Does a Deficiency of the Signal Recognition Particle (SRP)-Pathway Affect the Biosynthesis of Its Components in Saccharomyces cerevisiae and Escherichia coli?
O. N. Kovalskaya (O. N. Avdeeva)*, P. V. Sergiev, A. A. Bogdanov, and O. A. Dontsova
Faculty of Chemistry, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 932-8846; E-mail: avdeeva@genebee.msu.su* To whom correspondence should be addressed.
Received March 23, 2006; Revision received March 31, 2006
We studied the behavior of the signal recognition particle (SRP) components in Saccharomyces cerevisiae upon deficiencies of the protein transport caused by the absence of the SRP membrane receptor alpha-subunit. A decrease in the concentration of the SRP membrane receptor alpha-subunit in the cell significantly decreased the level of an SRP component, protein SRP72, as well as the levels of mRNAs of SRP protein components and the SRP receptor beta-subunit. But the amount of 7SL RNA remained unchanged. In contrast, in Escherichia coli cells the gradual decrease in the level of the protein FtsY (a homolog of the SRP membrane receptor alpha-subunit) was not associated with changes in the Ffh protein level.
KEY WORDS: signal recognition particle (SRP), cotranslational protein transport, transcription regulationDOI: 10.1134/S0006297906070042
Abbreviations: SRP) signal recognition particle; SR) SRP-receptor.
Membrane and secretory proteins are cotranslationally transported with
involvement of signal recognition particles (SRPs). SRPs were first
found in animal cells [1]. They were shown to
consist of six proteins (SRP9, SRP14, SRP19, SRP54, SRP68, and SRP72)
and the 7SL RNA molecule of about 300 nucleotide residues in length [1, 2].
SRP interacts with the so-called signaling sequence of the protein synthesized on the ribosome after it has left the ribosomal tunnel and also with the ribosome itself, and this decelerates the synthesis of the peptide chain. The resulting complex (consisting of SRP, the growing peptide, and the ribosome) is bound to the membrane due to the interaction of the SRP with the membrane SRP-receptor (SR). Thereafter, SRP loses its affinity for the signaling peptide and ribosome, the ribosome binds to the translocational region of the endoplasmic reticulum membrane (the so-called translocon), the peptide is transposed into the translocon, and the growing peptide is cotranslationally translocated across/into the membrane at the normal rate. The SRP/SR complex dissociates, and the SRP is released into the cytoplasm. This sequence of the events presents the so-called SRP-pathway of protein transport. The SRP-mediated protein cotranslational transport of proteins is regulated by three GTPases, one of which is a constituent of SRP (SRP54) and two are components of the membrane receptor (the SR alpha- and beta-subunits) [3].
The discovery of SRP in yeast [4] and bacterial cells [5, 6] suggested that a universal pathway of protein cotranslational transport should exist in all organisms and also allowed the structure and functioning of SRP from different sources to be compared. In the SRP from Saccharomyces cerevisiae, the 7SL RNA is larger in size (its length is 519 residues) than that isolated from animal cells, the SRP9 protein is absent, the SRP19 protein is represented by the homologous protein Sec65, and the SRP14 protein is present as a homodimer. Moreover, SRP from S. cerevisiae includes the protein SRP21 specific only for yeast [7]. The bacterial SRP isolated from Escherichia coli cells includes only two components: the protein Ffh and a 4.5S RNA molecule. The protein Ffh and the 4.5S RNA are structural and functional analogs of protein SRP54 and domain IV of the 7SL RNA molecule, respectively [5, 6]. In E. coli cells, the role of the SR alpha-subunit is played by its structural and functional homolog, the protein FtsY [8].
In addition to structural differences, there are also some differences in functioning of SRP from S. cerevisiae and E. coli. But the general scheme of functioning of SRP from S. cerevisiae is identical to that of SRP from animal cells: the yeast SRP can recognize the growing signaling peptide exposed on the ribosome, interact with the ribosome, arrest the translation, and GTP-dependently interact with the SR alpha-subunit, with the subsequent release of the peptide and recovery of the protein translation on the ribosome [3, 9].
