Two Soluble Pyrophosphatases in Vibrio cholerae: Transient Redundancy or Enduring Cooperation?
A. Salminen1*, M. Ilias2, G. A. Belogurov1, A. A. Baykov3, R. Lahti1, and T. Young2*
1Department of Biochemistry, University of Turku, Turku, Finland; fax: +358-2-333-6860; E-mail: anusalmi@utu.fi; geobel@utu.fi; reijo.lahti@utu.fi2School of Biosciences, University of Birmingham, Birmingham, UK; fax: +44-121-414-3982; E-mail: ilias14@yahoo.com; t.w.young@bham.ac.uk
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-3181; E-mail: baykov@genebee.msu.su
* To whom correspondence should be addressed.
Received March 10, 2006
Soluble pyrophosphatases (PPases), which are essential for cell life, comprise two evolutionarily unrelated families (I and II). Prokaryotic genomes generally contain a single PPase gene encoding either family I or family II enzyme. Surprisingly, four Vibrionales species, including the human pathogen Vibrio cholerae, contain PPase genes of both families. Here we show that both genes are transcriptionally active in V. cholerae, and encode functional PPases when expressed in Escherichia coli. In contrast, only the family I PPase protein is detected in V. cholerae under our experimental conditions. Phylogenetic analyses indicate that family II enzymes are not native to gamma-proteobacteria, but are of benefit to the marine species of this bacterial class. In this context, we favor the hypothesis that in the course of evolution, family II PPase was laterally transferred to the Vibrionales ancestor and partially degenerated due to functional redundancy, but nevertheless remained fixed as an adjunct to the family I enzyme.
KEY WORDS: gamma-proteobacteria, phylogeny, lateral gene transfer, functional redundancy, non-orthologous displacementDOI: 10.1134/S0006297906090057
Abbreviations: CHES) N-cyclohexyl-2-aminoethanesulfonic acid; PPase) inorganic pyrophosphatase; ppa) gene encoding PPase; vcPPase1) V. cholerae family I PPase; vcPPase2) V. cholerae family II PPase.
Soluble inorganic pyrophosphatase (PPase, EC 3.6.1.1) catalyzes the
cation-dependent hydrolysis of inorganic pyrophosphate to
orthophosphate. This reaction provides a thermodynamic pull for many
biosynthetic reactions [1, 2]
and is essential for life [3, 4]. Soluble PPases are divided into two non-homologous
families with completely different 3D structures. Family I PPases,
which have been known for about 70 years, are among the best
characterized phosphoryl transfer enzymes [5, 6]. They are homodimers (in eukaryotes) or
homohexamers (in prokaryotes) of compact, one-domain subunits. Family
II PPases were only recently found [7, 8] and have thus been only partially characterized.
All known family II PPases are homodimers of two-domain subunits, which
interact through N-terminal domains [9, 10]. The N- and C-terminal domains are connected by a
flexible linker, and the active site is located between the domains.
Family II PPases are an order of magnitude more active than family I
PPases and exhibit maximal activity in the presence of Mn2+,
whereas Mg2+ is the most effective activator of family I
PPases [11, 12].
Family I PPases predominate in eukaryotes, archaea, and most eubacterial phylums. In contrast, family II PPases predominate only in Bacilli and Clostridia and are found only sporadically in other prokaryotic lineages. Inspection of 283 completed genome sequences of prokaryotes shows that they generally contain a single gene encoding either a family I or a family II soluble PPase. The only exceptions are, interestingly, four species from the order Vibrionales, including the human pathogen Vibrio cholerae, which have genes for both PPase families.
In this report, we show that active enzymes (vcPPase1 and vcPPase2) are generated from both V. cholerae ppa genes when they are overexpressed in E. coli. Using RT-PCR and Western blotting, we also examined the expression of both PPase genes when V. cholerae is cultured under different conditions. Finally, we used phylogenetic analyses to infer the history of PPase transmission in gamma-proteobacteria and to assess the significance of PPase dichotomy in Vibrionales.
