
|
The Language of Methylation in Genomics of EukaryotesP. VolpeDepartment of Biology, University of Rome “Tor Vergata”, Via della Ricerca Scientifica 1, 00133 Rome, Italy; fax: (+39-06) 7259-4244; E-mail: volpe@bio.uniroma2.it
|
Received October 13, 2004
Background studies have shown that 6-methylaminopurine (m6A) and 5-methylcytosine (m5C), detected in DNA, are products of its post-synthetic modification. At variance with bacterial genomes exhibiting both, eukaryotic genomes essentially carry only m5C in m5CpG doublets. This served to establish that, although a slight extra-S phase asymmetric methylation occurs de novo on 5´-CpC-3´/3´-GpG-5´, 5´-CpT-3´/3´-GpA-5´, and 5´-CpA-3´/3´-GpT-5´ dinucleotide pairs, a heavy methylation during S involves Okazaki fragments and thus semiconservatively newly made chains to guarantee genetic maintenance of -CH3 patterns in symmetrically dimethylated 5´-m5CpG-3´/3´-Gpm5C-5´ dinucleotide pairs. On the other hand, whilst inverse correlation was observed between bulk DNA methylation, in S, and bulk RNA transcription, in G1 and G2, probes of methylated DNA helped to discover the presence of coding (exon) and uncoding (intron) sequences in the eukaryotic gene. These achievements led to the search for a language that genes regulated by methylation should have in common. Such a deciphering, initially providing restriction minimaps of hypermethylatable promoters and introns vs. hypomethylable exons, became feasible when bisulfite methodology allowed the direct sequencing of m5C. It emerged that, while in lymphocytes, where the transglutaminase gene (hTGc) is inactive, the promoter shows two fully methylated CpG-rich domains at 5´ and one fully unmethylated CpG-rich domain at 3´ (including the site +1 and a 5´-UTR), in HUVEC cells, where hTGc is active, in the first CpG-rich domain of its promoter four CpGs lack -CH3: a result suggesting new hypotheses on the mechanism of transcription, particularly in connection with radio-induced DNA demethylation.
KEY WORDS: maintenance and de novo post-synthetic modification, demethylation, Okazaki fragments, parental and daughter chains, eukaryotic gene structure, coding (exon) and uncoding (promoter and intron) regions, unique, repeated, foldback, viral and mitochondrial sequences, restriction minimaps, hTGc gene, m5C sequencing of bisulfite-converted sequences, CpG-rich domains, regulation of transcription
Evolution of DNA has been accompanied by that of its post-synthetic modification implying formation, along double helix, of two classes of methylated bases, namely m6A and m5C in prokaryotes [1, 2] and m5C alone in eukaryotes [3, 4]. In concomitance with the disappearance of the system of deoxyadenylate (dam) and deoxycytidylate (dcm) DNA methylases (met) [5] and that of restriction-modification (RM) endonucleases [6-8], both characterizing the world of bacteria [9], molecular selection led in eukaryotes to the loss of m6A not only in nuclear [10] but also in mitochondrial [11] and chloroplast [12] DNAs. The m5C base was conserved in these three nucleic acids by a novel met family which likely evolved through alternate splicing [13-15]. The presence of m6A in bacterial DNA was shown to be due partly to the activity of dam met [5, 16, 17] and partly to the met activity of the type I, type II, and type III RM systems involved in the digestion of infecting m6A-free phage DNAs [6-8, 18]. Alternatively, the presence of m5C in the same bacterial DNA was shown to be due partly to the activity of dcm met [5, 19, 20] and partly to the met activity of the sole type II RM system involved in the digestion of infecting m5C-free phage DNAs [6]. In bacterial DNA the m6A and m5C modified bases were related not only to the RM anti-phagic defense [21-23]: by complementing the function of the induction-repression operon machinery [24], they were also associated with an auxiliary mechanism to regulate transcription [25]. This association was in harmony with the hypothesis that in higher cells the concentration of m5C in promoters and introns [10, 26, 27] would also be associated with mechanisms modulating gene expression [10, 28-32].