SRP from E. coli can recognize the signaling peptide and interact with the FtsY protein, and this interaction also depends on GTP [8, 10]. The bacterial SRP can also interact with protein L23 of the E. coli ribosome [11, 12]. Whether SRP from E. coli can interrupt translation is still unclear.
The discovery of SRP in yeast and bacteria has allowed genetic approaches to be used for studies on the role of the cotranslational transport of proteins and SRP components in the vital activity of the cell and for investigations of the cell response to disturbance in SRP function. Gene deletions of some SRP components in the yeast Yarrowia lipolytica and Schizosaccharomyces pombe are lethal [3]. And although the yeast S. cerevisiae can survive notwithstanding a gene deletion of any SRP component [7], the absence of one of the SRP or SR components results in a significant reduction in the cell growth rate (the cell duplication time increases four- to six-fold) and disorders in the transport of secretory and membrane proteins to the endoplasmic reticulum.
It is also interesting to trace changes in the levels of individual components of the SRP-pathway upon deletion of one of the genes encoding them. The SRP assemblage partially occurs in the nucleolus. The proteins SRP14, SRP21, SRP68, SRP72, and Sec65 are transported into the nucleus and bound with 7SL RNA producing the so-called pre-SRP. The latter is exported from the nucleus into the cytoplasm, and there the last component, the SRP54 protein, is bound [13, 14]. The deletion of the 7SL RNA gene has been shown [7] to result in a significant decrease in the levels of the proteins SRP14, SRP54, SRP68, and Sec65 in the cell. The gene deletion of one of the so-called core proteins (SRP14, SRP21, SRP68, and SRP72) is also accompanied by a decrease in the level of all other SRP components. However, the gene deletion of the Sec65 or SRP54 protein does not lead to decrease in the levels of the core proteins and 7SL RNA. In work [15], changes in the transcription level of all genes have been studied by hybridization of mRNA with DNA probes under conditions of gradual decrease in the cell level of SRP54 or the SR beta-subunit. The removal of these proteins at least twofold changed the transcription degree of about 700 genes, which were approximately 10% of their total number in the yeast genome. These changes concerned only proteins involved in all crucial functions of the cell, including protein synthesis, protein modification/degradation, protein folding, RNA processing, DNA replication, recombination, etc. But analysis of changes in expression of the genes of other SRP-pathway components remained beyond the limits of this research.
Deletion of any gene encoding the proteins Ffh, FtsY, or the 4.5S RNA molecule is lethal [10]; therefore, the involvement of these components in the cotranslational transport of proteins can be studied only under conditions of a gradual decrease in the expression of the corresponding genes. In particular, accumulation in the cell of unprocessed forms of secretory and membrane proteins is a consequence of such a decrease [16-19]. However, it remained unclear whether the genes encoding Ffh, FtsY, and 4.5S RNA were expressed in coordination or independently. Note that these three genes do not constitute an operon and are located in different regions of the E. coli genome.
The purpose of this work was to determine whether a disturbance in formation of a valid SR affects the synthesis of other components of the SRP-pathway in S. cerevisiae and E. coli cells. To answer this question, we developed a system that allowed us to control the arrest of the synthesis of the yeast SR alpha-subunit or its bacterial analog and follow the fate of other components of the SRP-pathway.
MATERIALS AND METHODS
Creation of yeast strains ONA11 and ONA72. The yeast strains used in the work are listed in the table. Strain ONA11 was created based on strain SOY46 by integrating the PCR-fragment I into the genomic DNA [20]. The PCR-fragment I amplified from the plasmid pProtA/HIS5 [21] was a sequence encoding 4.5 of Z-domain of the A protein of the Staphylococcus aureus envelope, the HIS5 marker, and flanking sequences of 50-52 nucleotides in length homologous to the integration regions in the srp72 gene. The SOY46 strain cells were transformed with PCR-fragment I as described in [20]. The presence in strain ONA11 genomic DNA of the sequence encoding an affinity tag on the C-end of the SRP72 protein was confirmed by PCR with the genomic DNA. The presence in the cell of the tagged protein SRP72 was shown by immunoblotting using secondary rabbit immunoglobulins to sheep immunoglobulins conjugated with horseradish peroxidase (Imtek, Russia). Strain ONA72 was produced similarly based on strain W303alpha.