MATERIALS AND METHODS
Cloning, expression, and purification. Genomic DNA was extracted from the A-B- vaccine strain JBK 70 of V. cholerae O1, an attenuated strain of El Tor N16961 [13]. The family I and family II PPase open reading frames (GenBank accession numbers NP 232173 and NP 231323, respectively) were amplified by PCR. The amplified ppa genes were sequenced in both directions and were shown to be identical to the published data [14]. The family I ppa gene was cloned into the pET23a vector (Novagen, Germany) using NdeI and HindIII restriction enzymes. lambdaDE3 prophage was integrated into an E. coli MC1061/YPPA1(Deltappa) [15] chromosome using a lambdaDE3 lysogenization kit (Novagen), and the resulting strain MC1061/YPPA1(Deltappa) (DE3) was used as the expression host. Family I PPase was purified by heat treatment (20 min at 70°C) in the presence of 50 mM MgCl2 followed by DEAE-Sepharose fast flow ion-exchange chromatography (Amersham Biosciences AB, Sweden). The family II ppa gene was cloned into the pET11c vector (Novagen) using NdeI and BamHI restriction enzymes, expressed in E. coli BL21(DE3), and purified as described by Parfenyev et al. [11]. The purity of the enzymes was checked by electrophoresis on 8 to 25% gradient polyacrylamide gels using the Phast System (Amersham Biosciences).
Enzyme assay. Rates of PPi hydrolysis were determined from continuous recordings of Pi liberation obtained using an automatic Pi analyzer [16]. Values for kcat and Km were determined at 25°C in 0.1 M Tris-HCl (pH 7.2) containing 20 mM Mg2+ (for family I PPase) or 0.083 M TES-KOH (pH 7.2) containing 17 mM KCl, 20 mM Mg2+, and 0.05 mM MnCl2 (for family II PPase).
Antisera to PPases. Polyclonal antisera were obtained from guinea pigs using 1 mg/ml purified vcPPases as antigens. Serum samples taken prior to immunization showed no reaction with either vcPPase1 or vcPPase2. The immunization protocol was essentially as described by Glover et al. [17]. We found that vcPPase2 was a more potent antigen than vcPPase1. Family I and family II proteins were immunologically distinct, and no cross reactivity was observed.
Growth of V. cholerae and preparation of total RNA and cell lysates. Cultures were grown with shaking at 37°C in LB broth buffered at pH 9 (50 mM CHES-NaOH), pH 7 (50 mM MOPS-NaOH), or unbuffered containing 2% w/v NaCl. Total RNA was stabilized and released from cells using the Qiagen RNA Protect Bacteria Reagent and extracted from lysates using the RNeasy Mini Protocol (Qiagen, UK). The ratio of 23S/16S RNA in samples was 2.6 ± 0.2, and the concentration of RNA was between 180 and 302 µg/ml.
For Western blotting cells were grown to OD600 = 1.0, concentrated 100× by centrifugation and resuspension in 100 mM Tris-HCl (pH 8.0), and analyzed on a 12% SDS-polyacrylamide gel. Protein transfer to polyvinylidene fluoride (PVDF) membranes and immunodetection were carried out essentially as described by Glover et al. [17]. Membranes were probed with either anti-vcPPase1 or anti-vcPPase2 antiserum as the primary antibody. Anti-guinea pig IgG-peroxidase conjugate (Sigma-Aldrich, USA) at 1 : 25,000 was used as the secondary antibody. The peroxidase conjugate-treated immunoblot was developed for 5 min in 6 ml of SuperSignal→ West Pico Chemiluminescent Substrate (Pierce Biotechnology, USA). Immunoreactive bands were detected using Kodak Bio Max MR-1 film.
Phylogenetic analyses. Proteins were aligned using MUSCLE version 3.52 [18]. Ambiguous sections and regions containing gaps in more than 10% of the sequences were removed. The resulting alignment of family I and family II PPases and concatenated beta and beta´ RNA polymerase subunits displayed 170, 275, and 2534 amino acid positions, respectively. Phylogenetic trees were generated with MrBayes 3.1.1 [19] using the Whelan and Goldman model of substitution [20]. Rate heterogeneity was modeled by discrete gamma-distribution [21] with four categories, and by allowing invariable sites. To evaluate the differences between PPase and RNA polymerase subunits trees, we employed the Shimodaira-Hasegawa test [22] implemented in TREE-PUZZLE 5.2 [23]. We introduced a lateral gene transfer event in cases where PPase alignments rejected the topology recommended by RNA polymerase subunit alignment at a 5% level of significance.
