Rapid Production of Human KIR2DL4 Extracellular Domain and Verification of Its Interaction with HLA-G
Yan-Rong Yu, Xing-Hui Tian, Yun Wang, and Mei-Fu Feng*
State Key Laboratory of Biomembrane and Membrane Biotechnology, Institute of Zoology, Chinese Academy of Sciences, No. 25, BeiSiHuanXi Road, Beijing, 100080, P. R. China; fax: +86-10-62571017; E-mail: fengmf@ioz.ac.cn; yuyr@ioz.ac.cn* To whom correspondence should be addressed.
Received December 22, 2004; Revision received March 29, 2005
Killer cell immunoglobulin-like receptor (KIR) 2DL4 is the only KIR member reported to be expressed by all human natural killer (NK) cells. It differs from other KIR members in both structure and function. Its specific interaction with HLA-G, a non-classical MHC class I molecule, has been suggested to play an important role in regulating NK cell-mediated cytotoxicity. However, this interaction is still in doubt. In addition, the soluble KIR2DL4 extracellular domain used in many studies was produced by eukaryotic expression, which is less efficient than prokaryotic expression. In this study, we describe a method of rapid production a large amount of soluble KIR2DL4 extracellular domain based on a prokaryotic expression system. With this soluble KIR2DL4, we verified the interaction between KIR2DL4 and HLA-G1.
KEY WORDS: KIR2DL4, HLA-G1, natural killer cell, protein expressionDOI: 10.1134/S0006297906130104
Abbreviations: NK) natural killer (cells); KIR) killer cell immunoglobulin-like receptor; ITIM) immunoreceptor tyrosine-based inhibition motif.
Natural killer (NK) cells are a fundamental component of the immune
system, which provide a link between innate and adaptive immunity. They
can lyse tumor and virally transformed target cells without prior
sensitization in the early stages of an immune response. However, the
molecular mechanism(s) involved in their cytotoxic function remained
mysterious for many years, until Ljunggren and Karre proposed the
“missing self” hypothesis in 1990 [1].
According to this hypothesis, the activity of NK cells is regulated by
the balance between inhibitory and activating signal and the inhibitory
signal is provided by the interaction of MHC class I molecules
expressed on normal self cells with their receptors expressed on NK
cell membrane. Since many tumor cells and virally transformed cells
have lost or express insufficient amounts of MHC class I molecules,
they will be recognized and lysed by NK cells as non-self cells.
Human NK cells express killer cell immunoglobulin-like receptors (KIR) and lectin-like receptors (CD94/NKG2) that can recognize MHC class I molecules and subsequently either inhibit or activate NK cytotoxicity [2]. KIR2DL4 (CD158d, 103AS) is a member of KIR family and shares structural features with both activating and inhibitory receptors [3, 4]. It has a single cytoplasmic immunoreceptor tyrosine-based inhibition motif (ITIM) that is found to be required for inhibitory function, and a positively charged amino acid (arginine) in the transmembrane region that is often related to activating function. Besides this distinct feature, KIR2DL4 also differs from other KIR members in its unique immunoglobulin (Ig) domain configuration: its two Ig domains are arranged in a D0-D2 configuration whereas the two or three Ig domains of other KIR members are arranged in the order D1-D2 or D0-D1-D2, respectively.
Although KIR2DL4 was reported to be an HLA-G-specific receptor expressed on all NK cells, their interaction was questioned by another study in which the authors failed to detect their interaction by both ELISA assay and surface plasmon resonance assay [5, 6]. Since the binding of HLA-G ligands to KIR2DL4 receptors may play an important role in reproduction, tissue transplantation, and tumor immune escape [5, 7, 8], it is important to verify their interaction or to search for new ligands for KIR2DL4.
The rapid production of sufficient amounts of functional molecules is an important step to perform functional and structural analysis of KIR2DL4, such as co-crystallization of its extracellular domain and its ligand. Thus far, all studies investigating the interaction of KIR2DL4 and HLA-G have used KIR2DL4 molecules expressed on NK cells or soluble KIR2DL4 extracellular domain secreted from transfected eukaryotic cells. However, eukaryotic expression systems are usually less productive and more laborious than prokaryotic expression. Here we report a method to produce a GST-KIR2DL4 extracellular domain fusion protein in E. coli with high yield. After refolding and purification, the expressed protein was used to confirm the interaction between KIR2DL4 and HLA-G1.