Strains used in this work

Creation of bacterial strain XDPBADFtsY. Escherichia coli strains used in the work are listed in the table. The strain XDPBADFtsY was created by integrating into strain X90 DE3 genomic DNA of the PCR-fragment II amplified from the plasmid pET23b-Py-kanR-PBAD-FtsY´ and containing the arabinose promoter, selective marker kanR (the gene responsible for resistance to kanamycin), and flanking sequences of 300-400 nucleotides in length homologous to the integration regions in the gene ftsY. Creation of the plasmid pET23b-Py-kanR-PBAD-FtsY´ included the successive cloning into the vector pET23b(+) (Novagen, Germany) of the following sequences: Py (the sequence of 300 nucleotides located in strain X90 DE3 genomic DNA ahead of the gene ftsY encoding sequence), kanR, PBAD (the arabinose promoter), and FtsY´ (the first 400 nucleotides of the sequence encoding the protein FtsY). The PCR-fragment II was integrated into strain X90 DE3 genomic DNA with involvement of Red-recombinase [22]. Upon electroporation, the cells were plated onto a solid nutritional medium LB (10 g tryptone, 5 g yeast extract, 10 g sodium chloride, 15 g agar per liter of medium) containing 100 mM arabinose and 15 mM kanamycin. The presence in the resulting strain XDPBADFtsY of the arabinose promoter ahead of the encoding sequence of the gene fstY was confirmed by PCR with the genomic DNA and also by the inability of the strain to produce cell colonies on an arabinose-free solid nutritional medium.
Northern blotting. To detect mRNA of the SRP protein components and 7SL RNA, strain ONA11 cells were grown on synthetic medium SD containing 2% galactose, precipitated by centrifugation at 6000g, washed twice in water (1/2 of the initial volume of the cell culture), and resuspended in SD medium supplemented with 2% glucose. The cells were replated and sampled 0, 2, 5, 10, 16, 25, and 40 h after the replating. Cell sampling was performed with consecutive stepwise dilution of the growing cell culture to provide for constant logarithmic growth phase of the cells and retention of the optical density of the cell culture on the sampling within the limits of A600 = 0.6-0.9. The control strain ONA72 was also cultured on SD medium supplemented with 2% glucose until the optical density of the cell culture reached the value of A600 = 0.6-0.8. From each sample, a cell portion was taken corresponding to 150 OD600, and the total cell RNA was isolated by hot phenol deproteinization [23].