RESULTS AND DISCUSSION
Heterologous expression and catalytic characteristics of V. cholerae PPases. Both V. cholerae PPases could be expressed in E. coli with a yield of 30 to 50 mg per liter of medium. The purified enzymes were homogenous based on SDS-PAGE. Based on the Michaelis-Menten parameters, the Vibrio PPases closely resemble their E. coli and B. subtilis counterparts (table). Thus, the turnover number (kcat) of vcPPase2 measured in the presence of Mn2+ (the most effective metal cofactor for family II PPases) is about 15-fold greater than that of vcPPase1 measured in the presence of Mg2+ (the most effective metal cofactor for family I PPases). The absolute values of kcat are smaller by a factor of two for the Vibrio PPases. Omitting Mn2+ from the reaction medium reduced kcat for vcPPase2 greater than 20-fold. The specificity constants (kcat/Km) are nearly identical for vcPPase1, vcPPase2, and E. coli PPase, and they differ by only 2.4-fold from that for B. subtilis PPase. These results indicate that both vcPPase1 and vcPPase2 genes encode active proteins with catalytic properties typical for their corresponding enzyme families. This is consistent with the finding that all the active site residues directly involved in ligand binding [6, 9, 10] are completely conserved in both V. cholerae PPases.
Michaelis-Menten parameters for PPases from the two families
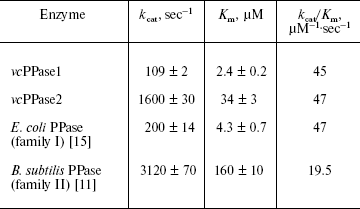
RT-PCR of V. cholerae total RNA extracts. Oligonucleotide primers were prepared to amplify fragments of 234 and 345 bp from the vcPPase1 and vcPPase2 genes, respectively. Figure 1 shows agarose gel electrophoresis of the RT-PCR products from total RNA extracts of V. cholerae grown under different conditions. Omission of the reverse transcriptase step yielded no products (data not shown), demonstrating that there was no DNA contamination of extracts, and omitting the RNA extract (lane 8) gave no products. RT-PCR using extracts from cells grown in LB medium at pH 7, pH 9, or unbuffered containing 2% NaCl, with primers for either the vcPPase1 gene (lanes 2, 4, 6) or the vcPPase2 gene (lanes 3, 5, 7) generated products of the expected sizes. Also, appropriate products were produced by PCR of Vibrio DNA using both primer sets (lane 9). These results suggest that transcription products for both genes were present under the conditions tested.
Western blotting of V. cholerae extracts. Extracts of V. cholerae were analyzed by Western blotting using an antiserum raised against purified overexpressed vcPPase1 (Fig. 2). As a control, E. coli extract was also analyzed. Western blotting revealed three immunoreactive bands in the E. coli extract (lane 1). The faintest of these (marked by an arrow) had the same molecular mass as the purified vcPPase1, as observed by Western blotting (lane 5) and SDS-PAGE followed by staining with Coomassie brilliant blue (lane 6). Presumably, this faint band is due to cross-reaction of the antiserum with E. coli family I PPase. The low intensity of the reaction is consistent with a low concentration of E. coli PPase present in the extract. The other two bands in lane 1 showed strong reactions but were absent from all of the V. cholerae extracts and, hence, represent highly abundant E. coli proteins that have epitopes recognized by the antiserum. Single intense bands were obtained in extracts of all three V. cholerae cultures (lanes 2-4). The electrophoretic mobility of these three bands as well as of the immunoreactive vcPPase1 and the major band of the Coomassie-stained antigen are the same. These results clearly indicate that V. cholerae synthesizes a family I PPase.Fig. 1. RT-PCR of total RNA extracts from V. cholerae cells grown under different conditions. Lanes: 2, 4, 6) cells were grown in LB medium buffered at pH 7 or 9 or in unbuffered LB medium containing 2% NaCl, respectively, and total RNA was amplified by RT-PCR using primers for the vcPPase1 gene; 3, 5, 7) cells were grown in LB medium buffered at pH 7 or 9 or in unbuffered LB medium containing 2% NaCl, respectively, and total RNA was amplified by RT-PCR using primers for the vcPPase2 gene; 1) DNA size markers; 8) no RNA; 9) Vibrio DNA.
