REVIEW: The Oxygenase-Peroxidase Theory of Bach and Chodat and Its Modern Equivalents: Change and Permanence in Scientific Thinking as Shown by Our Understanding of the Roles of Water, Peroxide, and Oxygen in the Functioning of Redox Enzymes
P. Nicholls
Department of Biological Sciences, Essex University, Colchester, Essex CO 4 3SQ U. K.; E-mail: pnicholl@essex.ac.uk
Received May 15, 2007
Alexander Bach was both revolutionary politician and biochemist. His earliest significant publication, “Tsar-golod” (“The Tsar of Hunger”), introduced Marxist thought to Russian workers. In exile for 30 years, he moved to study the dialectic of the oxidases. When his theory of oxidases as combinations of oxygenases and peroxidases was developed (circa 1900) the enzyme concept was not fully formulated, and the enzyme/substrate distinction not yet made. Peroxides however were then and remain now significant intermediates, when either free or bound, in oxidase catalyses. The aerobic dehydrogenase/peroxidase/catalase coupled systems which were studied slightly later clarified the Bach model and briefly became an oxidase paradigm. Identification of peroxidase as a metalloprotein, a key step in understanding oxidase and peroxidase mechanisms, postdated Bach's major work. Currently we recognize catalytic organic peroxides in flavoprotein oxygenases; such organic peroxides are also involved in lipid oxidation and tryptophan radical decay. But most physiologically important peroxides are now known to be bound to transition metals (either Fe or Cu) and formed both directly and indirectly (from oxygen). The typical stable metalloprotein peroxide product is the ferryl state. When both peroxide oxidizing equivalents are retained the second equivalent is held as a protein or porphyrin radical. True metal peroxide complexes are unstable. But often water molecules mark the spot where the original peroxide decayed. The cytochrome c oxidase Fe-Cu center can react with either peroxide or oxygen to form the intermediate higher oxidation states P and F. In its resting state water molecules and hydroxyl ions can be seen marking the original location of the oxygen or peroxide molecule.
KEY WORDS: peroxidase, oxygenase, cytochrome oxidase, heme proteins, oxygen intermediates, BachDOI: 10.1134/S0006297907100021
THE LEGACY OF ALEXANDER BACH
Alexander Bach spent his first thirty years being educated and becoming a revolutionary with the Narodnya Volya (People's Will) party in Czarist Russia, the next thirty years as an imaginative research worker while in exile in France and Switzerland, and the final thirty years as laboratory manager and science administrator following the Soviet revolution in 1917 [1, 2]. Some might consider this the perfect career pattern. Bach produced published work in all three phases of his career. In the first phase his most significant work was “Tsar-golod” (“The Tsar of Hunger”) [3], referring to Nekrasov's poem [4], which apparently represented an introduction to Marxist ideas for Russian workers [5]. In the second phase he will be remembered for the theory of biological oxidation developed with Chodat, the “oxygenase-peroxidase” theory [6]. In his final phase he published and copublished some further work on oxidases, including perhaps most significantly a commentary on Warburg's terminal oxidative enzyme [7], but was primarily engaged in the great enterprise of developing scientific and engineering capability for the new Soviet state [8]. He seems to have missed the development of the cytochrome/cytochrome oxidase theory and the separate identification of peroxidases and catalases, although further research on this point is desirable.
This paper will ask three questions:
- what was the nature of Bach's ideas about biological oxidation?
- what current themes on oxidative enzymes echo the early Bach-Chodat theory?
- were there any links between Bach's political and scientific ideas?
BACH'S IDEAS ABOUT OXIDASES
Oxygen metabolism (especially by plants) was seen by Bach as a set of linked oxygenase-peroxidase processes. But this statement means something quite different today, for us, from the way it would have been understood 100 years ago, by Bach and his contemporaries then (although he of course lived into the modern era). The first process or “reaction” was an “oxygenation” to give rise to some kind of peroxy species. The second process or “reaction” was the utilization of this species to oxidize an organic species such as a phenol or quinol. The initial absence of anything resembling a molecular scheme in Bach's writings--chemical formulas or equations of the simplest kind--is notable; these only came later. One of the earliest reviews with atomic formulas is that of Kastle [9], but Kastle does not know the structure of hydrogen peroxide (variously given as OH or H2O →_O) and non-canonical entities such as ozone (O-) and antozone (O+) are entertained. Later accounts of Bach's work [1] provide modern summaries of his ideas in current style, but these of course both simplify his thoughts and underplay the tremendous difficulties in creating biochemical ideas before atomic theory itself was well established.
The uses of the suffix “ase” (as in the German “oxydase”, “oxygenase”, and “peroxydase”) suggest to us enzymes. That today implies a protein catalyst. But in 1904 it was not known that enzymes were either proteins or catalysts [10]; the category of “proteins” was itself ill-defined and catalysts were seen in the context of reactive surfaces (as Wieland and Warburg later debated the relative validity of blood, charcoal, and platinum as model systems). The oxygenase process could therefore include peroxidation of lipids and still count as enzymatic for example. Gallagher [11] thought so even 20 years later. The absence of a clear enzyme/substrate distinction was also notable (absence of specificity led to failure to define the oxidized substrate as such). The antiquity of peroxidase [12] and its ready assay concealed the conceptual difficulties in using it as enzyme and especially oxidase paradigm. Invertase proved easier despite its apparent substrate structural complexity and difficulty of assay [13].
