Nickase and a Protein Encoded by an Open Reading Frame Downstream from the Nickase BspD6I Gene Form a Restriction Endonuclease Complex
A. K. Yunusova1, E. A. Rogulin1, R. I. Artyukh1, L. A. Zheleznaya1, and N. I. Matvienko2*
1Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: (4967) 790-5532Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: (495) 924-0493; E-mail: nikmatv@vega.protres.ru
* To whom correspondence should be addressed.
Received January 30, 2006; Revision received March 29, 2006
We are the first to have isolated a protein (186 amino acid residues) encoded by the open reading frame adjacent to the end of the BspD6I nickase (N.BspD6I) gene. Cleavage of both DNA strands near the sequence recognized by nickase (5´-GAGTC/5´-GACTC) occurs when this protein is added to the reaction mixture containing N.BspD6I. The protein encoded by the open reading frame and the nickase are suggested to be subunits of heterodimeric restriction endonuclease R.BspD6I.
KEY WORDS: restriction-modification system, restriction endonuclease, site-specific DNA nickase, heterodimeric restriction endonucleaseDOI: 10.1134/S0006297906070157
Abbreviations: bp) base pairs; dNTP) deoxynucleoside triphosphates; DTT) dithiothreitol; IPTG) isopropyl-beta-D-thiogalactoside; N) nickase; ORF) open reading frame; PCR) polymerase chain reaction; RE, R) restriction endonuclease.
Type II restriction endonucleases (REs) comprise one of the largest
enzyme classes (more than 3500 enzymes). These enzymes recognize a
short (4-8 bp) specific sequence in double-stranded DNA and cleave both
DNA stands inside or near the recognition sequence using
Mg2+ as a cofactor. However, in spite of common
functionality, different REs use various mechanisms for cleavage of
both DNA strands depending on the character of the recognition sequence
and number of their catalytic centers (one or two). So, cleavage of
palindromic sequence (top and bottom strands of which are identical) is
provided by dimerization of identical monomers, each containing a
single catalytic center. X-Ray structural analysis shows that the
dimers not only interact with DNA, but also form multiple contacts with
each other [1]. Formation of dimer-dimer contacts
provides additional control for recognition of the predetermined
sequence, because recognition of “wrong” sequence is lethal
for the cell. It is probably formation of these contacts that activates
the catalytic centers of monomers, because substitution of an amino
acid residue that is involved in specific recognition of the base in
only one subunit influences cleavage of both strands [2]. When REs recognize asymmetric sequence (and, as a
rule, split DNA beyond the recognized sequence), cleavage of both DNA
strands occurs in different ways. Some REs require interaction with two
copies of recognition sequences. The restriction endonuclease
FokI is the best studied one [3, 4] for which the cleavage of both DNA strands follows
dimerization of monomers bound with two copies of recognition
sequences, and the catalytic domains of the monomers are involved in
dimerization. Another type of two-strand DNA cleavage is realized by
heterodimeric REs [5, 6]. These
endonucleases are composed of two different subunits, each containing
one catalytic center and splitting one DNA strand, but the strand
cleavage requires heterodimer formation. Finally, some REs contain two
catalytic centers in one monomeric molecule [7, 8].
Heiter and coauthors [6] supposed the existence of yet another type of heterodimeric REs dissociating in the process of their isolation. Nickases (endonucleases cleaving only one strand) isolated from natural sources are supposed to be just subunits of these REs. In fact, an open reading frame (ORF) is located downstream of the nickases' genes encoding a protein displaying high homology with the nickase catalytic domain.
In the present work we aimed to elucidate whether the protein encoded by the ORF adjacent to the BspD6I nickase gene is just the subunit forming a heterodimeric RE together with nickase and dissociating from the protein-protein complex during its isolation.