Equal aliquots of the total cell RNA were separated in formaldehyde-containing agarose gel, and a capillary transfer of RNA onto a Hybond N+ membrane (Amersham, USA) was performed. Both mRNA and 7SL RNA were detected using a DIG Nucleic Acid Detection Kit (Roche, Switzerland). PCR fragments from 170 to 370 nucleotides in length complementary to the encoding regions of mRNA and 5´-terminal region of 7SL RNA were used as probes. Probe amplification with yeast genomic DNA was performed using the following primers: for protein SRP14 mRNA (gene srp14) srp14-for 5´-ATGGCAAATACTGGCTGTTTATCACC-3´ and srp14-rev 5´-TGTCATGCGAGCCATAAGACA-3´, for protein SRP21 mRNA (gene srp21) srp21-for 5´-ATGTCTGTGAAACCCATTGACAACTAC-3´ and srp21-rev 5´-CGCTCTTTTCGACATCTGTGTTGA-3´, for protein Sec65 mRNA (gene sec65) sec65-for 5´-ATGCCTAGATTAGAAGAGATTGACGATTTC-3´ and sec65-rev 5´-TCCGAACGTTCAACCTTACCAG-3´, for the SR alpha-subunit mRNA (gene srp101) srp101-for 5´-CAATTAGCAGTCTTTACCCC-3´ and srp101-rev 5´-AAAATAGTGCATTAAATGAAGGAG-3´, for the SR beta-subunit mRNA (gene srp102) srp102-for 5´-ATGCTTAGTAATACACTTATTATTGCCTG-3´ and srp102-rev 5´-GGTTAGCAGCGTAAGCAAGC-3´, for carboxypeptidase Y mRNA (gene prc1) prc1-for 5´-ATGAAAGCATTCACCAGTTTACTATGTGG-3´ and prc1-rev 5´-TCAATGCCCAGGATTTTAGGGTCC-3´, and for 7SL RNA (gene scr1) scr1-for 5´-GGTGGGATGGGATACGTTGAG-3´ and scr1-rev 5´-TAGCCGGGACACTTCAGAAC-3´.
Western blotting. To detect protein SRP72-4.5Z, cell strains ONA11 and ONA72 were grown, and samples were taken similarly to those for detecting mRNA of the SRP protein components and 7SL RNA. A cell portion from each sample corresponding to 2.5 OD600 was used to prepare the cell extract as described in [24]. The resulting cell extract was separated in 10% denaturing polyacrylamide gel, the extract from 0.25 OD600 of cells was placed into one lane, and then the samples were electrotransferred from the gel onto a PVDF membrane (Amersham). Protein SRP72-4.5Z was detected with rabbit immunoglobulins conjugated with horseradish peroxidase (Imtek) and using an ECL Western Blotting Detection Kit (Amersham).
To detect protein FtsY, cells of strains XDPBADFtsY and N4156::pAra14-FtsY´ were cultured on LB nutritional medium (10 g tryptone, 5 g yeast extract, 10 g sodium chloride per liter of medium) supplemented with 100 mM arabinose and 15 mM kanamycin, or 15 mM arabinose and 100 mM ampicillin, respectively. After the optical density of the cell culture achieved the value of A600 = 0.5, the cells were precipitated by centrifugation at 5000g, washed twice in LB medium (volume of the washings was 1/2 of initial volume of the cell culture), and resuspended in tenfold volume of LB nutritional medium supplemented with 15 mM kanamycin (strain XDPBADFtsY) or 100 mM ampicillin (strain N4156::pAra14-FtsY´). Upon cell replating onto arabinose-free nutritional medium, samples were taken at 0, 1, and 4 h. The total cell extract was separated in 12% denaturing polyacrylamide gel, the extract from 0.3 OD600 cells was placed into one lane, and then the samples were electrotransferred from the gel onto a PVDF membrane (Amersham). Protein FtsY was detected with primary antibodies to protein FtsY [18] isolated from rabbit blood serum. Sheep immunoglobulins conjugated with horseradish peroxidase (Imtek) to rabbit immunoglobulins were used as secondary antibodies. The secondary antibodies were detected using an ECL Western Blotting Detection Kit (Amersham).
Protein Ffh was detected similarly to detection of protein FtsY using monoclonal antibodies to protein Ffh, which were kindly presented by Prof. A. G. Tonevitskii.
RESULTS AND DISCUSSION
Cells lacking the SR alpha-subunit have defects in the transport of some secretory and membrane proteins to the endoplasmic reticulum [25]. However, what occurs with the SRP itself remained unclear. To answer this question, it was necessary to create a yeast strain with regulated expression of the SR alpha-subunit providing the SRP components to be detected. As the original strain, we used strain SOY46 [25] deprived of the chromosomal copy of the gene encoding the SR alpha-subunit (gene srp101) and with the SR alpha-subunit expressed from the plasmid pSO400 under the control of the GAL1-promoter (table). Upon the transfer of the cell culture onto a synthetic medium with glucose instead of galactose as the source of carbon, the GAL1-promoter was cut off and the expression of the gene encoding the SR alpha-subunit was interrupted. To monitor the synthesis of SRP proteins, we introduced onto the C-end of the core protein SRP72 of the complex an affinity tag which represented 4.5 of the Z-domain of protein A from the staphylococcus aureus envelope. The resulting strain was called ONA11. In the control experiments, strain ONA72 was used which contained the chromosomal copy of the gene encoding the SR alpha-subunit and the tagged protein SRP72 (strain ONA72).