MATERIALS AND METHODS
Cell lines and antibodies. NK92 cells were maintained in alpha-MEM supplemented with 12.5% (v/v) horse serum, 12.5% (v/v) fetal bovine serum (FBS), 2 mM L-glutamine, 1.5 g/liter sodium bicarbonate, 0.2 mM inositol, 0.1 mM 2-mercaptoethanol, 0.2 mM folic acid, 100-200 U/ml IL-2, and K562 cells transfected with pLPCX-HLA-G1/EGFP or pLPCX-EGFP were maintained in RPMI1640 supplemented with 10% FBS and 5 µg/ml puromycin. Both cell lines were grown in a 37°C humidified atmosphere containing 5% CO2.
Mouse monoclonal anti-GFP primary antibody was purchased from Abcam Ltd. (UK) and rabbit polyclonal anti-GST primary antibody was purchased from Invitrogen (USA). Horseradish peroxidase conjugated secondary antibodies were purchased from Santa Cruz (USA).
Cloning of KIR2DL4 cDNA. Total cellular RNA of NK92 cells was prepared with TRIZOL (Invitrogen) and was reversely transcribed with ThermoscriptTM (Invitrogen) to produce total cellular cDNAs. The KIR2DL4 cDNA was amplified with the forward primer 5´-ATGTTGCTCATGGTCGTCAGCATGGCGTGT-3´ and the reverse primer 5´-GGCAGACATGTTTATTTGAAGAGGAGA-3´ with Platinum pfx DNA polymerase (Invitrogen) and was ligated into pGEM-T Easy vector (Promega, USA). The construct was transformed into DH5alpha cells and positive colonies were sequenced. After sequencing, we compared the sequence that we cloned with other reported sequences from GenBank with vector NTI suite 6.0 software (InforMax, Inc., USA).
Construction and expression of GST-KIR2DL4 extracellular domain fusion protein. The encoding sequence of extracellular domain of KIR2DL4 was amplified using pGEM-T Easy/KIR2DL4 as the DNA template with the forward primer 5´-AATGGATCCCAGAGTGTGTGGGCAC-3´ containing a BamHI site, and the reverse primer 5´-CATCTCGAGTCAGAGTACCTAATCACAGC-3´ containing an XhoI site. The PCR product was digested with BamHI and XhoI (New England Biolabs Inc., USA) and was ligated into pGEX-4T2 vector (Amersham Biosciences, USA).
The GST-KIR2DL4 extracellular domain fusion construct was transformed into E. coli GJ980 competent cells (kindly provided by Prof. Gong Yi, Shanghai Research Center of Biotechnology, Chinese Academy of Sciences) and the transformants were selected on agar plates with 50-100 µg/ml ampicillin. A positive transformant was then grown in Luria-Bertani (LB) medium at 37¡ãC with vigorous shaking. Isopropyl-1-thio-beta-D-galactopyranoside (IPTG, 0.1-0.4 mM) (Promega, USA) was added when the OD600 value reached 0.6-1 to induce protein expression and the E. coli were harvested 3-4 h later.
Refolding and purification of the fusion protein. The E. coli was collected by centrifugation and was resuspended in 0.1 volume of 0.1 M phosphate buffer (PB, pH 8.0). After sonication, the lysate was centrifuged at 5000g for 30 min and the supernatant was removed to another tube. Both the supernatant and the pellet were examined to detect the presence of a fusion protein. After confirming that the fusion protein was mainly in the pellet fraction, the pellet was washed with 0.1 M PB containing 0.5% Triton X-100, 2 M urea, and 1 M NaCl, respectively, and then was dissolved in 0.1 volume of denaturing solubilization buffer (0.1 M PB, 8 M urea, pH 8.5). After being agitated for 15-30 min, the solution was centrifuged at 10,000g for 15 min and the supernatant was collected for further refolding.
The fusion protein was refolded by dialyzing the supernatant in alkaline buffer (20 mM Tris and 0.3 M NaCl, pH 9.6) at 4¡ãC for 48 h with three changes of the alkaline buffer. Then the dialysis buffer was changed to alkaline buffers with pH value equal to 9.0 or 8.5, and then to a normal phosphate buffered saline (PBS), with 12 h dialysis at 4¡ãC for each buffer. Finally, the protein was further purified with glutathione-Sepharose 4B (Pharmacia Biotech, USA). The protein concentration, before and after each step, was determined by Bradford assay.