MATERIALS AND METHODS
Escherichia coli BL21(DE3) strain and expression vector pET28c with T7 phage RNA-polymerase promoter (Novagen, USA) were used in the study. Plasmids pBR322 and pHC624 [9], M13mp19 phage DNA, restriction endonucleases Bli736I (BsaI isoschizomer), RspLKI (SphI isoschizomer), BspLU4I (AvaI isoschizomer), E. coli DNA polymerase I Klenow fragment, phage T4 DNA-ligase, and phage T4 polynucleotide kinase were isolated in our laboratory. Restriction endonucleases XhoI, NcoI, and HinfI are commercial products of MBI Fermentas (Lithuania). N.BsmI enzyme is the commercial product of New England Biolabs (USA). DNA-polymerase Pwo was kindly provided by Dr. I. E. Granovsky (Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences). Oligonucleotides were synthesized by Syntol (Russia).
Preparation of the strain producing the ORF product. The ORF sequence was synthesized via polymerase chain reaction (PCR) using the following primers to the termini of the sequence:
forward - GCAGGTCTCACATGCAAGATATTCTGGACT,
reverse - GTCCTCGAGTTTTAAATTGTACTCAATGAAAGTT
(sequences typed in bold letters comprise the recognition sites of Bli736I and XhoI, respectively). Chromosomal DNA of Bacillus species D6 cells was used as a template. The purified PCR product was treated with the restriction endonucleases Bli736I and XhoI and ligated with pET28c plasmid cleaved with NcoI and XhoI. Escherichia coli BL21(DE3)/pMSsc cells were transformed with the recombinant plasmid pET28/ORF. The recombinant plasmid was isolated from the transformed cells, and unchanged nucleotide sequence of the ORF after the PCR process was confirmed by sequencing.
Isolation of the ORF product. Escherichia coli BL21(DE3)/pMSsc, pET28/ORF cells expressing the ORF product were grown to the middle of log phase at 37°C, and the recombinant protein synthesis was induced by addition of isopropyl-beta-D-thiogalactoside (IPTG). The duration of the induction was 3 h at 37°C. The cells were precipitated by centrifugation. The pellet was suspended in buffer A (20 mM Tris-HCl, pH 8.0, 300 mM NaCl), and subsequent manipulations were carried out at 4°C. The suspended cells were sonicated, and the lysate was cleared by centrifugation. Imidazole was added to the supernatant to 10 mM concentration and the solution was applied to a column with Ni-NTA agarose (Qiagen, USA) equilibrated with buffer B (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10 mM imidazole). Then the column was washed with buffer B. The protein was eluted with stepwise concentration gradient of imidazole (30, 50, 100, and 250 mM) in buffer A. Electrophoretic analysis of the resulting fractions in 13% polyacrylamide gel with SDS demonstrated that the ORF product eluted at 100 mM imidazole concentration. The fractions containing the ORF product were combined, and imidazole was depleted by dialysis against buffer containing 10 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, and 1 mM dithiothreitol (DTT). The imidazole-free protein was transferred to the storage buffer (10 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, 1 mM DTT, 50% glycerol) by dialysis and stored at -20°C. This protein preparation (with the concentration of 0.2 mg/ml) was used in further work.
Isolation of BspD6I nickase. The nickase was isolated from the E. coli strain-producer of recombinant nickase obtained earlier [10]. The purification was carried out on a column with Ni-NTA agarose, and further manipulations were carried out according to the same scheme as for the ORF product.
Analysis of endonuclease activity. Nickase at the concentration of 2 mg/ml and activity of 400 U/µl was used in our work. The reaction mixture (40 µl) contained the buffer for nickase (10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 150 mM KCl, 1 mM DTT), 1 µg of pHC624 plasmid, 1 µl nickase (at various concentrations), and 1 µl of the ORF product (also at various concentrations). The reaction mixture was incubated for 1 h at 55°C. The reaction was stopped by addition of EDTA to final concentration of 20 mM. Optimum conditions for the activity of the complex formed by nickase and the ORF product were determined at varying pH from 7 to 10 and KCl concentrations from 50 to 300 mM.