Strain ONA11 cells were replated from liquid nutritional medium with galactose onto glucose-containing medium, and samples were taken beginning from 0 and up to 40 h thereafter. Strain SOY46 has the level of SR alpha-subunit protein about 50-fold higher than the wild-type cells, and it decreases to the wild-type level about 16 h after replating [25]. After 16 h of growth in the presence of glucose, the level of the SR alpha-subunit continued to fall, and this gradually increased the time of cell division. The cell division time reached a maximum (about 5 h) 20-25 h after the replating, and the SR alpha-subunit became undetectable by immunoblotting. On replating onto the synthetic medium with glucose, strain ONA11 created by us behaved similarly to strain SOY46. Figure 1a shows the results of detection of SR alpha-subunit mRNA with a specific DNA probe. Even 2 h after the cell replating onto the glucose-containing medium, the level of SR alpha-subunit mRNA is sharply decreased and continues to gradually decrease during further incubation of the cells. And this decrease in the level of the SR alpha-subunit mRNA in strain ONA11 correlated with the decrease in the level of the SR alpha-subunit protein in strain SOY46 upon replating onto glucose-containing medium [25].
To assess the SRP component levels under conditions of the decrease in cell concentration of the SR alpha-subunit, we determined the levels of mRNA of the SRP protein components, 7SL RNA, and the tagged protein SRP72. Figure 1b shows the results of northern blotting of 7SL RNA isolated from strain ONA11 cells with different levels of the SR alpha-subunit, and no correlation is observed between the level of 7SL RNA and the transcription degree of the SR alpha-subunit gene.Fig. 1. Levels of the SRP-pathway components in S. cerevisiae cells. a) Results of northern blotting of SR alpha-subunit mRNA. Lanes 0-40 present analyses of total RNA isolated from cells of strain ONA11 sampled 0, 2, 5, 10, 16, 25, and 40 h after cell replating onto glucose-containing synthetic medium; C - analysis of total RNA isolated from cells of the control strain ONA72. Total RNA level in each lane is equivalent to that of the total RNA isolated from 20 (a, d-f) and 5 (b, g) OD600 of cells. b) Results of northern blotting of 7SL RNA. Designations are the same as in Fig. 1a. c) Results of immunodetection of protein SRP72-4.5Z. The order of application of the cell extract samples from cells of strains ONA11 and ONA72 is the same as the order of application of the total cell RNA on detecting the SR alpha-subunit mRNA (Fig. 1a). d-g) Results of northern blotting of mRNA of proteins SRP14, SRP21, Sec65, and carboxypeptidase Y, respectively. Designations are the same as in Fig. 1a.
