible that it would be reasonable to use such genes only at a lower taxonomical level.
4. The last class includes mainly the group of RPO genes. Phylogenetic trees constructed on their basis have a great number of correctly resolved nodes and a small number of false ones, which makes them the main candidate for the “lucky gene”.
DISCUSSION
We have found that the best approximation to the result of analysis of the complete set of sequences is observed in the RPO group that combines genes rpoA, rpoB, rpoC1, and rpoC2, encoding different subunits of plastid RNA polymerase. The tree inferred from such a set has completely resolved topology (unlike the tree inferred from complete sets and having one unresolved node) and just a single node that does not coincide with the tree inferred from a complete set. However, two genes of this group (rpoB and rpoC1) belong to class 1 genes that are not ideal for phylogenetic analysis (although they are the best in this class). To check whether there is a more appropriate combination of individual genes, we studied phylogenetic trees inferred from the gene groups including genes rpoA and rpoC2 as well as the best genes from other groups. All studied combinations gave worse results than those obtained with a combination of all genes of the RPO group. Thus, the tree based on the combination rpoA, rpoC2, ccsA, and cemA gives 33 correctly resolved nodes and the combination rpoA + rpoC2 + matK produces 36 such nodes.
A sufficiently good result (30 correctly resolved nodes and three incorrect ones) was obtained with the combination of RPS (ribosomal proteins of the small ribosomal subunit). However, this group cannot be considered “lucky genes” because in technical aspect obtaining the sequences of these genes is much more complicated than in the case of RPO genes. This is so because there are only four RPO genes and their arrangement is quite compact (rpoB, rpoC1, and rpoC2 follow one another, making up a single operon), whereas the chloroplast genome contains 11 genes of ribosomal proteins of the small ribosomal subunit, and they are dispersed: eight in the large single copy region, one in the small one, and three in the inverted repeat (for gene rps12, trans-splicing is characteristic; its 5´ end is located in a large single copy region and its 3´ end is in the inverted repeat--therefore, it is counted twice).
It should be noted that although the combination of genes rpoA, rpoB, rpoC1, and rpoC2 is proposed for the first time as a phylogenetic marker, separate genes from this group have already attracted attention of researchers in molecular systematics and phylogeny. Thus, in a recent work by Qiu et al. [34] the multigene phylogeny of flowering plants was analyzed with involvement of rpoC2. Attention was paid to rpoB and rpoC1 in searching for a universal DNA barcode (the genome region that makes possible species identification) for land plants [35]. Thus, the representativeness of at least some of these genes in the GenBank database is sufficiently high and will probably increase, which is of no small importance for a phylogenetic marker.
However, it is known that such event as RNA editing is characteristic of the chloroplast gene transcripts. This process was demonstrated experimentally in bryophytes [36] and some other spore-bearing plants [37], as well as in flowering plants [38], whereas in the latter genes of the RPO group are among transcripts most susceptible to editing. Thus, in a member of Orchidaceae family Phalaenopsis, 15 sites of editing (non-synonymous) were found in these genes, which results in differences between real and predicted protein sequences. The effect of RNA editing on phylogeny reconstruction is still poorly studied, although Qiu et al. [34] report that the editing site inclusion or exclusion only slightly influenced some weakly supported and unreliable nodes. Further investigation of the RPO group genes, carried out for different groups of flowering plants and with account of their peculiarities (such as RNA editing), will make possible better understanding of their possibilities and limitations as phylogenetic markers, able to replace completely or in part the information on complete chloroplast genomes.
The authors express their gratitude to V. V. Aleshin and D. D. Sokoloff for discussion of the results and comments on the article and to Yu. M. Logachev for compiling the program for analysis of random samples.
This work was supported by the Russian Foundation for Basic Research (grants 06-04-48484 and 06-04-96315), GK 02.512.11.2069, and MK grant 920-2007-4.
REFERENCES
1.Borsch, T., Hilu, K. W., Quandt, D., Wilde, V.,
Neinhuis, C., and Barthlott, W. (2003) J. Evol. Biol.,
16, 558-576.
2.Lohne, C., and Borsch, T. (2005) Mol. Biol.
Evol., 22, 317-332.
3.Chase, M. W., Soltis, D. E., Olmstead, R. G.,
Morgan, D., Les, D. H., Duvall, M. R., Price, R., Hills, H. G., Qiu,
Y., Kron, K. A., Rettig, J. H., Conti, E., Palmer, J. D., Clegg, M. T.,
Manhart, J. R., Sytsma, K. J., Michaels, H. J., Kress, W. J., Donoghue,
M. J., Clark, W. D., Hedren, M., Gaut, B. S., Jansen, R. K., Kim,
K.-J., Wimpee, C. F., Smith, J. F., Furnier, G. R., Straus, S. H.,
Xiang, Q., Plunkett, G. M., Soltis, P. S., Eguiarte, L. E., Learn, G.
H., Barrett, S. Ch., Graham, S., and Albert, V. A. (1993) Ann.
Missouri Bot. Gard., 80, 528-580.
4.Johnson, L. A., and Soltis, D. E. (1994) Syst.
Bot., 19, 143-156.
5.Hilu, K. W., and Liang, H. (1997) Am. J.
Bot., 84, 830-839.
6.Hilu, K. W., Borsch, T., Muller, K., Soltis, D. E.,
Soltis, P. S., Savolainen, V., Chase, M. W., Powell, M. P., Alice, L.
A., Evans, R., Sauquet, H., Neinhuis, T. C., Slotta, A. B., Rohwer, J.
G., Campbell, C. S., and Chatrou, L. W. (2003) Am. J. Bot.,
90, 1758-1776.