The present analysis is focused on one of the relevant questions in Genomics of Eukaryotes: is m5C actually a regulatory signal for gene expression? In animal genomes, 1 to 5 out of every 100 Cs are modified [4, 33-35]. In plant genomes, this concentration can significantly increase up to 10 or even 30% [36]. In this framework, and not only because the occurrence of methylation in Drosophila DNA still remains sub judice [37-40], a closer look at the main puzzles of the mosaic world of methylation seemed worthwhile. It was shown that a maintenance met system mostly recognizes hemimethylated 5´-CpG-3´/3´-Gpm5C-5´ to yield dimethylated 5´-m5CpG-3´/3´-Gpm5C-5´ dinucleotide pairs [41, 42]. These appeared as a function of genome organization, with an alternation of hypomethylated vs. hypermethylated regions [10, 27]. The idea of a possible role of m5C in the modulation of gene expression originated from the crucial finding, in synchronized cells, concerning an inverse correlation between the largest part of DNA methylation, taking place during phase S, and the largest part of RNA transcription, taking place during phases G1 and G2 [43-45]. Inverse correlation was confirmed for many housekeeping (HK) and tissue-specific (TS) genes whose “primary” sequences were already known [26, 46]. While this information suggested that m5C is not an error of Nature, since it is located at precise nucleotide sites and gene sequences, the innovative bisulfite reaction used to directly sequence m5C [47, 48] facilitated the search for a presumed “secondary” code which could be shared by genes characterized by m5C-dependent regulation [26, 27].
The quasi-universality of DNA methylation in pro- and eukaryotes and age-dependence. Following studies which demonstrated that both m6A and m5C are involved in RM to defend bacteria from phage infection [21-23], attempts at finding m6A in vertebrate DNA and an A met in vertebrate nuclei or mitochondria were unsuccessful [4, 49]. A small amount of m6A was shown, instead, to exist in DNA of higher plants [50, 51], protozoa [52-54], fungi [55], algae [16, 56, 57], and invertebrates [4]. The m5C base has been found in the DNA of all animals and higher plants [4, 33, 34, 36, 50, 58]. Analysis of base composition in the DNA of Drosophila was contradictory: one methodology showed complete absence of m5C [37]; other studies showed that in some sequences C can be still modified [38, 59]. The latter fact suggested that, while a differentiation-dependent DNA methylation was well documented in the animal kingdom [33-35, 49], its presence in insects, although slight, might even be correlated with lethality [39, 40, 60].
Cell-cycle methodology helped to reveal background rules of eukaryotic DNA methylation. DNA methylation appeared as an intriguing phenomenon in living matter: some mets and de-methylases, two DNA de-aminases, and two DNA re-aminases were taken into consideration [28]. However, only the existence of mets was in the realm of reality, since they were characterized in vivo and in vitro [13, 14, 61]. Borek considered the hypotheses brought forward on the probable role of DNA methylation and made the following dramatic remark: “Model building of differentiating systems by developmental biologists can be stimulating; but we must bear in mind that even though out of necessity the interaction of macromolecules must be invoked, Biochemistry is still the ultimate arbiter of validity of models” [62]. Holliday and Pugh replied: “While we can agree with Borek's last sentence, we must point out that it would greatly impede biological research if every theory or hypothesis was discounted because of the lack of direct biochemical evidence” [28]. New facts had therefore to be discovered, and this was achieved by exploiting cell-cycle methodology.
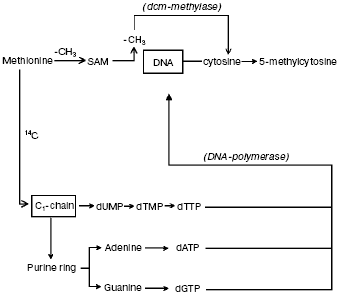
The question concerning parallelism between DNA synthesis and methylation. It was suggested that DNA methylation may continue for some hours after DNA synthesis is completed [63, 64]. Hence, by incubating synchronized HeLa cells with [14C]methyl-L-methionine, used as common tracer for both DNA synthesis and methylation [10, 41], it became possible to verify that DNA methylation does follow DNA synthesis [41] since the labeled carbon atom from -CH3 of L-methionine did not enter the pyrimidine ring but pierced via C1-chain the purine ring of A and G and the -CH3 of T; on the other hand, the whole -CH3 of L-methionine was transferred via S-adenosyl-L-methionine (SAM) to DNA C (Fig. 1). In conclusion, the maximal labeling emerging from the 14C of L-methionine -CH3 was always found in S in four hydrolyzed bases: in m5C, signifying methylation, and in A, G and T, implying synthesis (Fig. 2a).