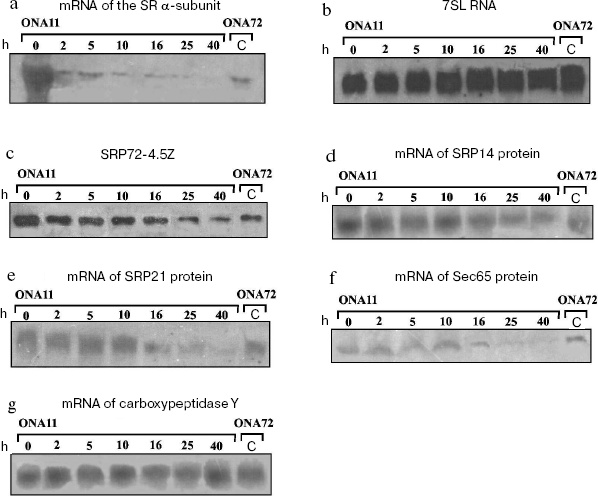
Figure 1c shows that high concentration of the SR alpha-subunit in the cell is associated with two-threefold higher level of SRP72-4.5Z protein in the ONA11 strain than in the control strain ONA72. Upon replating the strain ONA11 cells onto glucose-containing medium, the level of SRP72-4.5Z protein within 2 h decreases about twofold. During further incubation of the cells on the glucose-containing synthetic medium, the protein SRP72-4.5Z level gradually decreases, and by 40 h it becomes about fivefold lower than at the initial point. Thus, increased level of the SR alpha-subunit in the cell leads to increase in the level of protein SRP72-4.5Z. The disappearance of the SR alpha-subunit is also associated with a decrease in the level of SRP72-4.5Z protein, although the latter does not disappear entirely: its level in strain ONA11 cells becomes two-threefold lower than that in the cells of the control strain ONA72 (Fig. 1c).
The decrease in the level of the SR alpha-subunit in strain ONA11 cells was accompanied by gradual decrease in the levels of mRNAs encoding proteins SRP14, SRP21, and Sec65 (Figs. 1d, 1e, and 1f). Between 10 and 16 h of the cell incubation on the glucose-containing synthetic medium, the levels of mRNAs of these proteins decrease to the mRNA levels in the control strain ONA72. During further incubation of strain ONA11 cells, the levels of mRNAs of these proteins continue to decrease, and by 40 h become 10-12-fold lower than at the initial point.
The behavior of the SR beta-subunit was followed, and gradual decrease in the cellular content of the SR alpha-subunit was shown to be accompanied by decrease in the mRNA level of the SR beta-subunit (data not presented).
We have performed northern blotting using mRNA of carboxypeptidase Y as the control, because disorders in SRP function have no influence on the transport of this protein to the endoplasmic reticulum [25]. Figure 1g shows that the level of carboxypeptidase Y mRNA does not change under conditions of either excess level of the SR alpha-subunit in the cell (lanes 0 and C) or its shortage (lanes 40 and C).
Our data suggest that the absence from the cell of the SR alpha-subunit results in a decrease in the SR beta-subunit mRNA level and also in the levels of SRP72 protein and mRNAs of proteins SRP14, SRP21, and Sec65. This decrease seems to be caused by reduced transcription of the genes encoding the SRP proteins. The level of 7SL RNA is unchanged, possibly, because 7SL RNA, as discriminated from mRNAs of the SRP proteins, is encoded by the gene transcribed by RNA polymerase III [26] and seems to be regulated otherwise. Moreover, 7SL RNA is strongly structured and seems to be more stable than mRNA.
The core protein SRP72-4.5Z (although in a decreased concentration) was found in the cell 40 h after strain ONA11 cells had been replated onto the glucose-containing medium; therefore, it was suggested that the full-value SRPs should be present in the cell during the period of measurements. Possibly, SRP can be also responsible for other functions in the cell, e.g., be involved in both the co- and posttranslational transport of proteins [27], or act as a chaperon interacting with the full-size protein and maintaining the transportability of the bound signaling peptides.
But how are the syntheses of the SRP components and its receptors coordinated? At first glance, the receptor alpha-subunit should activate the transcription of genes of the SRP protein components. But this protein is incorporated in the membrane, and it is difficult to imagine how it can be transferred into the cell nucleus. It is more likely that disorders in formation of the full-value SR induce an accumulation in the cytoplasm of proteins that normally should be transported to the endoplasmic reticulum by the SRP-pathway. As a result of changes in the folding and processing, one or more of these proteins acquires the ability to penetrate into the nucleus and suppress the transcription of certain genes, in particular, those encoding the SRP proteins. Similar examples, although concerning activation of certain gene transcription, have been described [28].