Preparation of HLA-G1 solution. HLA-G1 cDNA (GenBank No. AF226990) was cloned from peripheral blood mononucleated cells of a healthy Chinese [9] and was first ligated into pEGFP-N1 vector. The HLA-G1/EGFP fragment was then integrated into pLPCX retroviral vector (Clontech, USA) and was packaged in 293T cells. K562 cells were then infected with the 293T supernatant and were selected by puromycin (5 µg/ml) for two weeks or more. After successful transfection was confirmed by flow cytometry and microscopy showing that more than 85% of the cells display green fluorescence, the transfected K562 cells were lysed with the lysis buffer (1% NP-40, 10 mM Tris, pH 7.5, 140 mM NaCl, 50 mM NaF, 1 mM EDTA, 0.4 mM sodium orthovanadate, 0.1 mM phenylmethylsulfonyl fluoride) at 4¡ãC for 30 min followed by centrifugation at 10,000g for 30 min. The supernatant was then pre-cleared with lysis buffer-equilibrated glutathione beads and was used as the HLA-G solution for ligand-binding assay.
Ligand-binding assay. Purified GST-KIR2DL4 fusion protein was first incubated with HLA-G solution at 4¡ãC for 30 min in the absence and then for another 2 h in the presence of lysis buffer-equilibrated glutathione beads. After washing the beads with lysis buffer three times, the protein complex was recovered by boiling the beads in 2× SDS-PAGE sample buffer for 5 min and was subjected to immunoblot analysis with anti-GFP antibody and anti-GST antibody.
RESULTS
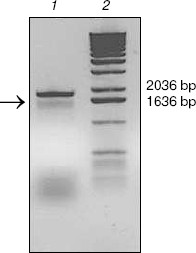
KIR2DL4 full-length cDNA cloning from NK92 cells. The cDNA of KIR2DL4 gene with a full length of 1596 bp was cloned from NK92 cells (Fig. 1) and was submitted to GenBank (GenBank No. AY601812). The sequencing result showed that our cDNA was a new variant of the KIR2DL4 gene, sharing the identical extracellular and transmembrane domains and a similar cytoplasmic domain with other reported cDNA variants in GenBank except that our clone had an extra fragment in its cytoplasm domain that encodes 21 amino acid residues (Fig. 2, see color insert, p. 4).
Fig. 1. Analysis of cDNA of KIR2DL4 cloned from NK92 cells in 1% agarose gel. Lanes: 1) PCR product, a fragment of expected length of 1596 bp (arrow indicated); 2) molecular weight markers (1 kb DNA Extension Ladder).
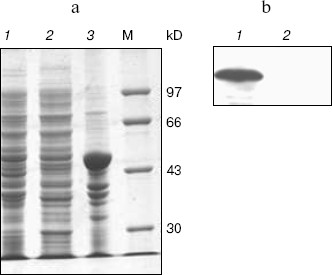
Expression, refolding, and purification of GST-KIR2DL4 fusion protein. The successful construction of a GST-KIR2DL4 extracellular domain fusion protein expression vector was confirmed by restriction digestion analysis with BamHI and XhoI (Fig. 3). The recombinant was expressed in E. coli GJ980 strain with IPTG induction. As shown in Fig. 4a, the most abundant protein, mainly present in the pellet fraction and occupying about 30% of total cellular protein determined by Quantity One software (Bio-Rad, USA), has an apparent molecular weight of about 51 kD, which is consistent with the expected molecular weight of GST-KIR2DL4 fusion protein. Immunoblotting with anti-GST antibody confirmed its identity as GST-KIR2DL4 fusion protein (Fig. 4b). During the process of refolding, there were tiny aggregates and, as a result, a part of protein was lost and the total refolding efficiency was 20%. After purification by affinity chromatography, another 40% of total protein was lost and the purity of protein of interest reached above 90% (Fig. 5). The yield of the fusion protein at the end was as high as 0.5 mg per 100 ml of E. coli culture.Fig. 2. Homology comparison of KIR2DL4 protein sequences (blue indicates 100% homology; yellow 75% homology; white 50% homology).
Fig. 3. Identification of pGEX-KIR2DL4 recombinant plasmid by BamHI and XhoI digestion. Lanes: 1) molecular weight markers; 2) restriction digestion products including a KIR2DL4 extracellular domain fragment of about 700 bp and a pGEX-4T-2 vector fragment of 4970 bp.