The concurrent discovery of catalase in the tobacco plant and the definition of its activity as a specific enzyme reaction by Loew [14], in contrast to the vagueness of Schonbein's ideas about peroxide decomposition, was a significant step; unlike peroxidase, catalase is highly substrate specific (exceptions came much later); catalase became important for understanding how metabolic processes may be subdivided into simpler steps (cf. the debates between Liebig and others on fermentation as a single or multiple step process). Its significance was recognized by Bach and Chodat [15]. Hydrogen peroxide decomposition is catalyzed by catalase; organic peroxide decomposition is not. As we now know even the partial reactivity of catalase with alkyl peroxides is quite slow compared to that with hydrogen peroxide, a function, as we now know of the catalase protein substrate channel (cf. below). If a process is blocked by catalase therefore hydrogen peroxide and not an organic peroxide is an intermediate. To get to this place took quite a while.
Can we (now) write a Bach model which is in some sense “historically” testable? With all the above caveats in mind we may as well adopt the Shamin formulations [1] (see below) (probably never written by Bach):
A + O2 → AO2,
AO2 + A(B) → AO + AO (or BO) or A + BO2
(the regeneration of A is a key uncertainty).
WHAT CURRENT AND EARLY MODERN THEMES INVOLVING OXIDATIVE ENZYMES
ECHO THE EARLY BACH-CHODAT THEORY?
Flavoprotein Oxidases
The first clearly defined double enzyme oxidase systems were probably Sylva Thurlow's “aerobic dehydrogenase” plus peroxidase or catalase “coupled oxidation” systems with hydrogen peroxide as a clearly defined intermediate [16, 17].
Any process involving oxygen uptake and peroxide production replaced Bach's oxygenase, including both non-enzymatic autoxidations such as that of cysteine or glutathione and enzymatic peroxide-generating reactions such as xanthine oxidase. Keilin and Hartree [18] later elaborated the coupled oxidation system into a way of comparing the peroxidatic activity of different heme proteins. It is remarkable that their paper, published over half a century after Bach's work, contained an elaborate historical introduction in which Bach and Chodat [19] played the central theoretical role. As the writer joined the Keilin laboratory the year after the publication of this paper it represented his introduction to Bach and he was impressed throughout his graduate work with Keilin's careful consideration of this and other historical predecessors. For Keilin, whose own key work [20] had followed that of the early Bach by only 20 years and was contemporaneous with some of Bach's later contributions [7], the past was far from the foreign country it was and remains to me. Simple coupled oxidations (Thurlow, Keilin) can be described as follows:
AH2 + O2 → (A H2O2) → A + H2O2,
H2O2 + BH2 → B + 2H2O.
The theory of coupled oxidation did not define a role for a flavoprotein peroxide intermediate (bracketed). The action of Keilin and Hartree's favorite oxidase, “notatin” or glucose oxidase from Penicillium notatum, can be analyzed without postulating such an intermediate [21]. Its role in generating peroxide or coupled oxidation systems can be treated assuming that only reduced and oxidized forms of the flavin occur in its catalytic cycle, the reaction of the reduced form with oxygen generating free peroxide directly [22].
Free reduced flavins do form peroxide intermediates when they react with molecular oxygen [23]. And another class of flavoproteins, the monooxygenases, in their reduced states do react with oxygen to give a relatively stable bound organic peroxide [24]. A single monooxygenase molecule therefore acts as both of the enzymatic species postulated by Bach:
AH + O2 → AOOH,
AOOH + BH → BOH + AOH,
AOH + CH2 → AH + C + H2O.
The enzyme in fact is a three-substrate system, reacting with oxygen, with a hydroxylatable species, and a reductant capable of returning the flavin to its reactive reduced state. In addition to the flavin peroxide the system involves intermediate formation of a hydroxyflavin, an unstable hydrated form of oxidized flavin. In such systems the second enzyme or reductant needed is the reducing species (C) that can regenerate reduced flavin. In some cases this step involves the dissociation of the oxidized or hydroxyflavin from the enzyme protein and its reduction by a separate flavin reductase. Such cases include bacterial luciferase [25] and the hydroxyphenylacetate hydroxylase family [26]. In these enzymes the riboflavin is a true coenzyme, shuttling between oxygenase and NADH-dependent reductase. The structure of the Acetobacter baumannii p-hydroxyphenylacetate hydroxylase has recently been determined [26].
Metalloenzymes Reacting with O2: Binuclear and Mononuclear Copper Enzymes
The enzymes whose activities Bach and Chodat were usually examining were in fact metalloenzymes although this was not known until much later. In addition to plant peroxidases, plant oxidases giving rise to obvious browning products included the copper-containing tyrosinases and catechol (polyphenol) oxidases (monooxygenases and dioxygenases). After the oxygenase-peroxidase work with Chodat, Bach himself began studying tyrosinase, publishing several papers between 1908 and 1914 [27]. It was bound to create difficulties for the theory, as tyrosinase cannot be separated into even notional oxygenase and peroxidase components and plant peroxidases oxidize tyrosine rather poorly.
The copper-dependence of these oxidases was shown in the first reconstitution experiments of Kubowitz [28] and Keilin and Mann [29]. Later it was realized that the copper center is binuclear. Again we have a single enzyme capable of two types of reaction [30]--an oxygenase step and an oxidase step for tyrosinase (monooxygenase-type):
CuI OH-CuI + O2 → CuII(O22-)CuII + OH-,
H+ + CuII(O22-)CuII + BOH → CuII(OH-)CuII + B(OH)2,
CuII(OH-)CuII + B(OH)2 → CuI OH-CuI + BO2 + H2O,
or a peroxidase-like step and an oxidase step for catechol oxidase (oxidase-type (diphenolase)):
CuI OH-CuI + O2 → CuII(O22-)CuII + OH-,
CuII(O22-)CuII + B(OH)2 → CuII(OH-)CuII + BO2 + OH-,
CuII(OH-)CuII + B(OH)2 → CuI OH-CuI + BO2 + H2O.
