New Viral Vector for Efficient Production of Target Proteins in Plants
T. V. Komarova, M. V. Skulachev, A. S. Zvereva, A. M. Schwartz, Yu. L. Dorokhov, and J. G. Atabekov*
Faculty of Biology and Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, Russia; fax: (495) 938-0601; E-mail: atabekov@genebee.msu.su* To whom correspondence should be addressed.
Received April 13, 2006; Revision received May 5, 2006
A new potato virus X (PVX)-based viral vector for superproduction of target proteins in plants has been constructed. The triple gene block and coat protein gene of PVX were substituted by green fluorescent protein. This reduced viral vector was delivered into plant cells by agroinjection (injection of Agrobacterium tumefaciens cells, carrying viral vector cDNA within T-DNA, into plant leaves), and this approach allowed to dramatically reduce the size of the vector genome. The novel vector can be used for production of different proteins including pharmaceuticals in plants.
KEY WORDS: potato virus X, viral vector, agroinjection, protein superexpressionDOI: 10.1134/S0006297906080049
Abbreviations: CP) coat protein; crTMV) crucifer infecting tobamovirus; GFP) green fluorescent protein; MP) movement protein; PVA) potato virus A; PVX) potato virus X; RdRp) RNA-dependent RNA-polymerase; SGP) subgenomic promoter; sgRNA) subgenomic RNA; TBSV) tomato bushy stunt virus; T-DNA) a fragment of Ti (tumor inducing) plasmid which is transferred into plant cell nucleus; TGB) triple gene block; TMV) tobacco mosaic virus; 3´-UTR) 3´-untranslated region; VIGS) virus-induced gene silencing.
A phytovirus-based vector is a useful tool for efficient expression of
target proteins in plants. It can provide production of target protein
up to 40% of total soluble protein of the cell, which corresponds to
4 g per kg of plant material [1]. Plant
expression systems have a significant advantage compared to other
methods of recombinant protein production since plants are much cheaper
and easier in cultivation than cell cultures (e.g., they do not require
sterile conditions). Plant viral vectors are being successfully applied
for production of pharmaceuticals [2] including
hepatitis B virus and Yersinia pestis antigens [3, 4].
Several approaches to construction of viral vectors have been developed to date. Target protein expression can be provided via duplication of viral subgenomic promoter recognized by viral polymerase. As a result, the vector synthesizes an additional subgenomic RNA (sgRNA) that directs target protein translation. The two most popular plant viral vectors are constructed by this principle--tobacco mosaic virus (TMV)-based 30B [5] and potato virus X (PVX)-based PVX 201 [6]. Both vectors provide efficient expression of target proteins (up to 10% of total soluble protein) and can systemically infect a plant. The disadvantage of this approach is the inevitable presence of direct repeats (sequences of subgenomic promoter) within the vector genome. This leads to relative instability of the vector and provides the possibility of reversion to wild-type virus. Moreover, such vectors produce the target protein together with all viral proteins (some of them are produced with efficiency comparable to the target protein). This can also reduce the effectiveness of the vector.
Plant viral vector can be also constructed by replacement of a viral gene with a target protein gene. Obviously, such a viral vector cannot produce one of its own proteins and, consequently, lacks some features of the original virus. This approach was used to create viral vectors based on tomato bushy stunt virus (TBSV) where the coat protein (CP) gene was partially or completely replaced with a reporter protein gene [7, 8]. Another efficient viral vector pIC4351 [9] was constructed on the basis of crucifer infecting tobamovirus (crTMV) [10] by the replacement of the CP gene with the green fluorescent protein (GFP) gene. Without CP this vector cannot spread systemically, but this weakness can be successfully compensated if the vector genome is delivered into a plant by the agroinfiltration technique. It implies cloning of the vector cDNA fused to plant promoter into binary vector--a plasmid that can be replicated in both E. coli and Agrobacterium tumefaciens and contains a fragment of T-DNA transferable into the plant cell nucleus. When a plant cell is infected with an agrobacterium strain carrying such a plasmid, the vector's cDNA is transferred into the cell nucleus within the T-DNA. Then the genomic RNA of the vector is transcribed and the replication starts.
An important advantage of agroinjection is high efficiency of target DNA transfection--almost 100% of cells become infected when agrobacterium suspension is injected into a plant leaf [11]. Agroinfiltration allows infection of the whole plant by immersing it into an agrobacterium suspension, and the bacteria penetrate into plant tissues by vacuum infiltration. Thus, application of these techniques makes virus cell-to-cell and systemic transport dispensable. Therefore, viral genes responsible for these functions can be removed from the viral vector genome.