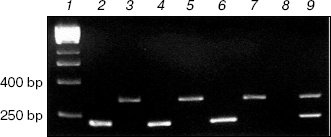
Figure 3 shows the results of Western blotting of extracts using the antiserum raised against purified overexpressed vcPPase2. The immunodetection system gave a strong positive reaction with only 83 ng of vcPPase2 (lane 5). However, immunoreactive proteins were not detected in the extracts from V. cholerae grown in unbuffered medium containing 2% NaCl (lane 2) or in medium buffered at pH 9 (lane 3) or pH 7 (lane 4). The weak band of about 29 kD observed in the E. coli extract (lane 1) apparently represents an E. coli protein that cross-reacts with the antiserum. The failure to detect family II PPase in the extracts from V. cholerae may be due either to the absence of translation or protein instability under the experimental conditions employed.Fig. 2. Western blotting analysis of vcPPase1 levels in extracts of V. cholerae cells grown under different conditions. Western blotting was carried out with anti-vcPPase1 serum (210 µg/ml) followed by 1 : 25,000 secondary antibody. Lanes: 1) E. coli BL21(DE3) extract; 2, 3, 4) extracts from V. cholerae cells grown in unbuffered LB medium containing 2% NaCl or in LB medium buffered at pH 9 or 7, respectively; 5, 6) 0.2 and 2.0 µg of vcPPase1, respectively; 7) protein size markers.
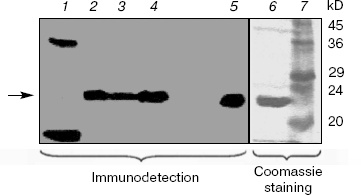
Evolution and distribution of PPases in gamma-proteobacteria. Family I PPases are widespread among gamma-proteobacteria with sequenced genomes. The molecular phylogenetic analysis (results not shown) revealed that the majority of gamma-proteobacterial family I PPases form a monophyletic group and are therefore possibly inherited from the common ancestor of this bacterial class. However, two closely related PPases from Haemophilus influenzae and Mannheimia succiniciproducens and that of Nitrosococcus oceani represent two distinct ancient lineages that cannot be classified with any other known family I enzymes. In these species, native family I PPase was possibly displaced by another enzyme of the same family that was transferred from as yet unsequenced lineages. Finally, the presence of two closely related family I PPases in Pseudomonas syringae indicates recent duplication of the gene in this lineage.Fig. 3. Western blotting analysis of vcPPase2 levels in extracts of V. cholerae cells grown under different conditions. Western blotting was carried out with anti-vcPPase2 serum (18.4 µg/ml) followed by 1 : 25,000 secondary antibody. Lanes: 1) E. coli BL21(DE3) extract; 2, 3, 4) extracts from V. cholerae cells grown in unbuffered LB medium containing 2% NaCl or in LB medium buffered at pH 9 or 7, respectively; 5, 6) 0.083 and 2.5 µg of vcPPase2, respectively; 7) protein size markers.
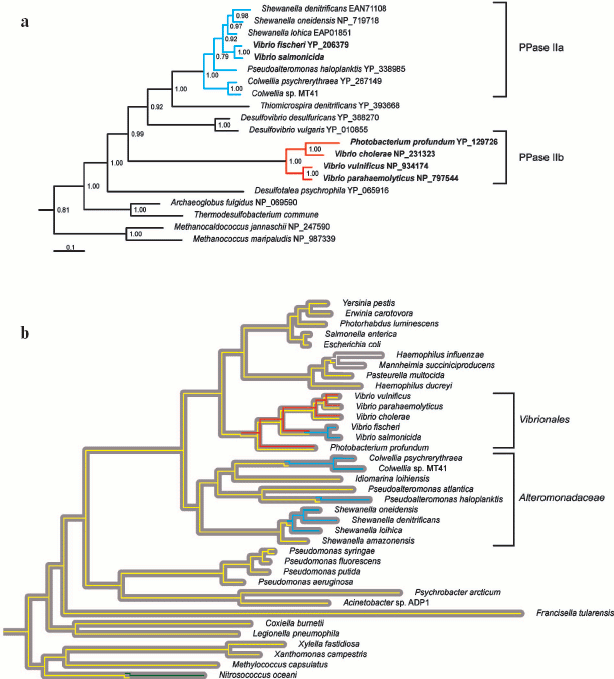
Family II PPase-like proteins have been identified solely in the orders Alteromonadaceae and Vibrionales of gamma-proteobacteria. Molecular phylogenetic analysis supports three distinct lineages among these proteins, further designated PPase IIa, IIb, and IIc. All three lineages possess the active site residues directly involved in ligand binding, including the signature sequences, DHH and SRKKQ [7, 8]. PPase IIa and IIb form two distinct clades that group with the delta-proteobacterial enzymes (Fig. 4a, see color insert), whereas PPase IIc (not shown in Fig. 4) represents an ancient lineage that is not classified with any other known family II PPases. PPase IIb was possibly acquired by lateral gene transfer from delta-proteobacterium to the common ancestor of Vibrionales (Fig. 4b, see color insert), and has coexisted with family I PPase ever since. The transmission history of PPase IIa is less evident. Lateral gene transfer from a delta- or epsilon-proteobacterium to the common ancestor of Vibrionales and Alteromonadaceae would necessitate the coexistence of PPase IIa with family I PPase, since the common ancestor is observed up to the branching points of Shewanella