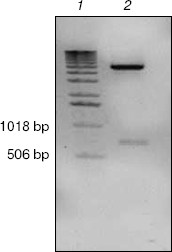
Fig. 4. SDS-PAGE and western blotting analysis of GST-KIR2DL4 fusion protein expression. a) SDS-PAGE (12% separating gel) detection of induced GST-KIR2DL4 fusion protein: 1) before IPTG induction; 2) supernatant after IPTG induction; 3) inclusion bodies after IPTG induction; M) protein molecular weight markers. b) Western blotting detection of induced protein: 1) after IPTG induction; 2) before IPTG induction.
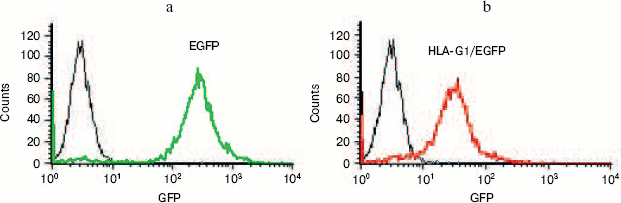
Expression of HLA-G1/EGFP fusion protein on K562 cell membrane. In order to get the natural HLA-G protein for ligand-binding assay, we transfected human erythroleukemia HLA-A, -B, -C, and -G-negative K562 cells with HLA-G1/EGFP fusion protein. The expression of HLA-G1/EGFP fusion protein on K562 cell membrane was verified by flow cytometry analysis (Fig. 6, see color insert, p. 5) and confocal imaging (Fig. 7). The functional activity of the fusion protein was confirmed by cytotoxicity assay, which showed that HLA-G1/EGFP could protect K562 cells from killing by NK92 cells (data not shown).Fig. 5. SDS-PAGE analysis of GST-KIR2DL4 fusion protein purification. Lanes: M) protein molecular weight markers; 1) before IPTG induction; 2) supernatant after IPTG induction; 3) inclusion bodies after IPTG induction; 4) purified protein after refolding.
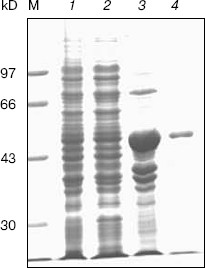
Fig. 6. Flow cytometry analysis of EGFP (a) and HLA-G1/EGFP protein expression (b) in K562-transfected cells. 93% and 85% of K562 cells express EGFP and HLA-G1/EGFP, respectively, after puromycin selection for two weeks.
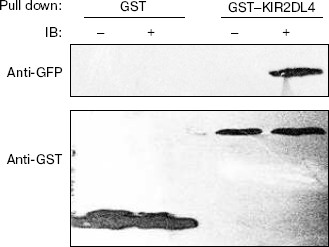
Binding activity of GST-KIR2DL4 fusion protein with HLA-G1. HLA-G1/EGFP transfected K562 cells were lysed and the lysate was pulled down with the refolded GST-KIR2DL4 fusion protein. Western blotting analysis of the precipitates showed that the refolded GST-KIR2DL4 fusion protein could specifically bind to HLA-G1. The controls of native GST protein or EGFP alone showed that the GST or EGFP part of the fusion protein did not contribute to their specific interaction (Fig. 8).Fig. 7. Confocal images of HLA-G1 on K562-transfected cells. a) EGFP mainly expresses in cytoplast and karyon of K562 cells; b) HLA-G1/EGFP located on the membrane of K562 cells.
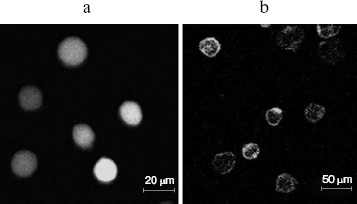
Fig. 8. Direct interaction of HLA-G1 and KIR2DL4. Western blotting was performed with GFP and GST antibodies. GST indicates samples pulled down by native GST protein; GST-KIR2DL4 indicates samples pulled down by refolded GST-KIR2DL4 fusion protein; symbol “-” indicates pLPCX-EGFP transfected K562 cells lysate; symbol “+” indicates pLPCX-HLA-G1/EGFP transfected K562 cells lysate.