It must be noted that viral vector replication elicits a special protective mechanism of the plant cell--posttranscriptional virus-induced gene silencing (VIGS). It leads to inhibition of viral vector infection and reduction of its productivity. VIGS can be neutralized with the help of protein suppressors, for example TBSV p19 [12] or potato virus A HcPro [13]. Simultaneous expression of a viral vector and VIGS-suppressing protein can be accomplished by co-injection of corresponding agrobacterium strains into a plant leaf. Efficiency of the vector replication and production of target protein can be significantly increased in this way.
PVX is a type member of the Potexvirus genus. Its genomic RNA encodes RNA-dependent RNA polymerase and CP genes (5´- and 3´-proximal genes, correspondingly). They flank the so-called triple gene block (TGB) encoding three genes (TGB1, TGB2, and TGB3) responsible for viral transport. PVX CP is also required for virus cell-to-cell movement. By deletion of the TGB and CP gene, we have constructed a viral vector that is completely incapable of the transport of infection. The vector's genome was reduced more than 2-fold and considerable increase in target protein production level was achieved when the vector was delivered into plant cells by agroinjection (together with the VIGS-suppressor gene).
MATERIALS AND METHODS
PVXdt vector construction. Several intermediate constructs (IC) were made to obtain plasmid containing viral vector PVXdt-GFP.
IC 1. Fragment from the plasmid PVX-201 (nt 3452-4949 of the viral genome) was inserted into SK+ vector (Stratagene, USA) via sites HindIII-ApaI. This construct was used as a matrix for PCR with primers “25KXba(m)” (CTCTTCTAGACTTATTCAAATC TCTAAGGTAAC) and “T3” (Promega, USA) for obtaining the first fragment for IC2.
IC 2. The first fragment (nt 3452-4485 of the viral genome) for this construct included part of the PVX polymerase gene and a region of subgenomic promoter, the second fragment was the GFP gene flanked with XbaI and SalI sites of restriction on 5´- and 3´-ends, correspondingly. The third fragment held the 3´-end of the CP gene and 3´-UTRPVX. All these fragments were cloned into pGEM3Z vector (Promega) via HindIII and SacI sites.
PVX-BIN19 plasmid construction. A fragment, which contained 35S promoter, PVX replicon with duplicated subgenomic promoter, and nos-terminator of transcription, was transferred from the plasmid PVX-201 into binary vector BIN19 between HindIII and EcoRI sites.
PVXdt-GFP plasmid construction. A fragment from IC2 was cloned into PVX-BIN19 construct using AvrII and SacI sites.
PVXdt-GFP (POL-mut) construct was obtained via endonucleolytic cleavage of 35S-PVXdt-GFP plasmid by restrictase AvrII, blunting by Klenow fragment, and self-ligation.
All stages were performed using Fermentas (Lithuania) enzymes.
Agroinjection. A 500-µl sample of overnight Agrobacterium tumefaciens (GV-3101 strain) culture was sedimented at 2300g for 5 min, and the pellet was resuspended in solution containing 10 mM MES (pH 5.5) and 10 mM MgCl2 to final OD600 0.2 (if the mixture of different strains was used, then final OD600 of each was 0.2). Leaves of greenhouse grown N. benthamiana plants were injected with this suspension using a syringe without needle.
Biolistic plant transfection. Particle bombardment was performed with a high-pressure helium-based device for biolistic transfection PDS-1000/He (BioRad, USA). For each shot, DNA (~5 µg of plasmid DNA) was precipitated on gold particles with spermidine (0.1 M) and calcium chloride (2.5 M) solutions according to the manufacturer's protocol. DNA-coated particles were resuspended in cold absolute ethanol and placed immediately on plastic flying disks. Bombardment was performed at a distance of about 4 cm in a vacuum chamber at helium pressure 900 psi.
GFP was visualized under UV illumination, lambda = 366 nm. Fluorescent microscopy was performed using a Zeiss Axiovert microscope.
Protein extraction from agroinjected spots and SDS-PAGE analysis. On the third day after infection, injected spots (~10 mg) were ground in the presence of celite and extraction buffer (40 µl) (60% glycerol, 5 mM beta-mercaptoethanol, 10% SDS, 250 mM Tris-HCl, pH 6.8) for total protein extraction, and in case of soluble protein extraction GFP-buffer (250 µl) (50 mM NaH2PO4, 10 mM Tris-HCl, pH 8.0, 200 mM NaCl) was used. Protein extracts were fractionated using SDS-PAGE followed by Coomassie staining.