7.Wolf, P. G. (1997) Am. J. Bot., 84,
1429-1440.
8.Soltis, P. S., Soltis, D. E., and Chase, M. W.
(1999) Nature, 402, 402-404.
9.Samigullin, T. H., Martin, W. F., Troitsky, A. V.,
and Antonov, A. S. (1999) J. Mol. Evol., 49, 310-315.
10.Alverson, W. S., Whitlock, B. A., Nyffeler, R.,
Bayer, C., and Baum, D. A. (1999) Am. J. Bot., 86,
1474-1486.
11.Buck, W. R., Goffinet, B., and Shaw, A. J. (2000)
Mol. Phylogenet. Evol., 16, 180-198.
12.Cranfill, R. B. (2001) Am. J. Bot., 87
(Suppl. 6), 121.
13.Korall, P., Pryer, K. M., Metzgar, J. S.,
Schneider, H., and Conant, D. S. (2006) Mol. Phylogenet. Evol.,
39, 830-845.
14.Nei, M., Kumar, S., and Takahashi, K. (1998)
Proc. Natl. Acad. Sci. USA, 95, 12390-12397.
15.Rosenberg, M. S., and Kumar, S. (2001) Proc.
Natl. Acad. Sci. USA, 98, 10751-10756.
16.Rokas, A., Williams, B. L., King, N., and
Carroll, S. B. (2003) Nature, 425, 798-804.
17.Rokas, A., and Carroll, S. B. (2005) Mol.
Biol. Evol., 22, 1337-1344.
18.Goremykin, V. V., Hansmann, S., and Martin, W. F.
(1997) Plant Syst. Evol., 206, 337-351.
19.Martin, W., Deusch, O., Stawski, N.,
Grünheit, N., and Goremykin, V. (2005) Trends Plant Sci.,
10, 203-209.
20.Degtjareva, G. V., Samigullin, T. H., Sokoloff,
D. D., and Valiejo-Roman, C. M. (2004) Bot. Zh., 89,
896-907.
21.Soltis, D. E., Albert, V. A., Savolainen, V.,
Hilu, K., Qiu, Y. L., Chase, M. W., Farris, J. S., Stefanovic, S.,
Rice, D. W., Palmer, J. D., and Soltis, P. S. (2004) Trends Plant
Sci., 9, 477-483.
22.Bapteste, E., Brinkmann, H., Lee, J. A., Moore,
D. V., Sensen, C. W., Gordon, P., Durufle, L., Gaasterland, T., Lopez,
P., Muller, M., and Philippe, H. (2002) Proc. Natl. Acad. Sci.
USA, 99, 1414-1419.
23.Corneli, P. S., and Ward, R. H. (2000) Mol.
Biol. Evol., 17, 224-234.
24.Miya, M., and Nishida, M. (2000) Mol.
Phylogenet. Evol., 17, 437-455.
25.Antonov, A. S. (2006) Plant Gene
Systematics [in Russian], Akademkniga, Moscow.
26.Chang, C. C., Lin, H. C., Lin, I. P., Chow, T.
Y., Chen, H.-H., Chen, W.-H., Cheng, C.-H., Lin, C.-Y., Liu, S.-M.,
Chang, C.-C., and Chaw, S.-M. (2006) Mol. Biol. Evol.,
23, 279-291.
27.Doyle, J. J., Doyle, J. L., and Palmer, J. D.
(1995) Syst. Bot., 20, 272-294.
28.Edgar, R. C. (2004) Nucleic Acids Res.,
32, 1792-1797.
29.Swofford, D. L. (2000) PAUP: Phylogenetic
Analysis Using Parsimony and Other Methods, Version 4,
Sinauer Associates, Sunderland, Massachusetts.
30.Simmons, M. P., and Miya, M. (2004) Mol.
Phylogenet. Evol., 31, 351-362.
31.Jansen, R. K., Kaittanis, C., Saski, C., Lee, S.
B., Tomkins, J., Alverson, A. J., and Daniell, H. (2006) BMC Evol.
Biol., 6, 32.
32.Nalapalli, S., Bausher, M. G., Lee, S. B.,
Jansen, R. K., and Daniell, H. (2007) Plant Biotechnol. J.,
5, 339-353.
33.Hansen, D. R., Dastidar, S. G., Cai, Z.,
Penaflor, C., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2007)
Mol. Phylogenet. Evol., doi: 10.1016/j.ympev.2007.06.004.
34.Qiu, Y.-L., Li, L., Hendry, T. A., Li, R.,
Taylor, D. W., Issa, M. J., Ronen, A. J., Vekaria, M. L., and White, A.
M. (2006) Taxon, 54, 837-856.
35.Chase, M. W., Cowan, R. S., Hollingsworth, P. M.,
van den Berg, C., Madrinan, S., Petersen, G., Seberg, O., Jorgsensen,
T., Cameron, K. M., Carine, M., Pedersen, N., Hedderson, T. A. J.,
Conrad, F., Salazar, G. A., Richardson, J. E., Hollingsworth, M. L.,
Barraclough, T. G., Kelly, L., and Wilkinson, M. (2007) Taxon,
56, 295-299.
36.Kugita, M., Yamamoto, Y., Fujikawa, T.,
Matsumoto, and Yoshinaga, K. (2003) Nucleic Acids Res.,
31, 2417-2423.
37.Wolf, P. G., Rowe, C. A., and Hasebe, M. (2004)
Gene, 339, 89-97.
38.Zeng, W.-H., Liao, S.-Ch., and Chang, Ch.-Ch.
(2007) Plant Cell Physiol., 48, 362-368.