Fig. 1. Biosynthetic and methylasic pathways of DNA in eukaryotic cells. In cytosol, the carbon atom coming from -CH3 of methionine through the C1-chain enters the purine ring of A and G and the -CH3 of T (it does not enter the pyrimidine ring) [10, 41]. After formation of the corresponding dATP, dGTP, and dTTP and after release of a PPi from each, in the nucleus, the DNA polymerase alpha enzyme introduces, during S, the dAMP, dGMP, and dTMP into semiconservatively newly synthesized chains. The met system transfers via SAM the entire -CH3 of methionine on given C residues located along these same semiconservatively newly synthesized chains [10, 41].
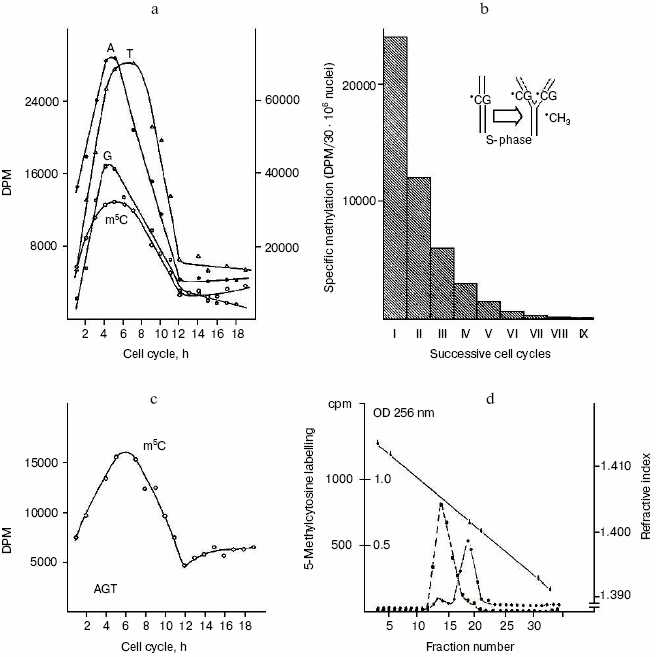
The question of whether or not newly replicating DNA chains are semiconservatively methylated. It was known that in isolated nuclei, in the absence of triphosphonucleosides (supplied by cytosol), DNA cannot be synthesized. On this basis, by employing nuclei in vitro labeled with [14C]methyl-SAM, one could verify whether or not the absence of DNA replication would influence DNA methylation. The result was unexpected: in HeLa nuclei, isolated in S, among the other DNA bases, only C continued to be methylated in the absence of DNA synthesis (Fig. 2c). This demonstrated that the two pathways of DNA replication and methylation (Fig. 1) can be separated from each other. In whole cells, DNA methylation followed DNA replication during S (Fig. 2a); in isolated nuclei, DNA methylation proceeded during S in the absence of DNA synthesis (Fig. 2c). By itself the occurrence of DNA methylation in S-phase nuclei was not an absolute demonstration that it involved new chains formed just before their isolation; but the correlations between in vivo DNA synthesis and in vivo and in vitro DNA methylation strongly suggested this possibility:Fig. 2. Semiconservative transmission of m5C. a) During the mitotic cycle [90] HeLa cells were labeled with [14C]methyl-L-methionine, and their genomic DNA was hydrolyzed to bases and chromatographed: radioactivity in A, G, and T (right ordinate) showed synthesis, while that in m5C (left ordinate) showed methylation [10, 41]. b) During a whole S, HeLa cells were labeled with [14C]methyl-L-methionine; after 14 h of growth in fresh medium -- to the end of a cycle in the absence of radioisotope -- their genomic DNA was hydrolyzed to bases and chromatographed: the measurement of radioactivity in m5C was repeated for 10 cycles (the insert suggested how, in the replication fork, methylation semiconservatively follows synthesis, since the labeled m5C per cell systematically decreased by half) [102]. c) During the mitotic cycle, nuclei isolated from HeLa cells were labeled with [3H-methyl]SAM and their DNA was hydrolyzed to bases and chromatographed: radioactivity in A, G, and T, accounting for synthesis, was negligible, whereas that in m5C showed methylation [41]. d) Ultracentrifugation of genomic HeLa cell DNA in alkaline CsCl gradient: the dashed line shows the originating in [14C]methyl-L-methionine radioactivity of m5C along the semiconservatively newly replicating chains made heavier by a previous incorporation of BrdUrd; the solid line shows the OD at 256 nm of the separated lighter parental chains [73].