Total RNA extraction and northern-blot analysis. Total RNA was extracted according to [14] from leaves on the third day after injection, fractionated in 1.5% agarose gel under denaturing conditions (10% formaldehyde), and transferred to a Hybond-XL nylon membrane (Amersham, UK). Membranes were probed with a denatured single-stranded anti-GFP DNA fragment labeled with [alpha-32P]dATP and then washed and exposed with X-ray film (Retina, Germany).
RESULTS
Construction of the optimized viral vector on the basis of PVX genome. A “minivariant” of the PVX201-GFP vector was constructed on the basis of the PVX genome--PVXdt-GFP (Fig. 1). PVXdt-GFP genomic RNA cDNA was cloned under control of the 35S promoter. The RNA is structurally bicistronic and contains PVX 5´- and 3´-untranslated regions (UTRs), RNA-dependent RNA-polymerase (RdRp) gene, first subgenomic promoter (SGP), and GFP gene. The whole construction cloned into binary vector pBIN19 [15] and can be transferred into plant cells by agrobacteria. It is noteworthy that the described viral vector contained 60 nt long 3´-terminal part of the CP PVX gene between the GFP gene and 3´-UTR. Deletion of this 60 nt region drastically decreased the efficiency of vector accumulation (data not shown).
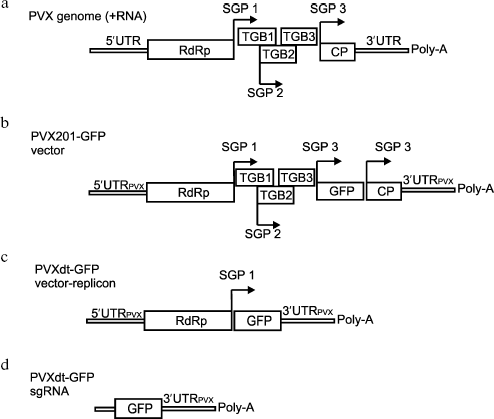
Agroinjection of the described construction into N. benthamiana leaves led to accumulation of substantial amounts of GFP. According to fluorescent microscopy (data not shown), GFP was produced in almost all cells in the agroinjected area of the leaf.Fig. 1. a) Schematic representation of PVX and PVX-based vectors. RdRp - gene of viral RNA-dependent RNA polymerase; TGB1, TGB2, TGB3 - triple gene block; CP - coat protein. Subgenomic promoters are indicated by arrows (SGP). b, c) Schematic representation of PVX-201 GFP (b) and PVXdt-GFP (c) vectors. The constructs are based on PVX cDNA fused to cauliflower mosaic virus 35S promoter and cloned into binary vector to be delivered into plant cells by agrobacteria. d) Schematic representation of subgenomic RNA produced by PVXdt-GFP.
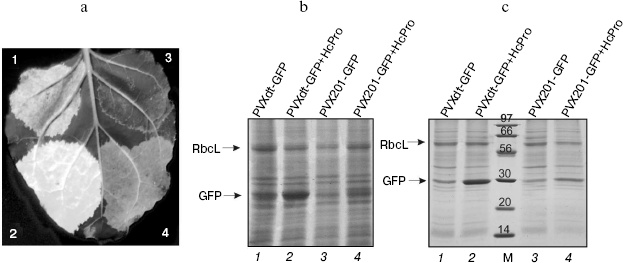
GFP superproduction could be detected on the third day after injection under UV illumination (Fig. 2a) and also on the Coomassie stained SDS-PAGE gels (total protein, Fig. 2b; soluble protein, Fig. 2c). Efficiency of GFP production by the PVXdt-GFP vector can be estimated by comparison with accumulation of RbcL (large subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase, Rubisco; this protein comprises 30-50% of total soluble protein [16]).