As predicted by Bach there is a peroxide-forming step followed by a peroxidase-like step and then a recovery step. In contrast to the original oxygenase-peroxidase model all three steps are catalyzed by the same enzyme, either in phenolase (monooxygenase) mode or in diphenolase (oxidase) mode. Both catechol oxidase [31] and tyrosinase [32] have been crystallized and their X-ray structures determined. The location of bridging waters and peroxide can be seen (cf. discussion below). In addition to the binuclear Cu oxidases known functionally to Bach there are also mononuclear Cu oxidases [33], including galactose oxidase and the amine oxidases [33]. These enzymes are “aerobic dehydrogenase” types, generating hydrogen peroxide as a redox end product and containing both metal (Cu) and organic redox cofactors. A different peroxide-like intermediate is therefore part of their catalytic mechanisms.
How Enzymes React with O2 to Generate Oxy, Peroxy, and Ferryl Species
Oxygen reacts with many ferrous heme proteins to give an oxyferro species. Sometimes this is almost stable (a lifetime of hours or even days) as with hemoglobin and myoglobin. It can decay to a superoxide-ferric form (when the Fe is mononuclear) or to a peroxy form (when the Fe is binuclear, as with hemerythrin, or part of a mixed binuclear complex, as with cytochrome c oxidase). The superoxide-ferric species can then be reduced to a ferric-peroxy species. Unlike the product of the oxygen reaction with hemocyanin (where the binuclear cuprous center becomes a binuclear cupric center with stably bound and doubly ionized peroxide) the Fe peroxy species are rarely stable; they commonly decay to a ferryl state and a radical. The typical sequence is therefore that given in the equation:
Fe2+ + O2 → Fe2+O2 → Fe3+O20- [+ e- + H+] → Fe3+HO2- → FeO2+ (+ OH0) → Fe3+.
The final ferryl state is formed at the expense of losing one oxidizing equivalent, notionally represented as a hydroxyl radical [34], but in a heme protein system this is a radical species either on the porphyrin (pi-cation) or on a protein residue.
Oxy and ferryl heme species: cyclooxygenase and cytochrome P450. The cytochrome P450 family members are monooxygenases with a single metal center (heme iron). Their specialty is adding an oxygen atom to an organic substrate [35]. As with the monoxygenases their reaction cycle therefore involves three substrates, oxygen, the hydroxylatable species, and a reductant, typically NADPH. The initial ferrous oxygen complex has to be activated by reduction and protonation [36] to give a reactive compound I-like species capable of donating an O atom to the second substrate and regenerating ferric enzyme:
Fe2+ + AH + O2 → Fe2+O2AH (+ e- + 2H+) → FeO3+AH + H2O,
FeO3+AH → Fe3+ + AOH,
Fe3+ + e- + H+ → Fe2+.
Cyclooxygenase-peroxidase [37] represents an enzyme complex about which Bach knew nothing but which formally conforms perhaps closest to his vision with an enzyme (oxygenase) and peroxidizable substrate interacting in the first step and the peroxidized substrate itself acting catalytically in the second (peroxidase) step [38]:
EH - H0 → E0 (+ 2O2 + A) → E + AOO(OO0) (+ e- + H+) → AOO(OOH),
Fe3+ + AOO(OOH) → FeO3+ + A(OO)OH,
FeO3+ + 2e- + 2H+ → Fe3+ + H2O.
Peroxy and ferryl heme species: myoglobin, hemoglobins, catalase, and peroxidases. But how do the enzymes that Bach did know about now fit in? Catalase and plant peroxidases normally cycle between the ferric state and higher oxidation states with the ferrous states difficult to attain (in the case of mammalian catalase not directly accessible by simple reduction). The oxyferrous state is normally reached indirectly, by peroxide attack on the compound II (ferryl) state. When oxyperoxidase is formed directly from ferroperoxidase and oxygen it is unstable, reacting with the unliganded ferrous species to generate ferric peroxidase [39, 40], presumably via compound II as in the equation:
Fe2+ + O2 → Fe2+O2,
Fe2+ + Fe2+O2 + 2H+ → 2FeOH3+,
FeOH3+ + Fe2++ 2H+ → 2Fe3+ + H2O.
Monomolecular decay possibilities to the ferryl state (with release of hydrogen peroxide) or to the ferric state (with release of superoxide) have also been postulated [41, 42]. Monomolecular decay to form compound I seems not to occur although a dismutation with compound II is a thermodynamically plausible route. The oxyferrous (compound III) state is populated during oxidase activity of peroxidase towards hydrogen donor substrates such as dihydroxyfumarate [43, 44]. Peroxidase can therefore itself act as both oxidase (oxygenase) and peroxidase in Bachian mode although the mechanistic details of the oxidatic reaction are still not completely determined:
Fe2+ + O2 → Fe2+O2,
Fe2+O2 + AH2 + H+ → FeO3+ + AH0 + H2O,
FeO3+ + AH2 → Fe3+ + A + H2O,
Fe3+ + AH0 → Fe2+ + A + H+.
Oxy species can thus be obtained in a relatively stable state. But true peroxy species cannot. The redox accessibility of stable free radicals is the reason. This heuristically important idea was unavailable to Bach and his contemporaries at the turn of the XIX/XX century. Michaelis' later struggle to gain acceptance of the stable organic free radical concept is well known [45]. The idea of radicals within protein structure was slower to arrive [46, 47]. Finally it became evident that plant peroxidase and catalase primary peroxide compounds were themselves radical species--porphyrin pi-cation radicals plus ferryl iron [48]. In metmyoglobin peroxide as well as yeast cytochrome c peroxidase the radical is protein-based. But wherever the radical resides the initial and true heme-peroxide complex thus generates compound I (radical + ferryl) within milliseconds if not faster. The precursor peroxide complex (compound 0 of horseradish peroxidase) has been identified by using special solvent systems [49] or by radiolytic reduction of oxyperoxidase [50].