[DeltaDNA*CH3/Deltat]in vivo = K1 * [DeltaDNA]in vivo,
[DeltaDNA*CH3/Deltat]in vitro = K2 * [DeltaDNA]in vivo,
[DeltaDNA*CH3/Deltat]in vitro = K3 * [DeltaDNA*CH3/Deltat]in vivo.
These equations established that the cells entering S carry old chains inherited by parental cells, while targets of met both in vivo and in vitro should be nothing but nascent chains (several of them still remained in the nuclei to be isolated) [10]. A similar conclusion was achieved by Adams who thought that, while old and new DNAs are methylated in isolated nuclei [65], new DNAs are formed without m5C in whole cells [66]. As a consequence, during S, DNA methylation might be a prerequisite for gene expression or tissue differentiation [33-35, 49, 67]. Whatever the case may be, the preferential methylation of newly born chains [41] did not contrast with the discontinuous methylation of Okazaki fragments, between the short sequences polymerized ad hoc for their ligation: those fragments resulted methylated before being ligated as soon as they were formed in the replication fork [68-71]. The semiconservativity of DNA methylation [41, 72], revealed for the first time by treating synchronized cells with [14C]methyl-L-methionine (Fig. 2b), was later confirmed by using restriction endonucleases [42] and through a direct separation, in alkaline CsCl, of heavy BrdUrd-containing new chains and light non-containing BrdUrd old chains [73] (Fig. 2d).
The question of differential methylation of euchromatic vs. heterochromatic DNAs. Evidence suggested that the S phase is subdivided into two parts with respect to the characteristics of DNA replicons, since early replicating euchromatic DNA tended to be GC-rich and late replicating heterochromatic DNA tended to be AT-rich [74]. Moreover, in Chinese hamster cells, DNA extracted in early S was methylated to a greater extent when compared to that extracted in late S [75]. This was also true for HeLa cells [10]. For this reason, one was led to suppose (i) that in newly replicating chains there would be sequences not uniformly methylated during S and (ii) that the CG-rich sequences should be, in general, preferentially methylated [10], in agreement with the expectation [76].
The question of the biological clock in methylating specific replicating sequences. Since DNA did not appear to be uniformly methylated in S, investigations continued to focus on specifically methylated targets [10] rather than considering the larger subdivision of hypermethylated euchromatic vs. hypomethylated heterochromatic replicons [75]. With this purpose in mind, methylated DNA was fractioned through ultracentrifugation in Ag+/Cs2SO4 gradients [77, 78]: a small heavy GC-rich fraction and a large light AT-rich fraction were obtained (the first, containing genes for rRNA, was mainly expressed in early S; the second was mainly expressed in late S). As for the m5C concentration, in early S it was found to increase on the heavier peak, while in late S it was found to increase on the lighter peak [67, 77, 78]. All this suggested that, at the different stages of S, one dealt with GC- or AT-rich sequences probably polymerized in correspondence with the formation of specific hyper- or hypomethylated templates [67, 77, 78]. In other words, it seemed that the genes would be methylated according to a given order and intensity along newly replicating chains [67, 77, 78]. This idea was confirmed after the discovery of hypermethylation of foldback sequences: throughout S, in HeLa cells, the methylation wave chronologically involved palindromic and then highly repeated and moderately repeated sequences; the unique sequences were characterized by a minimal late methylation [79].
The question of whether or not methylated DNA sequences are repaired with each cell cycle. A finer analysis of the two nuclear DNA fractions separated in Ag+/Cs2SO4 gradient confirmed that m5C actually had a differential distribution in them [77, 78]. The highest concentration of methylated sequences was found in the denser side of the heavy fraction; in the light DNA the highest concentration of methylated sequences was found in the lighter side [77, 78, 80]. Such a specific distribution of m5C was found to depend on the S-phase stages [67, 77, 78]. This implied two generalizations: (i) hypermethylation on GC-rich sequences would offer no surprise because of its rather statistical character (for instance, GC-rich rRNA genes would be hypermethylated according to the size of their GC target); (ii) hypermethylation on AT-rich sequences would seem to work against the principle of GC target size, since a decrease in C residues would correspond to an increase in their methylation [67, 77, 78].