amazonensis, V. fischeri, and V. salmonicida. However, such coexistence is unlikely in view of the finding that in extant genomes, PPase IIa is never present in concurrence with other PPases. Accordingly, we suggest that PPase IIa invaded several Alteromonadaceae and Vibrionales lineages independently (Fig. 4b, see color insert) in the form of a highly contagious vector, e.g., phage or plasmid. This proposal is reinforced by the finding that all PPase IIa-harboring species share marine habitats where gene exchange is fairly common [24, 25]. Finally, PPase IIc is identified in Vibrio parahaemolyticus and Photobacterium profundum as the third PPase-like protein other than family I and PPase IIb enzymes. PPase IIc was possibly acquired by lateral gene transfer, but its rarity and uncertain functional identity preclude a more detailed inference at present.
Overall, our phylogenetic data suggest that family II enzymes are not native to gamma-proteobacteria, but are of benefit to the marine species of this bacterial class. This is illustrated by the highly efficient displacement of family I PPase by PPase IIa in Alteromonadaceae. Interestingly, Vibrionales retained family I PPase after acquisition of PPase IIb. We hypothesize that PPase IIb is partially degenerated shortly after acquisition, and fixed in Vibrionales as an adjunct, rather than a substitute for the family I enzyme. Consistent with this theory, PPase IIa displaced both family I PPase and PPase IIb in V. fischeri and V. salmonicida. Our results collectively suggest that PPase dichotomy in Vibrionales is of functional significance, and not a transient artifact of gene exchange between bacterial species.Fig. 4. Evolution and distribution of PPases in gamma-proteobacteria. a) Bayesian estimate of family II PPase phylogeny [19] was calculated for 85 non-redundant sequences, but only the clade incorporating gamma-proteobacterial enzymes is shown. Monophyletic groups defined as PPase IIa and IIb are depicted in blue and red, respectively. Vibrionales names are indicated in bold. Clade credibility values are shown above the nodes. Protein Genbank accession numbers are specified following the species names. b) Gray ribbons depict the phylogeny of proteobacteria calculated from concatenated alignments of RNA polymerase beta and beta´ subunits. The Bayesian tree [19] was calculated for an alignment of 77 proteobacterial sequences, but only the clade of gamma-proteobacteria is shown. The yellow, blue, and red lines represent the transmission routes for family I PPase, PPase IIa, and PPase IIb. In addition, white and green lines represent the transmission routes for family I PPases that are not native to gamma-proteobacteria. While the grouping of P. profundum and V. cholera family II PPases is inconsistent with the species tree, the topology recommended by the species tree was not rejected by PPase alignment using the Shimodaira and Hasegawa method [22]. Accordingly, we did not introduce a transfer or duplication event in this case. In contrast, tree topologies incorporating monophyly of Vibrionales family II PPases were rejected with the Shimodaira-Hasegawa test.
We thank University of Oslo bioportal for providing CPU time. Preliminary sequence data was obtained from the Institute for Genomic Research website at http://www.tigr.org, the US Department of Energy Joint Genome Institute http://www.jgi.doe.gov, and the Wellcome Trust Sanger Institute http://www.sanger.ac.uk.
This work was supported by the Academy of Finland grant 201611, ISB (the National Graduate School in Informational and Structural Biology), the Ministry of Education and the Academy of Finland, the Russian Foundation for Basic Research grant 06-04-48887, and the Association of Commonwealth Universities.
REFERENCES
1.Kornberg, A. (1962) in Horizons in Biochemistry
(Kasha, H., and Pullman, B., eds.) Academic Press, Inc., New York,
pp. 251-264.
2.Peller, L. (1976) Biochemistry, 15,
141-146.