The classical metmyoglobin/methemoglobin peroxide species [51, 52] is ferryl, with a single oxygen atom bound distally to the heme Fe. The second oxidizing equivalent in the peroxide generates one of a set of protein radicals [53, 54] and the second oxygen atom is lost as water. However with appropriately mutated protein both peroxy and compound I forms can be seen [55, 56]. Figure 1 (see insert) illustrates the heme pocket structure of horse heart ferrylmyoglobin as obtained by X-ray crystallography at pH 5.2 [57]. A single ferryl oxygen atom is present which may be able to form a hydrogen bond with a protonated distal histidine, or if itself a protonated ferryl, to form such a bond with neutral distal histidine. No other water molecules were visualized in the heme pocket or capable of H-bonding to heme pocket species. As this structure was obtained at a rather acid pH, it may represent a mixture of protonated and unprotonated ferryl states. The unprotonated ferryl is only weakly reactive as an oxidant. Protonation of ferrylmyoglobin at low pH markedly increases its oxidative activity [58]. For catalase and peroxidase the active protonated state may thus be the major ferryl species through the neutral pH range (see following discussion).
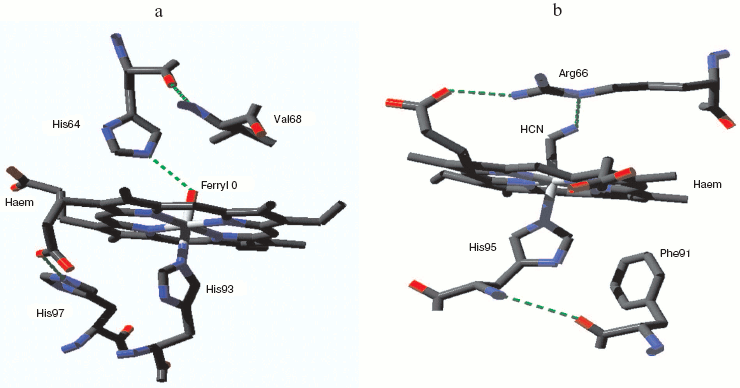
Mammalian myoglobin normally forms the ferryl state with loss of one oxidizing equivalent very rapidly. But one natural myoglobin showing a relatively stable compound 0 (true peroxy) state has been discovered, from the sea slug Aplysia limacina [59]. This myoglobin has no tyrosines capable of acting as radical sinks for oxidizing equivalents; it also lacks the distal histidine that can facilitate O-O bond breaking and stabilize the resulting ferryl species. The characteristic low spin peroxide complex has been identified by UV-visible and EPR spectroscopy [59] but a crystal structure is not yet available. Figure 1b shows the heme pocket structure for the Aplysia myoglobin cyanide complex, which as a two atom low spin species may be expected to resemble the peroxide form. The distal arginine, hydrogen-bonded to one heme porphyrin propionate group, may also be able to from a hydrogen bond with the terminal N of the bound cyanide and by analogy with the distal O of a bound peroxide.Fig. 1. Myoglobin ferryl and peroxide complexes. a) Metmyoglobin ferryl (horse heart): from pdb file 1GJN. cf. Hersleth et al. [57]. (Note this structure was obtained with crystals at pH 5.2 where the ferryl species may be a mixture of protonated and unprotonated forms: cf. refs. [57] and [58].) b) Aplysia limacina myoglobin cyanide complex (compound 0 analog): from pdb file 2FAL. (Note the Aplysia myoglobin low spin peroxy complex is a species for which the stable cyanide complex may be expected to be structurally similar.) Atomic coordinates from the Protein Data Bank. Structure visualized with Deepview Swiss-PdbViewer v3.7 and the figure was created with POV-Ray v3.6.
Oxy, peroxy, and ferryl species: cytochrome c oxidase. Of course the major route of oxygen reduction in aerobic organisms is through some type of terminal oxidase, cytochrome c oxidase in the case of nearly all eukaryotes and many prokaryotes. The need for such an entity was understood long before its actual discovery. The insight that it must exist surely belongs to Otto Warburg, an idea he entertained long before any way of examining the proposed ferment was available. The “Atmungsferment” was slowly seen as quite different from the plant oxidase systems examined by Bach and Chodat.
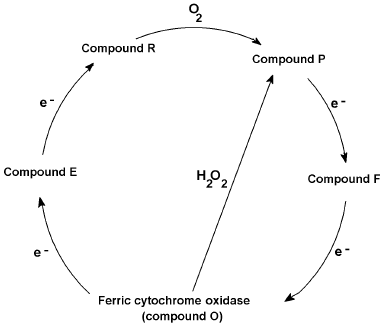
Like the presumably non-physiological peroxidase cycle from compound III to the other peroxide intermediates and ferric peroxidase, cytochrome c oxidase catalyzes a physiologically functional oxygen cycle with similar intermediates, formally peroxy (“P”) and ferryl (“F”), occurring between the initial oxyferro species and the ferric “ground” state. Conversely the oxidase can also undergo a non-physiological peroxide cycle [60, 61]--short-circuiting the mixed valence (E) and oxygenated (A) states but generating the peroxy (P) and ferryl (F) forms directly from the ferric state (Fig. 2). Protein radicals are components of the oxidase redox states, certainly some of the “P”, and possibly some of the “F” forms [61]. One equivalent intermediates, either radicals or transition metal redox changes, are an inevitable consequence of reducing a four-electron acceptor with four one-electron donor reactions. Electron transfer, as opposed to Warburg's and others' “activation” of oxygen, was a key idea in the development of the idea of a respiratory chain. Bach and Nikolajew [7] correctly noted the chemical equivalence but theoretical inequivalence of the “sauerstoffubertragendenferment” and the “wasserstoffubertragendenferment”. Ideas about biological oxidations did not look back after the focus changed to hydrogen (protonated electrons) rather than oxygen.