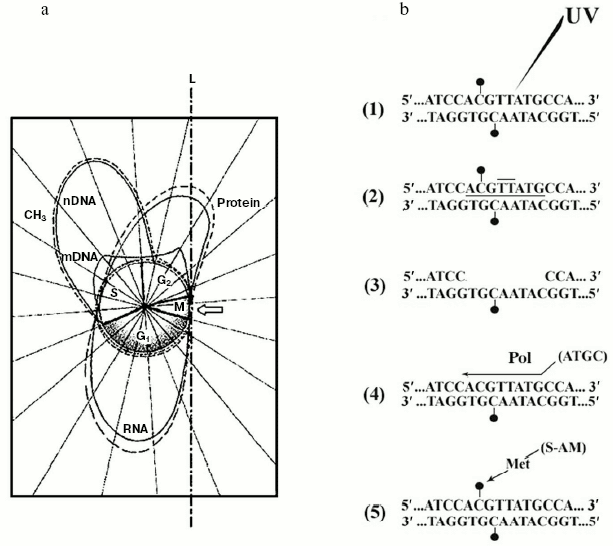
Therefore, while the extra-S time was characterized by a minimal, probably de novo DNA methylation [41], S exhibited maintenance DNA methylation [41]. For this reason, one assumed, the mechanisms discussed in (i) and (ii) should act in S [45]. But it was obvious that, with successive cell cycles, the amount of methylated sequences could not increase without limits (Fig. 3a). Consequently, changes in the amount of methylated DNA during development [81] and in differentiated tissues [34] were not so conspicuous. Thus, whatever the mechanism of DNA methylation was in S, statistical or specific, it was reasonable to suppose that, particularly during the extra-S time, each small wave of de novo DNA methylation might be followed by a corresponding small wave of DNA de-methylating repair [11]. This was fully confirmed by discovery of a “repair-modification” mechanism [82] which demonstrated that, after radio-damage of a methylated double helix, given genes may lose directly methylated groups, belonging to m5Cs, without participation of a DNA demethylase [11, 82] (Fig. 3b). Although the existence of DNA demethylating proteins should not be excluded [83], a non-enzymatic DNA demethylation could occur in the case of an “incomplete” repair of methylated double strands. Their “complete” repair would necessarily require a “re-methylation” and for this reason the participation of a met would be necessary both inside and outside S [11, 82, 84].
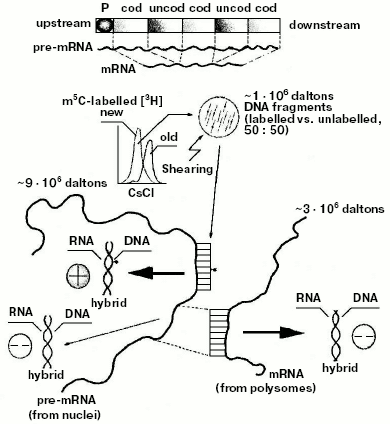
Discovery of the internal design of the eukaryotic gene. At the beginning of the 70s, while attention was paid to methylation of specific DNA sequences, a problematic discussion took on not only the size and origin of eukaryotic pre-mRNAs and mRNAs [85-88], but also the internal structure of the eukaryotic transcriptional unit [89]. The background experiment performed to elucidate both these points originated from previous studies regarding the timing of DNA methylation during the cellular cycle [41, 90] and from the nonrandom genetic scattering of m5C along a semiconservatively replicating DNA chain [10]. This experiment showed a preferential methylation of gene promoter [10] (as reviewed in [91]) and of all those regulatory and signal sequences that do not code for mRNAs [10] (as reviewed in [31]). To summarize, in HeLa cells, if half of the population of hybrids between genomic DNA fragments (used as probes) and pre-processed high molecular weight mRNAs (purified from nuclei) contained a large number of m5Cs, the whole population of hybrids between genomic DNA fragments (also used as probes) and processed low molecular weight mRNAs (purified from polysomes) contained few, if any, m5Cs (Fig. 4).Fig. 3. Inverse correlation between DNA methylation and gene expression [10, 45]. a) DNA methylation vs. DNA, RNA, and protein biosynthesis. The methylation “orbit” corresponds to labeling of m5C in Figs. 2a and 2c [41]. The transcription and translation orbits correspond to data from [44, 93]: duplication of genomic DNA (__) and its methylation (--) show an apogee in S [97]; duplication of mtDNA (__) shows maxima in S and G2 [97]; RNA (__) and protein (_ _) synthesis show apogees in G1 and G2 [44, 93]; repair synthesis of genomic DNA (--) is constant around the cycle [45, 73, 82]. The arrow shows a switch-off of macromolecular events in M [45]. L, line of asymmetry. b) Hypomethylation of repair patches (RPs) [73, 82]: a symmetrically dimethylated 5´-m5CpG-3´/3´-Gpm5C-5´ dinucleotide pair is flanking a radio-induced TT-dimer (2); after digestion of the damaged region (3), which previously included a m5CpG dinucleotide (1), “excision-repair” replaces its m5C nucleotide with a simple C nucleotide, because the repair DNA polymerase system does not find any methylated dCTP in the soluble pool of triphosphonucleosides [10, 11, 41]; this yields a hemi-methylated CpG/Gpm5C dinucleotide pair, namely an incompletely reconstructed RP (4); during S, met may add to C the lost -CH3 (“repair-modification”), providing a completely reconstructed RP (5).