DISCUSSION
We successfully cloned a new variant of the KIR2DL4 cDNA from NK92 cells. The full length of this variant was 1596 bp with an open reading frame of 1191 bp. Compared with other sequences from GenBank, the sequence we cloned from NK92 cells encodes a protein that contains the conserved extracellular and transmembrane domains, but has an extra fragment in its relatively polymorphic cytoplasm domain. This insert is produced by alternative splicing resulting from the lack of an A (adenine) in the pre-RNA sequence (Fig. 2, see color insert, p. 4). Since the extra 269-290 amino acid contains no ITIM or ITIM-like motif, which is required for inhibitory signal transduction, it is speculated that it will not affect signal transduction after ligand binding. However, further studies are needed to test this speculation.
The protein refolding of inclusion bodies has been a bottleneck for expressing recombinant protein in prokaryotic cells for a long time. E. coli thioredoxin reductase (trxB) mutant strain GJ980 allows the formation of disulfide bonds in the prokaryotic cell, which is believed to encourage correct folding and soluble expression of recombinant protein [10], but in our present study the protein of interest was still expressed mostly in inclusion bodies. We tried several methods to refold the recombinant protein, but mostly failed because of aggregation. Finally, we got the correctly folded protein by using alkaline buffer and stepwise reduction of the pH value of the alkaline buffer. The calculated pI of KIR2DL4 extracellular domain, based on its amino acid sequence, is 8.2, which may explain the ability of an alkaline buffer to enhance its refolding process. In addition, we found that washing the inclusion bodies with a high concentration of saline, detergent and 2 M urea could facilitate purification.
The ligand-binding assay in the present study supported the observation that KIR2DL4 was a HLA-G specific receptor [5]. Recognition of non-classical, rather than classical MHC class I molecules is consistent with KIR2DL4's divergent structure compared to KIRs that recognition HLA-A, -B, and -C. Unlike these KIRs, 2DL4 is phylogenetically conserved in humans and Old World primates, fitting well with recognition of conserved, nonpolymorphic class Ib, HLA-G [11, 12]. However, we have no explanation so far why this ligand-receptor interaction was not observed in another study [6]. We hope that the availability of the protocol reported here to rapidly produce a large amount of soluble KIR2DL4 extracellular domain could facilitate the co-crystallization of HLA-G1 and KIR2DL4 and, as a consequence, could promote the further study of their interaction.
REFERENCES
1.Ljunggen, H. G., and Karre, K. (1990) Immunol.
Today, 11, 237-244.
2.Middleton, D., Curran, M., and Maxwell, L. (2002)
Transplant. Immunol., 10, 147-164.
3.Borrego, F., Kabat, J., Kim, D. K., Lieto, L.,
Maasho, K., Pena, J., Solana, R., and Coligan, J. E. (2002) Mol.
Immunol., 38, 637-660.
4.Faure, M., and Long, E. O. (2002) J.
Immunol., 168, 6208-6214.
5.Rajagopalan, S., and Long, E. O. (1999) J. Exp.
Med., 189, 1093-1100.
6.Boyson, J. E., Erskine, R., Whitman, M. C., Chiu,
M., Lau, J. M., Koopman, L. A., Valter, M. M., Angelisova, P., Horejsi,
V., and Strominger, J. L. (2002) Proc. Natl. Acad. Sci. USA,
99, 16180-16185.
7.Algarra, I., Garcia-Lora, A., Cabrera, T.,
Ruiz-Cabello, F., and Garrido, F. (2004) Cancer Immunol.
Immunother., 53, 904-910.
8.Carosella, E. D., Moreau, P., Le Maoult, J., Le
Discorde, M., Dausset, J., and Rouas-Freiss, N. (2003) Adv.
Immunol., 81, 199-252.
9.Zhou, J. J., Qu, H., Wu, C. H., Han, W. D., Li, Z.
W., and Feng, M. F. (2002) Acta Sci. Natur. Univers. Pekinensis,
38, 212-220.
10.Tong, Q., Yang, Y. G., Zhang, H. T., Chen, Y.,
Yang, S. L., and Gong, Y. (2001) Acta Biochim. Biophys. Sin.,
33, 30-34.
11.Cantoni, C., Verdiani, S., Falco, M., Pessino,
A., Cilli, M., Conte, R., Pende, D., Ponte, M., Mikaelsson, M. S.,
Moretta, L., and Biassoni, R. (1998) Eur. J. Immunol.,
28, 1980-1990.
12.Rajalingam, R., Parham, P., and Abi-Rached, L.
(2004) J. Immunol., 172, 356-369.