Fig. 2. Cytochrome c oxidase reaction cycle. Proposed reaction cycle reducing oxygen to waters via O (oxidized), E (mixed valence binuclear center), R (reduced), P (ferryl + radical), and F (ferryl or radical) states, together with hydrogen peroxide short circuit.
STRUCTURAL LOCATIONS OF BOUND OXYGEN SPECIES--DIOXYGEN, PEROXIDE, FERRYL, HYDROXYL, AND WATER: VISIBLE, GHOSTLY, AND INVISIBLE
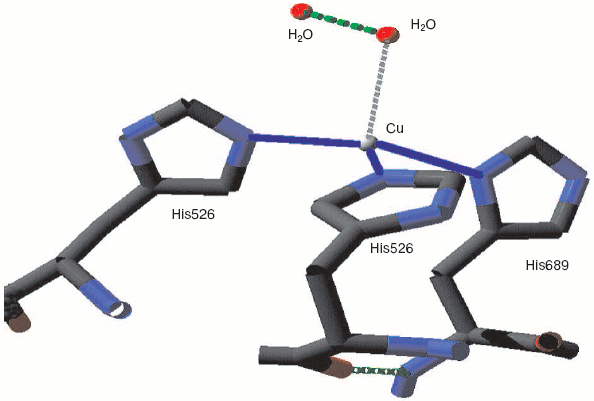
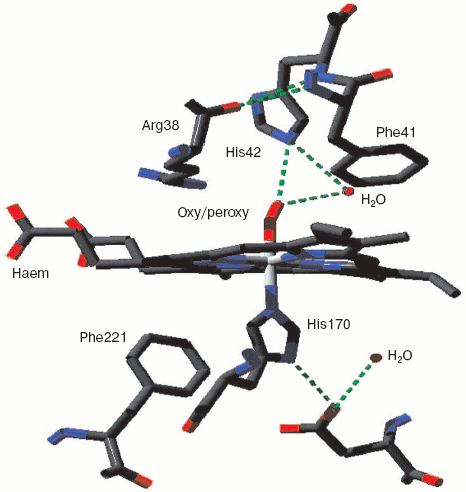
Monoamine oxidase is a mononuclear copper enzyme that reduces oxygen to hydrogen peroxide. Its active site contains a copper atom whose triple histidine (imidazole/imidazolate) ligation is closely similar to that of the CuB in the binuclear center of cytochrome c oxidase. As Whittaker points out [33], the crystal structure shows two associated water molecules that may be the ghost of oxygen or peroxide. Figure 3 (see insert) illustrates the location and arrangement of these two molecules. One can occupy a position almost perpendicular to the distorted plane defined by the three Cu-N bonds. The second is probably H-bonded to the first and creates a Cu-O-O angle rather similar to that seen for the two more closely associated waters in resting cytochrome c oxidase.
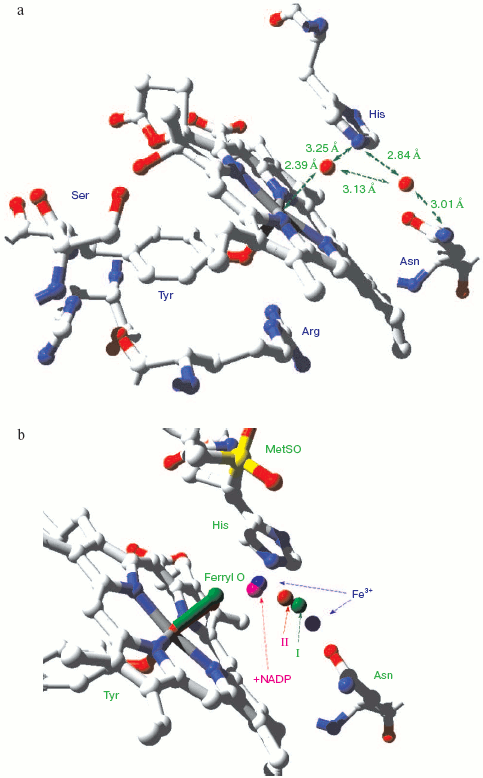
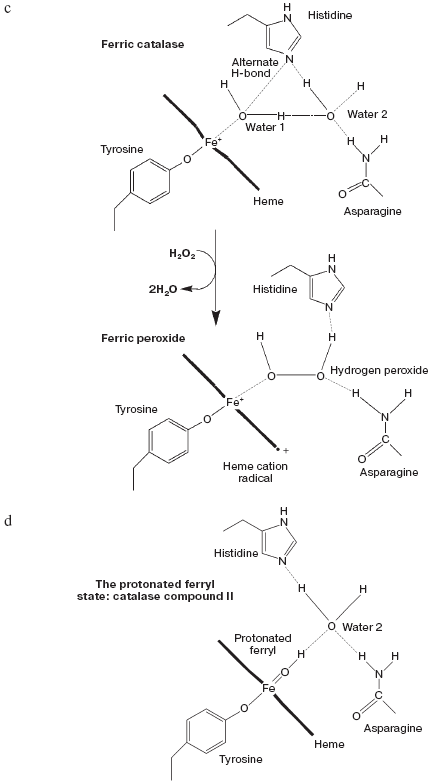
Catalase compound I (pi-cation radical/ferryl) binds peroxide and oxidizes it to molecular oxygen. Both hydrogens and two electrons are removed from the second peroxide molecule but the O-O bond remains intact as the (triplet) oxygen “diradical” molecule is released [62]. The catalase crystal structure also contains either one or two defined water molecules, one or both of which may be displaced by Fe ligands and at least one when the Fe is ligated to an oxygen atom to form a ferryl structure. Figure 4a (see insert) illustrates the locations and arrangement of these two molecules in the “resting” (ferric) state of the heme of Escherichia coli catalase HPII [63]. One water is about 2.4 Å from the heme Fe and can be weakly H-bonded either to the second water or to the distal histidine nitrogen. The second water bridges the distal gap between the conserved histidine and asparagine. Figure 4b (see insert) shows the overlaid structures of two ferric forms and two peroxide compounds with the associated water displacements in the homologous Proteus mirabilis enzyme [64]. Both the standard ferric enzyme and its NADP complex contain two heme pocket waters. Both compound I and compound II have a ferryl oxygen closer to the heme Fe than the first water molecule but displacing it. The second water molecule, bridging the two distal residues, is retained in the peroxide compounds, moving towards the ferryl oxygen and away from the position of the second water in ferric enzyme. Figure 4c (see insert) summarizes the possible H-bonding patterns in the heme pocket as ferric enzyme forms compound I. The reaction proton stoichiometry indicates that compound I ferryl is unprotonated [62]. But the compound II ferryl oxygen seems to be protonated; the retained water molecule can thus link ferryl O, histidine imidazole N and asparagine N, although perhaps not all three simultaneously (Fig. 4d; see insert). This water binding site may be the one occupied by the high spin ligands that react with catalase compounds I and II as well as ferric enzyme [62]. The retained water molecule may, of course, represent the distal peroxide oxygen left after the splitting of the O-O bond when compound 0 forms compound I. Compound 0, the true peroxide complex, however remains hypothetical for catalases.Fig. 3. Monoamine oxidase copper-associated water molecules (oxygen atoms). The “ghost” of a peroxide or oxygen molecule [33]. From pdb file 1JRQ. Structure obtained as in Fig. 1.