Localization of phosphodiester bonds cleaved by the mixture of nickase and the ORF product. The position of the hydrolyzed phosphodiester bonds in DNA strands was determined by the elongated primer method [11]. DNA of M13mp19 phage was used as a template for the synthesis of double-stranded DNA radioactive labeled in one strand, polylinker of which contained a sequence recognized by the nickase. 5´-Labeled oligonucleotide complementary to the sequence of 20 bp distant from the end of polylinker was used as a primer for the synthesis. The sequencing reaction was carried out according to the “fmolTM DNA Sequence System” protocol (Promega, USA). The products of the sequencing and cleavage of the labeled DNA were analyzed in 6% polyacrylamide gel in the presence of 7 M urea on a Macrophor device (LKB, Sweden).
RESULTS AND DISCUSSION
Earlier we isolated a Bacillus species D6 endonuclease, which has been identified as the site-specific nickase BspD6I [12]. N.BspD6I is an isoschizomer of BstSEI [13] and BstNBI [14] nickases. All three nickases recognize the same sequence (5´-GAGTC/5´-GACTC) and cleave only one DNA strand containing the sequence GAGTC at the distance of four nucleotides from the recognition site.
The similarity of nickases and restriction endonucleases regarding their properties such as high specificity of the recognition sequence, accuracy of cleavage of this sequence, and sensitivity to methylation formed a basis for the supposition that nickases represent restriction endonucleases that have lost their ability for dimerization [15, 16].
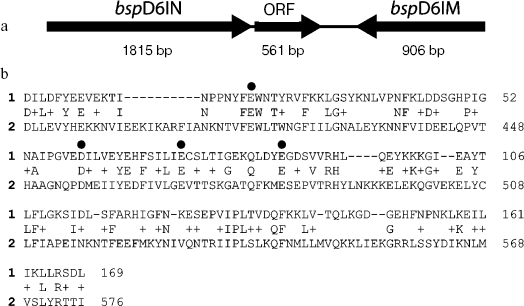
Apart from the fact that all known natural nickases are able to recognize the same sequence and cleave DNA identically, their gene complexes possess not only identical organization, but also totally coincident nucleotide sequences [15-17]. The gene complexes consist of the nickase gene, DNA-methyltransferase gene, and an ORF encoding a protein of 186 amino acid residues with earlier unknown function (Fig. 1a). The amino acid sequence of this protein is highly homologous to the C-terminus of nickase: 27% of the residues are identical and 20% are homologous (Fig. 1b). Moreover, the residues comprising the nickase catalytic center are coincident.
The ORF was amplified by PCR. The forward primer contained the recognition site for IIS Bli736I restriction endonuclease, and the reverse primer contained the XhoI recognition site. The position of the Bli736I recognition site in the primer enables formation of sticky end after cleavage of PCR product; this sticky end is identical to that formed by NcoI, and the second codon of the gene can remain unchanged. The resulting fragment was cleaved with the restriction endonucleases Bli736I and XhoI and ligated with pET28c vector cleaved with the restriction endonucleases NcoI and XhoI. Ligation of the PCR fragment and the vector was planned so that the result of expression was a protein carrying six histidines on the C-terminus. The E. coli BL21(DE3) strain was chosen as the recipient strain. Since a possibility could not be excluded that the ORF-encoded protein possesses a low nickase activity, pRARE/MSsc plasmid [10] was introduced into the cells in anticipation. The use of this plasmid constructed earlier in our work on the basis of the pRARE plasmid (Novagen, USA) is due to two reasons. The first, it carries the gene encoding M.SscL1I methyltransferase, which methylates adenine at the recognition site for nickase and protects DNA from hydrolysis by nickase [18]. Second, it is bearing the genes encoding rare for E. coli tRNAs, which is important because the gene encoding the small subunit contains many (22) codons rare for E. coli.Fig. 1. a) Organization of the gene complex containing the BspD6I nickase gene. Sizes of each gene (bp) are given under arrows. b) Alignment of amino acid sequences for the protein encoded by the ORF (1) and BspD6I nickase (2). Letters between the lines indicate identical residues, and the sign (+) indicates homological residues. Points indicate amino acid residues whose substitution for alanine results in the lost or substantial decrease in BstNBI nickase activity, but does not influence the DNA binding [15].