The efficiency of target protein (GFP) accumulation by PVXdt-GFP vector (lanes 1 and 2, Figs. 2b and 2c) was much higher than the one by PVX201-GFP (lanes 3 and 4, Figs. 2b and 2c). This difference is especially noticeable in case of co-agroinjection of viral vectors with the plasmid expressing the gene of the VIGS suppressor (HcPro PVA). In this case, efficiency of GFP production by PVXdt-GFP was approximately 2.5-fold higher than by PVX201-GFP.Fig. 2. GFP expression in N. benthamiana leaves agroinjected with PVXdt-GFP and PVX201-GFP vectors (3dpi). a) Photography of UV illuminated areas agroinjected with PVX201-GFP (sector 3) and PVXdt-GFP (sector 1) or co-agroinjected with HcPro gene-silencing suppressor and PVXdt-GFP (sector 2) or PVX201-GFP (sector 4). b) Coomassie stained SDS-PAGE gel loaded with total protein extracted from agroinjected areas. c) Coomassie stained SDS-PAGE gel loaded with soluble protein extracted from agroinjected areas. M, protein molecular weight markers (molecular weights are indicated in kD). Arrows indicate positions of RbcL and GFP bands.
PVXdt-GFP is a viral vector-replicon. In the above experiment, PVXdt-GFP vector expression was detected by the target protein (GFP) accumulation. High efficiency of its production and the viral vector structure imply that the target protein synthesis must be a result of genomic RNA replication and subsequent GFP sgRNA synthesis. In theory this process includes the following steps: 1) the viral vector cDNA containing binary plasmid is replicated in A. tumefaciens; 2) the T-DNA containing this cDNA is transferred into the plant cell during agrobacterium infection (agroinjection); 3) 35S promoter directs bicistronic genomic RNA transcription on the matrix of cDNA, resulting RNA is exported into cytoplasm; 4) genomic RNA translation results in replicase synthesis; 5) virus-vector RNA is replicated, (-) and (+) RNAs are synthesized; 6) GFP sgRNA synthesis and its subsequent translation resulting in GFP production.
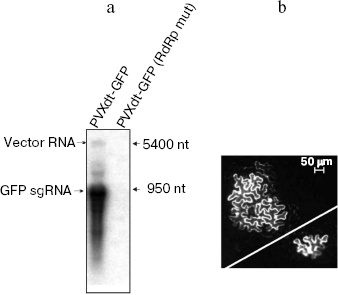
Previous studies and the current conception of the mechanism of agro-transformation imply that stages 1-4 really occur during agroinjection [17]. In our study, we confirmed that the other steps occur during the process. Indeed, mRNA corresponding to GFP sgRNA was detected by northern-blot analysis of total RNA extracted from leaves agroinjected with PVXdt-GFP vector (Fig. 3a). Minor amounts of the vector genomic RNA were also detected. Mutation of the replicase gene led to abolishing of both genomic and subgenomic RNAs. This result confirms that the replicase is essential for synthesis of both RNAs of the vector.
In another experiment, cDNA of PVXdt-GFP was delivered into plant cells by particle biolistic bombardment (see “Materials and Methods”). This method allows transfection of a small number of cells in a plant leaf. Therefore a viral vector deficient in cell-to-cell transport can be only expressed in separate cells. Indeed, such cells were observed by fluorescence microscopy in plant leaves bombarded with PVXdt-GFP cDNA (Fig. 3b, bottom, right).Fig. 3. PVXdt-GFP vector is a self-amplifying system. a) Northern-blot analysis of RNA extracted from N. benthamiana leaves agroinjected with PVXdt-GFP and analogous vector with mutated replicase (RdRp mut); 32P-labeled anti-GFP probe was used, positions of 5400- and 950-nt RNA molecular weight markers are indicated. b) Complementation of cell-to-cell movement by biolistic co-transfection of PVXdt-GFP and MP TMV expressing plasmid. GFP fluorescence of loci (in case of co-transfection) and a separate cell (in case of PVXdt-GFP only transfection) in UV light.
Biolistic particle bombardment is widely used in plant virology to study cell-to-cell movement of plant viruses [18]. For example, it was shown that TMV 30K movement protein (MP TMV) provides cell-to-cell spread of transport-deficient PVX mutant. This was demonstrated using co-bombardment of PVX-based vector together with a plasmid construct expressing the MP TMV gene (35S MP TMV [19]).
In our study, we performed an analogous experiment of particle bombardment of N. benthamiana leaves with a mixture of PVXdt-GFP cDNA and 35S-MP TMV construct. As a result, groups (loci) of GFP-expressing cells were observed under UV light in the fluorescent microscope (Fig. 3b, top, left). It is noteworthy that each locus contained several cells efficiently expressing GFP. In control experiments, co-transfection of GFP-expressing construct (expression was directed by 35S promoter) and 35S-MP TMV did not result in formation of GFP-expressing loci (data not shown). Thus it can be concluded that co-transfection of PVXdt-GFP and 35S-MP TMV led to transportation of the vector genomic RNA from the originally transfected cell to adjacent cells where GFP could be expressed only as a result of PVXdt-GFP replication and the subgenomic RNA synthesis.