Peroxidase compound 0 (described above), the initial low spin complex formed by reaction with peroxide, can be seen but is too unstable for a crystal structure to be obtained. However peroxidase compound III (oxyperoxidase) is relatively stable and also has a species (oxygen or superoxide, depending upon the redox state assigned to the Fe) with two oxygen atoms. Figure 5 (see insert) shows the location of the two Fe-linked O atoms in this superoxy/compound III species. As it decays the two bonded oxygen atoms are reduced and end up as two molecules of water that mark the spot where the superoxide oxygen used to be--the ghost of oxygen (and perhaps an analog of the ghost of peroxide)--as shown by Hajdu and coworkers using the spontaneous reduction induced by the X-rays themselves [65].Fig. 4. Catalase heme pocket water molecules. a) E. coli HPII catalase [63]. From pdb file 1GGH. Ferric state. Structure obtained as in Fig. 1. b) Proteus mirabilis catalase [64]. From pdb files 2CAE, 2CAH, 2CAG, 2CAF. 2CAE ferric state (blue) (cf. Fig. 4a); 2CAH ferric NADPH complex (magenta); 2CAG peroxide compound II (red); 2CAF peroxide compound I (green). Overlaid structures as in Fig. 1. c) Interpretation of ferric and compound I structures. ChemdrawTM diagram. d) Interpretation of compound II structure. ChemdrawTM diagram.
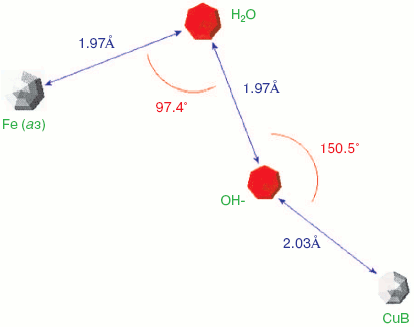
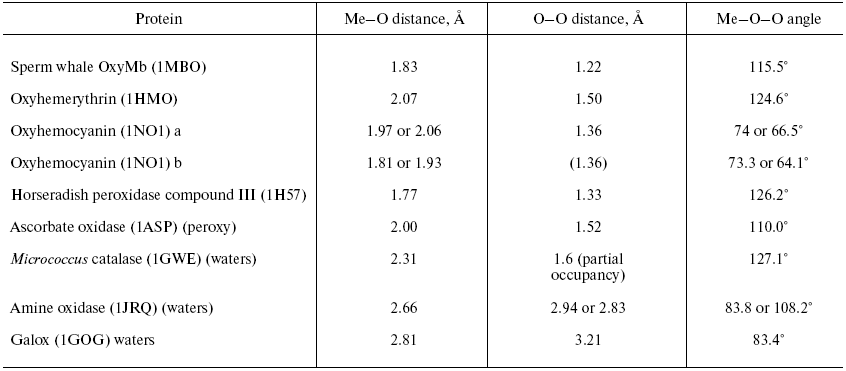
Two forms of cytochrome c oxidase have been obtained with crystal structures showing waters or peroxide “ghost” structures in the binuclear pocket bridging the Fe (heme) and CuB atoms, the Rhodobacter sphaeroides enzyme [66, 67] and its mammalian counterpart [68, 69]. The arrangement of metals (CuB and heme a3 Fe) and waters in the bacterial enzyme is shown in Fig. 6 (see insert). It differs significantly from its mammalian counterpart only in the relative position of the oxygen and Fe atoms--the maximum scattering density is at 2.22 Å from the Fe in beef heart enzyme (pdb 2OCC) and at 1.97 Å in the bacterial enzyme (pdb 2GSM). The apparent distance between the two oxygen atoms is therefore greater in the 2GSM structure (1.97 Å) than in the 2OCC structure (1.69 Å)--neither quite a peroxide nor a hydrogen-bonded water pair. Table lists the characteristic O-O distances as well as other structural information for the two oxygens of cytochrome c oxidase compared with other oxygen- and peroxide-binding metalloproteins.Fig. 5. Peroxidase heme pocket water molecules. Horseradish peroxidase peroxide complex III (produced with H2O2 + ferulic acid). Initial state (dioxygen/peroxide plus H-bonded water molecule). From pdb file 1H57. Progressive reduction in the X-ray beam induced reduction of bound oxygen to waters [65]. Structure obtained as in Fig. 1.