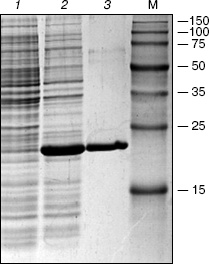
The protein yield from the recombinant strain after the induction by IPTG comprised about 40% of total cell protein (Fig. 2, lane 2). After purification on the column with Ni-NTA agarose a virtually homogeneous protein was eluted (Fig. 2, lane 3) with molecular mass close to the expected value (20 kD).
The pHC624 plasmid containing two recognition sites 5´-GASTC-3´ (S = G or C) separated by a distance of ~500 bp was used for the analysis of the obtained protein. Figure 3 demonstrates the fact that no alteration in plasmid DNA occurs when the ORF product alone is added to the reaction mixture (lane 2). Monomeric and dimeric isoforms of plasmid DNA convert into open circles, when nickase alone is added (lane 3), which is evidence for single strand cleavage made by the nickase. Two fragments, 500 and 1500 bp, are formed upon the addition of the nickase combined with the ORF product. The total length of the formed fragments corresponds to the plasmid size (2015 bp). As a control, the plasmid was cleaved by HinfI restriction endonuclease (lane 5) recognizing double-stranded sequence 5´-GANTC-3´ (N = A, G, C, T). The plasmid pHC624 bears only two GANTC sequences. They both have G/C-pair in the degenerate position; therefore, the plasmid sequences recognizable by the nickase and by HinfI are identical. The length of fragments formed under HinfI action are identical to those of fragments formed under the action of the nickase and the ORF product mixture.Fig. 2. Results of analysis of the ORF product by electrophoresis in 13% SDS-polyacrylamide gel: 1) cell lysate of E. coli strain-producer of the ORF protein before induction with IPTG; 2) lysate of the same cells after the induction with IPTG; 3) protein eluted with 100 mM imidazole. M, protein molecular mass marker; molecular masses (kD) are given on the right.
The following control experiment was carried out to prove the fact that just the complex of nickase with ORF product but not an admixture of nucleases cleaving DNA at the site of initial single-stranded cleavage produced by nickase participates in cleavage of the strand with GAGTC sequence (Fig. 3b). This experiment checked the ability of ORF product to cleave the second DNA strand at the single-stranded break-off produced by another nickase, N.BsmI, which recognizes the DNA site GAATGC.Fig. 3. Analysis of products of pHC624 (a) and pBR322 (b) plasmid hydrolysis by various enzymes (electrophoresis in 1% agarose gel). a) pHC624 plasmid (1 µg) was incubated: 1) without addition of proteins; 2) with 0.5 µg ORF protein; 3) with 0.05 µg nickase; 4) with 0.05 µg nickase and 0.015 µg ORF protein; 5) with 5 U of HinfI. b) pBR322 plasmid (1 µg) incubated (1) in buffer for N.BsmI at 65°C for 1 h; 2-7) products of plasmid hydrolysis after treatment with nickase, endonuclease, and 0.015 µg of ORF protein; 2) N.BsmI; 3) N.BsmI + ORF product; 4) RspLAI + BspLU4I; 5) RspLAI + BspLU4I + ORF product; 6) N.BsmI + RspLAI + BspLU4I; 7) N.BsmI + RspLAI + BspLU4I + ORF product. M, marker for DNA fragment length (numbers on the right give fragment lengths in bp). Plasmid isoforms are given on the left (S1, monomeric superhelix; S2, dimeric superhelix; O1, open monomeric ring; O2, open dimeric ring).
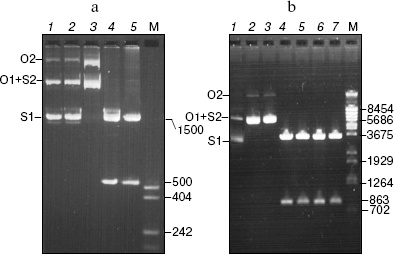
The pBR322 plasmid has one recognition site for N.BsmI nickase at base 1353. Monomeric and dimeric superhelical plasmid forms are converted into open rings under the treatment with N.BsmI nickase (lane 2). No linear plasmid forms are generated under subsequent treatment of the obtained specimen with the ORF product. Thus, the circle DNA molecules with single-stranded break-off generated by nickase different from N.BspD6I are not cleaved to linear molecules.