In general, our study demonstrates that PVXdt vector is able to replicate in plant cells, and it provides a high level of target protein expression in the majority of plant leaf cells when delivered by agroinjection or agroinfiltration (in this application cell-to-cell movement is not required). Approximate estimations show that the price of production and purification of recombinant protein in plants is about 50-100 $US per gram. This is 20-40 times less than the corresponding price for the same quality protein produced in cell cultures [20].
Thus the developed vector could be used in biotechnology for fast and efficient production of protein pharmaceuticals. It is also a good model system for molecular biology and molecular virology studies.
The authors would like to thank Dr. E. V. Skurat for construct PVX 201-GFP and for valuable discussion.
This work was supported by the Russian Foundation for Basic Research (grant Nos. 04-04-48490, 05-04-48674, and 05-04-08002), Federal Agency for Science and Innovations grant (contract numbers 02.435.11.3012 and 03-04-52008b), and the President of the Russian Federation grant for young PhD (No. MK-1115.2005.4).
REFERENCES
1.Gleba, Y., Klimyuk, V., and Marillonnet, S.
(2005) Vaccine, 23, 2042-2048.
2.Caòizares, M. C., Nicholson, L., and
Lomonossoff, G. P. (2005) Immunol. Cell Biol., 83,
263-270.
3.Mechtcheriakova, I. A., Eldarov, M. A., Nicholson,
L., Shanks, M., Skryabin, K. G., and Lomonossoff, G. P. (2006)
J. Virol. Meth., 131, 10-15.
4.Santi, L., Giritch, A., Roy, C. J., Marillonnet,
S., Klimyuk, V., Gleba, Y., Webb, R., Arntzen, C. J., and Mason,
H. S. (2006) Proc. Natl. Acad. Sci. USA, 103,
861-866.
5.Shivprasad, S., Pogue, G. P., Lewandowski, D. J.,
Hidalgo, J., Donson, J., Grill, L. K., and Dawson, W. O. (1999)
Virology, 255, 312-323.
6.Chapman, S., Kavanagh, T., and Baulcombe, D. (1992)
Plant J., 2, 549-557.
7.Scholthof, H. B., Morris, T. J., and Jackson, A. O.
(1993) Mol. Plant Microbe Interact., 6, 309-322.
8.Scholthof, H. B. (1999) J. Virol.,
73, 7823-7829.
9.Marillonnet, S., Giritch, A., Gils, M.,
Kandzia, R., Klimyuk, V., and Gleba, Y. (2004) Proc. Natl.
Acad. Sci. USA, 101, 6852-6857.
10.Dorokhov, Yu. L., Ivanov, P. A., Novikov, V. K.,
Agranovsky, A. A., Morozov, S. Yu., Efimov, V. A., Casper, R., and
Atabekov, J. G. (1994) FEBS Lett., 350, 5-8.
11.Marillonnet, S., Thoeringer, C., Kandzia, R.,
Klimyuk, V., and Gleba, Y. (2005) Nat. Biotechnol., 23,
718-723.
12.Voinnet, O., Pinto, Y. M., and Baulcombe, D. C.
(1999) Proc. Natl. Acad. Sci. USA, 96, 14147-14152.
13.Savenkov, E. I., and Valkonen, J. P. T. (2002)
J. Gen. Virol., 83, 2325-2335.
14.Napoli, C., Lemieux, C., and Jorgensen, R. (1990)
Plant Cell, 2, 279-289.
15.Bevan, M. (1984) Nucleic Acids Res.,
12, 8711-8721.
16.Parry, M. A., Andralojc, P. J., Mitchell, R. A.,
Madgwick, P. J., and Keys, A. J. (2003) J. Exp. Bot., 54,
1321-1333.
17.Tzfira, T., and Citovsky, V. (2003) Plant
Physiol., 133, 943-947.
18.Waigmann, E., Ueki, S., Trutnyeva, K., and
Citovsky, V. (2004) Crit. Rev. Plant Sci., 23,
195-250.
19.Morozov, S. Yu., Fedorkin, O. N., Jüttner,
G., Schiemann, J., Baulcombe, D. C., and Atabekov, J. G. (1997) J.
Gen. Virol., 78, 2077-2083.
20.Giddings, G., Allison, G., Brooks, D., and
Carter, A. (2000) Nat. Biotechnol., 18, 1151-1155.