Oxy, superoxy, peroxy, and double water complexes with metalloprotein metalsFig. 6. Cytochrome c oxidase binuclear center metals--resting state. Water molecules or hydroxide ions (more ghosts of peroxide or oxygen?) bridging the two metals CuB and heme Fe in the binuclear center of Rhodobacter sphaeroides cytochrome c oxidase. From pdb file 2GSM [67]. Structure obtained as in Fig. 1. Note that the two oxygens are too close to be H-bonded. An otherwise very similar resting structure for the mammalian enzyme (pdb file 2OCC, not shown) is interpreted [69] in terms of a closer 1.69 Å O-O distance and a longer O-Fe distance (cf. table) thus retaining some peroxide character.

Other O--O-metal or H2O--H2O-metal complexes
LINKS BETWEEN BACH'S POLITICAL AND SCIENTIFIC IDEAS
“Tsar-golod” was thought of by its author and supporters as offering “scientific knowledge” about the structure of society. One of the topics considered at some length is the division of labor [5]. Subdividing an apparently single but complex system into multiple simpler components can work for socioeconomic analyses as well as for biochemical model building. Finding out how nature makes molecular oxygen reactive and getting new political ideas accepted may also have some parallels. But the social side of science that most concerned Bach in his administrative career was its practical usefulness [8]. This may have tended to isolate him from later developments in biological oxidation mechanisms and bioenergetics. Nonetheless his earlier ideas continued to influence others after he had effectively left the field. They remain with us now but resonate more faintly. Such is the scientific life.
The author wishes to acknowledge his discussions on some of these topics with Dr. A. Konstantinov. He also thanks Dr. D. Svistunenko for help with some Russian language material. His continuing research at Essex University is carried out in cooperation with Profs. C. E. Cooper and M. T. Wilson, and he thanks them for the hospitality of their laboratories and for the technical facilities that permitted the preparation of this review and analysis.
REFERENCES
1.Shamin, A. N. (1970) A. N. Bach, in Dict.
Scient. Biography, Vol. 1, pp. 360-363 (Scribner's).
2.Popov, V. O., and Zvyagilskaya, R. A. (2007)
Biochemistry (Moscow), 72, 1029-1038.
3.Bach, A. N. (1895) The Tsar of Hunger
[in Russian], published by Narodnya Volya group.
4.Nekrasov, N. A. (1864) The Rail Road [in
Russian].
5.Pearl, D. L. (1991) Slavic Review,
50, 768-778.
6.Bach, A. N., and Chodat, R. (1902) Ber. Deutsch.
Chem. Gesell., 35, 2466-2470.
7.Bach, A. N., and Nikolajew, K. A. (1926)
Biochem. Zs., 169, 105-112.
8.Bach, A. N. (1939) Planning Science, Izd-vo
Inostrannykh Yazykov, Moscow.
9.Kastle, J. H. (1910) The Oxidases. Hygienic
Laboratory Bull., 59, Treasury Department, Washington.
10.Chodat, R., and Bach, A. (1904) Ber. Deutsch.
Chem. Gesell., 37, 36-43.
11.Gallagher, P. H. (1923) Biochem. J.,
17, 515-529.
12.Planche, L. A. (1810) Bull. de Pharmacie,
2, 578-580.
13.Michaelis, L., and Menten, M. L. (1913)
Biochem. Z., 49, 333.
14.Loew, O. (1901) Catalase, a New Enzyme of
General Occurrence, Report No. 68, US Dept. Agriculture,
Washington.
15.Bach, A., and Chodat, R. (1903) Ber. Deutsch.
Chem. Gesell., 36, 1756-1761.
16.Thurlow, S. (1925) Biochem. J., 19,
175-187.
17.Harrison, D. C., and Thurlow, S. (1926)
Biochem. J., 20, 217-231.
18.Keilin, D., and Hartree, E. F. (1955) Biochem.
J., 60, 310-325.
19.Chodat, R., and Bach, A. (1903) Ber. Deutsch.
Chem. Gesell., 36, 606-608.
20.Keilin, D. (1925) Proc. Roy. Soc. B,
98, 312-339.
21.Stankovich, M. Y., Schopfer, L. M., and Massey,
V. (1978) J. Biol. Chem., 253, 4971-4979.
22.Nicholls, P. (1964) Biochim. Biophys.
Acta, 81, 479-495.
23.Bruice, T. C. (1980) Acc. Chem. Res.,
13, 256-262.
24.Massey, V. (1994) J. Biol. Chem.,
269, 22459-22462.
25.Ghisla, S., and Massey, V. (1989) Eur. J.
Biochem., 181, 1-17.
26.Alfieri, A., Fersini, F., Ruangchan, N.,
Prongjit, M., Chaiyen, P., and Mattevi, A. (2007) Proc. Natl. Acad.
Sci. USA, 104, 1177-1182.
27.Bach, A. N. (1908) Ber. Dtsch. Chem.
Gesell., 41, 221-225.
28.Kubowitz, F. (1937) Biochemisch. Z.,
292, 221-229.
29.Keilin, D., and Mann, T. (1938) Proc. Roy.
Soc. B, 125, 187-204.
30.Siegbahn, P. E. M. (2004) J. Biol. Inorg.
Chem., 9, 577-590.
31.Klabunde, T., Eicken, C., Sacchettini, J., and
Krebs, B. (1998) Nature Struct. Biol., 5, 1084-1090.
32.Matoba, Y., Kumagai, T., Yamamoto, A., Yoshitsu,
H., and Sugiyama, M. (2006) J. Biol. Chem., 281,
8981-8990.