To compare the biogenesis of SRP upon a decrease in the level of the SR alpha-subunit in S. cerevisiae yeast and in bacteria, it was necessary to create a strain of E. coli with regulated expression of the gene encoding the protein FtsY homologous to the SR alpha-subunit. The strain XDPBADFtsY (table) was constructed with the ftsY gene in the genomic DNA under the control of the arabinose promoter, and this provides for the arabinose-dependent regulation of the expression of the gene ftsY. A similar strain N4156::pAra14-FtsY´ with the protein FtsY gene controlled by the arabinose promoter was described earlier [18, 19]. Strain N4156::pAra14-FtsY´ cell replating onto an arabinose-free liquid medium caused a gradual decrease in the FtsY protein level. Four hours after the cell replating, the protein FtsY became undetectable by antibodies, and this was associated with pronounced defects in the transport of the membrane proteins SecY and lac-permease [18, 19].
The behavior of strain XDPBADFtsY created by us was similar to that of strain N4156::pAra14-FtsY´: the protein FtsY levels in these two strains fell synchronously (Fig. 2a). Strain X90 DE3, which was the base for producing the XDPBADFtsY strain, was used as the control. The substitution of the natural promoter Py ahead the encoding region of the ftsY gene by the arabinose promoter did not noticeably change the protein FtsY level (Fig. 2a).
Immunodetection revealed that the protein Ffh levels in cells of strains XDPBADFtsY and N4156::pAra14-FtsY´ did not depend on the FtsY protein level (Fig. 2b); thus, it seemed that for the time given neither the ffh gene transcription decreased, nor the protein Ffh degraded. Therefore, the genes encoding the Ffh and FtsY proteins were supposed to be expressed independently. We did not determine the level of the other component of the bacterial SRP, 4.5S RNA, because it is known to execute in the cell a number of functions and be present in a fourfold excess as compared to Ffh protein [17].Fig. 2. Immunodetection of the FtsY (a) and Ffh (b) proteins. Lanes: 1-3) total extract from strain XDPBADFtsY cells sampled 0, 1, and 4 h after replating the cells onto arabinose-free nutritional medium; 4-6) total extract from strain N4156::pAra14-FtsY´ cells also sampled 0, 1, and 4 h after replating the cells onto arabinose-free nutritional medium; 7) extract from the cells of the control strain X90 DE3.
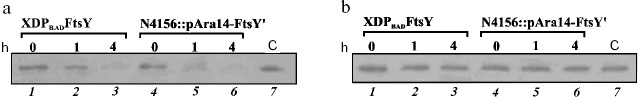
As opposed to yeast cells, the SR level in bacterial cells does not influence the level of SRP. It seems that this regulatory pathway appeared later in evolution. The need for cutting off the synthesis of the SRP protein components in the eukaryotic cell is likely to be a consequence of the ability of these cells to stop translation. Possibly, an accumulation of the “braked” SRP-ribosome complexes and a decrease in the pool of functioning ribosomes is more perilous for the cell than the accumulation of precursors of secretory and membrane proteins in the cytoplasm.
We are grateful to Prof. M. Rout and S. S. Dokudovskaya (Rockefeller University, USA) for their help in the SRP72 protein tagging, to Prof. A. G. Tonevitskii (Biological Faculty, Moscow State University, Russia) for the presented antibodies to the Ffh protein, to Prof. J. Luirink (Vrije Universiteit, Netherlands) for the presented strain N4156::pAra14-FtsY´ and antibodies to the FtsY, and to Prof. P. Walter (University of California San Francisco, USA) for the presented strain SOY46.
This work was supported by the Howard Hughes Medical Institute (HHMI 55005605), the Russian Foundation for Basic Research (project No. 04-04-49505), the Russian State Program Supporting the Leading Scientific Schools (5259.2006.4), and an individual grant for O. N. Avdeeva from the Federation of European Biochemical Societies (FEBS, Collaborative Experimental Scholarships for Central and Eastern Europe).