To prove the fact that linear DNA fragments with single-stranded break-offs are not subjected to further cleavage, the initial plasmid and plasmid treated with N.BsmI were subjected to cleavage with the RspLAI and BspLU4I restriction endonucleases with coordinates of sites 562 and 1425, respectively (lanes 3 and 5). The cleaved plasmids were incubated with the ORF product (lanes 5 and 7). The small fragment contains a single-stranded break in the plasmid treated with N.BsmI. If any admixture of unspecific endonucleases capable of cleaving DNA at single-stranded break would be present in the ORF specimen, the small fragment would be cleaved, and this could be seen under separation of the hydrolysis products in the gel. Since no such effect is observed, one can conclude that DNA fragments with single-stranded break-off made by N.BsmI nickase are not subjected to further hydrolysis.
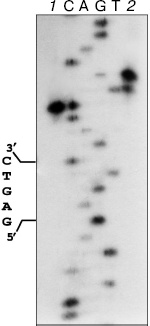
The formation of two fragments of pHC624 under the incubation with nickase and the ORF product is evidence for cleavage of two DNA strands. The hydrolysis of phosphodiester bounds in the strand containing GAGTC sequence occurs four nucleotides apart from the recognizable sequence (Fig. 4, lane 1), and the complementary strand is cleaved six nucleotides apart from the site with some wobble: approximately 10% of molecules are cleaved five nucleotides apart (lane 2).
Since the isolated nickase cleaves only one strand of DNA, it is reasonable to suppose the participation of the ORF product in the cleavage of the second strand. Nativity of the pHC624 plasmid under the incubation with excess of the ORF product is evidence for the absence of endonuclease activity in the ORF product specimen (in the ORF product itself and in possible admixtures as well). One could anticipate the ORF product would play a role of effector protein, which via binding with nickase could favor the activation of an additional catalytic site in the nickase. However, this explanation must be excluded: we have determined the 3D structure of the nickase at the resolution of 1.8 Å (accession code 2EWF in the Protein Data Bank; the work is in preparation for publication), in which only one single catalytic site is present.Fig. 4. Localization of phosphodiester bonds hydrolyzed by the mixture of nickase and ORF fragment. C, A, G, and T are the sequencing reaction products of M13mp19 phage DNA. 1) M13mp19 labeled DNA incubated with the mixture of N.BspD6I and ORF product; 2) M13mp19 labeled DNA, incubated with the mixture of N.BspD6I and ORF product, followed by the inactivation of enzymes (heating at 80°C for 20 min) and addition of Klenow fragment and dNTP mixture. Electrophoresis in 6% polyacrylamide gel in the presence of 7 M urea at 50°C.
As we have noted above, the amino acid sequence of the ORF exhibits high homology with the C-domain (catalytic) of nickase. Moreover, it comprises all amino acid residues forming the active center in the nickase. Thus, the ORF product must be considered as small subunit of a new heterodimeric restriction endonuclease, R.BspD6I, the large subunit of which is nickase.
The following data are evidence for the existence of such heterodimeric RE in Bacillus species D6 strain. A separate single RE peak, which we termed BspD6III, was eluted apart from the nickase from the first column at the initial steps of the nickase isolation directly from Bacillus species D6 strain [12]. This RE cleaved substrate DNA with the formation of the same set of fragments as PleI did (unpublished data). However, our attempts to purify further this RE led to the elution of nickase instead of PleI isoschizomer from the column. In the light of data obtained in the present work, the phenomenon of PleI isoschizomer transformation into nickase is easily explainable by the lost of one (small) subunit during purification.
As evident from the data obtained in this work, the small subunit being beyond the complex with nickase cannot make a single-strand cleavage into DNA. This is apparently due to its structure being devoid of a domain recognizing specific DNA sequence, and its interaction with DNA is realized apparently via interaction with nickase. This interaction is probably rather weak. The formed protein-protein complex is unstable, and its dissociation occurs as a result in the course of purification.