33.Whittaker, J. W. (1999) in Essays in
Biochemistry, Vol. 34, Metalloproteins, Portland Press,
London, pp. 155-172.
34.George, P., and Irvine, D. H. (1954) Br. J.
Radiol., 27, 131-137.
35.Hlavica, P. (2004) Eur. J. Biochem.,
271, 4335-4360.
36.Nagano, S., and Poulos, T. L. (2005) J. Biol.
Chem., 280, 31659-31663.
37.Smith, W. L., and Marnett, L. J. (1991)
Biochim. Biophys. Acta, 1083, 1-17.
38.Wu, G., Rogge, C. E., Wang, J. S., Kulmacz, R.
J., Palmer G., and Tsai, A. L. (2007) Biochemistry, 46,
534-542.
39.Wittenberg, J. B., Noble, R. W., Wittenberg, B.
A., Antonini, E., Brunori, M., and Wyman, J. (1967) J. Biol.
Chem., 242, 626-634.
40.Phelps, C. F., Antonini, E., Giacometti, G., and
Brunori, M. (1974) Biochem. J., 141, 265-272.
41.Adediran, S. A., and Lambeir, A. (1989) Eur.
J. Biochem., 186, 571-576.
42.Rodriguez-Lopez, J. N., Smith, A. T., and
Thorneley, R. N. F. (1997) J. Biol. Chem., 272,
389-395.
43.Chance, B. (1952) J. Biol. Chem.,
197, 577-589.
44.Nicholls, P. (1962) in The Oxygenases
(Hayaishi, O., ed.) Academic Press, New York, pp. 263-305.
45.Michaelis, L. (1932) J. Biol. Chem.,
96, 703-715.
46.Gibson, J. F., Nicholls, P., and Ingram, D. J. E.
(1958) Nature, 181, 1398-1399.
47.Yonetani, T., and Schleyer, H. (1966) J. Biol.
Chem., 241, 3240-3243.
48.Dolphin, D., Forman, A., Borg, D. C., Fajer, J.,
and Felton, R. H. (1971) Proc. Natl. Acad. Sci. USA, 68,
614-618.
49.Baek, H. K., and van Wart, H. E. (1989)
Biochemistry, 28, 5714-5719.
50.Denisov, I. G., Makris, T. M., and Sligar, S.
(2002) J. Biol. Chem., 277, 42706-42710.
51.Kobert, R. (1900) Arch. Ges. Physiol.
(Pflüger's Arch.), 82, 603-630.
52.George, P., and Irvine, D. H. (1952) Biochem.
J., 52, 511-517.
53.King, N. K., and Winfield, M. E. (1963) J.
Biol. Chem., 238, 1520-1527.
54.Svistunenko, D. (2005) Biochim. Biophys.
Acta, 1707, 127-155.
55.Egawa, T., Shimada, H., and Ishimura, Y. (2000)
J. Biol. Chem., 275, 34858-34866.
56.Egawa, T., Yoshioka, S., Takahashi, S., Hori, H.,
Nagano, S., Shimada, H., Ishimori, K., Morishima, I., Suematsu, M., and
Ishimura, Y. (2003) J. Biol. Chem., 278, 41597-41606.
57.Hersleth, H. P., Dalhus, B., Gorbitz, C. H., and
Andersson, K. K. (2002) J. Biol. Inorg. Chem., 7,
299-304.
58.Silaghi-Dumitrescu, R., Brandon, J., Reeder, B.
J., Nicholls, P., Cooper, C. E., and Wilson, M. T. (2007) Biochem.
J., 403, 391-395.
59.Svistunenko, D. A., Reeder, B. J., Wankasi, M.
M., Silaghi-Dumitrescu, R.-L., Cooper, C. E., Rinaldo, S., Cutruzzola,
F., and Wilson, M. T. (2007) Roy. Soc. Chem. Dalton Trans.,
840-850.
60.Wrigglesworth, J. M. (1984) Biochem. J.,
217, 715-719.
61.Fabian, M., and Palmer, G. (1995)
Biochemistry, 34, 13802-13810.
62.Nicholls, P., Loewen, P., and Fita, I. (2001)
Adv. Inorg. Chem., 51, 52-106.
63.Melik-Adamyan, W. R., Bravo, J., Carpena, X.,
Switala, J., Mate, M. J., Fita, I., and Loewen, P. C. (2001)
Proteins: Struct. Funct., 44, 270-280.
64.Gouet, P., Jouve, H. M., Williams, P. A.,
Andersson, I., Andreoletti, P., Nussaume, L., and Hajdu, J. (1996)
Nature Struct. Biol., 3, 951-956.
65.Berglund, G. I., Carlsson, G. H., Smith, A. T.,
Szoke, H., Henriksen, A., and Hajdu, J. (2002) Nature,
417, 463-468.
66.Svensson-Ek, M., Abramson, J., Larsson, G.,
Tornroth, S., Brezezinski, P., and Iwata, S. (2002) J. Mol.
Biol., 321, 329-339.
67.Qin, L., Hiser, C., Mulichak, A., Garavito, R.
M., and Ferguson-Miller, S. (2006) Proc. Natl. Acad. Sci. USA,
103, 16117-16122.
68.Yoshikawa, S., Shinzawa-Itoh, K., Nakashima, R.,
Yaono, R., Yamashita, E., Inoue, N., Yao, M., Fei, M. J., Libeu, C. P.,
Mizushima, T., Yamaguchi, H., Tomizaki, T., and Tsukihara, T. (1998)
Science, 280, 1723-1729.
69.Tsukihara, T., Aoyama, H., Yamashita, E.,
Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono,
R., and Yoshikawa, S. (1996) Science, 272, 1136-1144.