REFERENCES
1.Walter, P., and Blobel, G. (1980) Proc. Natl.
Acad. Sci. USA, 77, 7112-7116.
2.Walter, P., and Blobel, G. (1982) Nature,
299, 691-698.
3.Pool, M. R. (2005) Mol. Membr. Biol.,
22, 3-15.
4.Brennwald, P., Liao, X., Holm, K., Porter, G., and
Wise, J. A. (1988) Mol. Cell. Biol., 8, 1580-1590.
5.Poritz, M. A., Strub, K., and Walter, P. (1988)
Cell, 55, 4-6.
6.Romisch, K., Webb, J., Herz, J., Prehn, S., Frank,
R., Vingron, M., and Dobberstein, B. (1989) Nature, 340,
478-482.
7.Brown, J. D., Hann, B. C., Medzihradszky, K. F.,
Niwa, M., Burlingame, A. L., and Walter, P. (1994) EMBO J.,
13, 4390-4400.
8.Miller, J. D., Berstein, H. D., and Walter, P.
(1994) Nature, 367, 657-659.
9.Mason, N., Ciufo, L. F., and Brown, J. D. (2000)
EMBO J., 19, 4164-4174.
10.Muller, M., Koch, H. G., Beck, K., and Schafer,
U. (2001) Progr. Nucleic Acid Res. Mol. Biol., 66,
107-157.
11.Ullers, R. S., Houben, E. N., Raine, A., ten
Hagen-Jongman, C. M., Ehrenberg, M., Brunner, J., Oudega, B., Harms,
N., and Luirink, J. (2003) J. Cell Biol., 161,
679-684.
12.Gu, S. O., Peske, F., Wieden, H. J., Rodnina, M.
V., and Wintermeyer, W. (2003) RNA, 9, 566-573.
13.Grosshans, H., Deinert, K., Hurt, E., and Simos,
G. (2001) J. Cell Biol., 153, 745-762.
14.Ciufo, L. F., and Brown, J. D. (2000) Curr.
Biol., 10, 1256-1264.
15.Mutka, S. C., and Walter, P. (2001) Mol. Biol.
Cell, 12, 577-588.
16.Phillips, G. J., and Silhavy, T. J. (1992)
Nature, 359, 744-746.
17.Jensen, C. G., Brown, S., and Pedersen, S. (1994)
J. Bacteriol., 176, 2502-2506.
18.Luirink, J., ten Hagen-Jongman, C. M., van der
Wejden, C. C., Oudega, B., High, S., Dobberstein, B., and Kusters, R.
(1994) EMBO J., 13, 2289-2296.
19.Seluanov, A., and Bibi, E. (1997) J. Biol.
Chem., 272, 2053-2055.
20.Wach, A., Brachat, A., Alberti-Segui, C.,
Rebischung, C., and Philippsen, P. (1997) Yeast, 13,
1065-1075.
21.Rout, M. P., Aitchison, J. D., Suprapto, A.,
Hjertaas, K., Zhao, Y., and Chait, B. T. (2000) J. Cell Biol.,
148, 635-651.
22.Datsenko, K. A., and Wanner, B. L. (2000)
Proc. Natl. Acad. Sci. USA, 97, 6640-6645.
23.Köhrer, K., and Domdey, H. (1991) Meth.
Enzymol., 194, 398-405.
24.Kushnirov, V. V. (2000) Yeast, 16,
857-860.
25.Ogg, S. C., Poritz, M. A., and Walter, P. (1992)
Mol. Biol. Cell., 3, 895-911.
26.Dieci, G., Giuliodori, S., Catellani, M.,
Percudani, R., and Ottonello, S. (2002) J. Biol. Chem.,
277, 6903-6914.
27.Abell, B. M., Pool, M. R., Schlenker, O.,
Sinning, I., and High, S. (2004) EMBO J., 23,
2755-2764.
28.Kaufman, R. J. (2002) J. Clin. Invest.,
110, 1389-1398.