Optimum conditions for the activity of the nickase and subunit are identical to those for nickase alone (10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 150 mM KCl, 1 mM DTT), 55°C.
Restriction endonucleases PleI and MlyI recognize the same DNA sequence as R.BspD6I does, but cleave it at positions 4/5 and 5/5, respectively. Thus, R.BspD6I is a neoschizomer for these enzymes.
The genes encoding the nickase and the small subunit are apparently translated commonly and form a single operon, because the terminating codon of the nickase gene is separated from initiating codon of the small subunit by only one nucleotide. The transcription terminator, apparently common for these genes, the palindrome GGAACAATGCCAGTTCATTGTTCC surrounded on each side by AT-enriched sites, is located just downstream from the small subunit gene. This palindrome, as noted Dr. Gololobova and coworkers [16], can serve as a transcription terminator for DNA-methyltransferase gene. This tandem location of nickase and small subunit genes in a single operon presumably provides the synthesis of proteins encoded by these genes in stoichiometrically equal amounts.
This work was supported by grants from the Russian Foundation for Basic Research (06-04-48947, 05-04-48901) and 2004naukograd grant (04-04-97313).
REFERENCES
1.Pingoud, A., and Jeltsch, A. (2001) Nucleic
Acids Res., 29, 3705-3727.
2.Stahl, F., Wende, W., Wenz, C., Jeltsch, A., and
Pigoud, A. (1998) Biochemistry, 37, 5682-5688.
3.Bitinaite, J., Wah, D. A., and Schidkraut, I.
(1998) Proc. Natl. Acad. Sci. USA, 95, 10570-10575.
4.Vanamee, E. S., Santagata, S., and Aggarwal, A. K.
(2001) J. Mol. Biol., 309, 69-78.
5.Stankevicius, A., Lubys, A., Timinskas, A.,
Vaitkevicius, D., and Janulaitis, A. (1998) Nucleic Acids Res.,
26, 1084-1091.
6.Heiter, D. F., Lunnen, K. D., and Wilson, G. G.
(2005) J. Mol. Biol., 348, 631-640.
7.Samuelson, J. C., Zhu, Z., and Xu, S-Y. (2004)
Nucleic Acids Res., 32, 3661-3671.
8.Armalyte, E., Bujnicki, J., Gledriene, J., Gasluns,
G., Kosinski, J., and Lybus, A. (2005) J. Biol. Chem.,
280, 41584-41594.
9.Boros, I., Posfai, G., and Venetianer, P. (1984)
Gene, 30, 257-260.
10.Rogulin, E. A., Perevyazova, T. A., Zheleznaya,
L. A., and Matvienko, N. I. (2004) Biochemistry (Moscow),
69, 1123-1127.
11.Brown, N., and Smith, M. (1980) Meth.
Enzymol., 65, 391-404.
12.Zheleznaya, L. A., Perevyazova, T. A., Alzhanova,
D. V., and Matvienko, N. I. (2001) Biochemistry (Moscow),
66, 989-993.
13.Abdurashitov, M. A., Belichenko, O. A.,
Shevchenko, A. V., and Degtyarev, S. Kh. (1996) Mol. Biol.
(Moscow), 30, 1261-1267.
14.Morgan, R. D., Calvet, C., Demeter, M., Agra, R.,
and Kong, H. (2000) Biol. Chem., 381, 1123-1125.
15.Higgins, L. S., Besnier, C., and Kong, H. (2001)
Nucleic Acids Res., 29, 2492-2501.
16.Gololobova, N. S., Okhapkina, S. S.,
Abdurashitov, M. A., and Degtyarev, S. Kh. (2005) Mol. Biol.
(Moscow), 39, 840-844.
17.Perevyazova, T. A., Rogulin, E. A., Zheleznaya,
L. A., and Matvienko, N. I. (2003) Biochemistry (Moscow),
68, 984-987.
18.Zheleznaya, L. A., Perevyazova, T. A.,
Zheleznyakova, E. N., and Matvienko, N. I. (2002) Biochemistry
(Moscow), 67, 498-502.