3.Chen, J., Brevet, A., Fromant, M., Leveque, F.,
Schmitter, J. M., Blanquet, S., and Plateau, P. (1990) J.
Bacteriol., 172, 5686-5689.
4.Ogasawara, N. (2000) Res. Microbiol.,
151, 129-134.
5.Baykov, A. A., Cooperman, B. S., Goldman, A., and
Lahti, R. (1999) Progr. Mol. Subcell. Biol., 23,
127-150.
6.Heikinheimo, P., Tuominen, V., Ahonen, A. K.,
Teplyakov, A., Cooperman, B. S., Baykov, A. A., Lahti, R., and Goldman,
A. (2001) Proc. Natl. Acad. Sci. USA, 98, 3121-3126.
7.Young, T., Kuhn, N., Wadeson, A., Ward, S., Burges,
D., and Cooke, G. (1998) Microbiology, 144,
2563-2571.
8.Shintani, T., Uchiumi, T., Yonezawa, T., Salminen,
A., Baykov, A. A., Lahti, R., and Hachimori, A. (1998) FEBS
Lett., 439, 263-266.
9.Merckel, M. C., Fabrichniy, I. P., Salminen, A.,
Kalkkinen, N., Baykov, A. A., Lahti, R., and Goldman, A. (2001)
Structure (Camb.), 9, 289-297.
10.Ahn, S., Milner, A. J., Futterer, K., Konopka,
M., Ilias, M., Young, T. W., and White, S. A. (2001) J. Mol.
Biol., 313, 797-811.
11.Parfenyev, A. N., Salminen, A., Halonen, P.,
Hachimori, A., Baykov, A. A., and Lahti, R. (2001) J. Biol.
Chem., 276, 24511-24518.
12.Zyryanov, A. B., Vener, A. V., Salminen, A.,
Goldman, A., Lahti, R., and Baykov, A. A. (2004) Biochemistry,
43, 1065-1074.
13.Levine, M. M., Kaper, J. B., Herrington, D.,
Losonsky, G., Morris, J. G., Clements, M. L., Black, R. E., Tall, B.,
and Hall, R. (1988) Infect. Immun., 56, 161-167.
14.Heidelberg, J. F., Eisen, J. A., Nelson, W. C.,
Clayton, R. A., Gwinn, M. L., Dodson, R. J., Haft, D. H., Hickey, E.
K., Peterson, J. D., Umayam, L., Gill, S. R., Nelson, K. E., Read, T.
D., Tettelin, H., Richardson, D., Ermolaeva, M. D., Vamathevan, J.,
Bass, S., Qin, H., Dragoi, I., Sellers, P., McDonald, L., Utterback,
T., Fleishmann, R. D., Nierman, W. C., White, O., Salzberg, S. L.,
Smith, H. O., Colwell, R. R., Mekalanos, J. J., Venter, J. C., and
Fraser, C. M. (2000) Nature, 406, 477-483.
15.Salminen, T., Käpylä, J., Heikinheimo,
P., Kankare, J., Goldman, A., Heinonen, J., Baykov, A. A., Cooperman,
B. S., and Lahti, R. (1995) Biochemistry, 34,
782-791.
16.Baykov, A. A., and Avaeva, S. M. (1981)
Analyt. Biochem., 116, 1-4.
17.Glover, D. J., McEwen, R. K., Thomas, C. R., and
Young, T. W. (1997) Microbiology, 143 (Pt. 9),
3045-3054.
18.Edgar, R. C. (2004) Nucleic Acids Res.,
32, 1792-1797.
19.Ronquist, F., and Huelsenbeck, J. P. (2003)
Bioinformatics, 19, 1572-1574.
20.Whelan, S., and Goldman, N. (2001) Mol. Biol.
Evol., 18, 691-699.
21.Yang, Z. (1994) J. Mol. Evol., 39,
306-314.
22.Shimodaira, H., and Hasegawa, M. (1999) Mol.
Biol. Evol., 16, 1114-1116.
23.Schmidt, H. A., Strimmer, K., Vingron, M., and
von Haeseler, A. (2002) Bioinformatics, 18, 502-504.
24.Jiang, S. C., and Paul, J. H. (1998) Appl.
Environ. Microbiol., 64, 2780-2787.
25.Dahlberg, C., Bergstrom, M., and Hermansson, M.
(1998) Appl. Environ. Microbiol., 64, 2670-2675.