Since polysomal mRNAs were known to be much shorter than nuclear pre-mRNAs [89, 92, 93], these results suggested for the first time that, at variance with the structure of bacterial genes (whose cistrons in the operon were known to be constituted of coding sequences [24]), the eukaryotic gene had to be thought of as a repetition, after the promoter, of intermittent “coding” and “uncoding” regions (Fig. 4). Methylation did not significantly involve the coding regions: it involved the uncoding ones, complementary to parts of pre-mRNAs to be removed during processing [10]. This finding, crucial for genetic engineering, was supported five years later by Chambon's splicing theory [94, 95]. Based on electron microscopic observations, it explained how a cDNA intermittently excludes from hybridization segments of the corresponding pre-mRNA: the sequences of pre-mRNA, non-hybridized with cDNA in [94], were nothing but the uncoding sequences [10] (Gilbert and then Crick proposed calling them “introns” [96]); the sequences of pre-mRNA, hybridized with cDNA [94], were nothing but the coding sequences [10] (Gilbert and then Crick proposed calling them “exons” [96]).Fig. 4. Model of the eukaryotic transcriptional unit. Hybridization of post-synthetically methylated DNA chains, sheared to fragments (probes) of 1*106 daltons, with large pre-mRNA (purified from nuclei) yielded methylated (50%) and unmethylated (50%) DNA/RNA hybrids; the same probes were hybridized with small mRNA (purified from polysomes) yielding 100% of unmethylated DNA/RNA hybrids. These results suggested that statistically along the gene there would be, after the promoter P, an intermittence of hypermethylated uncoding and hypomethylated coding regions [10] (hybridization of DNA, containing the ovalbumin gene, with ovalbumin mRNA also showed intermittent hybridized coding, exon, and unhybridized uncoding, intron, regions [94-96]).
Assumption on the mechanism regulating gene activity in eukaryotes. It is worth emphasizing, with further information, the experimental circumstances that provided an additional key in revealing the biochemical basis of the regulation of gene activity in eukaryotes. The evidence that the uncoding sequences, promoter and introns, are hypermethylated took on particular interest when the cell-cycle dependence of bulk DNA methylation was compared with that of bulk gene expression: the maximal rate of DNA methylation [41] followed the maximal rate of DNA replication [90, 97] during S, but the maximal rates of transcription [10, 43-45] and translation [93, 97] took place mainly during G1 and G2 (Fig. 3a). This was also the first concrete suggestion as to a possible inverse correlation between gene methylation and pre-mRNA transcription [10, 44, 45] (as reviewed in [32]). Today there are many observations supporting this suggestion [28, 30, 98-100], those regarding the regulation of a large number of housekeeping (HK) and those concerning tissue-specific (TS) genes, for instance [26, 46]. Also, the methylation-dependent regulation of the integrated viral [29, 101-104] genes is of particular interest.
Search for specific targets of methylation along the double helix. The long list of genes that have to be methylated to be switched-off [26, 46] led to the search for a “code” that they would acquire in common through post-synthetic modification catalyzed by the met system [13, 14, 61, 105, 106]. The basic hypothesis was that m5C might function as a signal for regulation of transcription [10, 28], since the hypermethylated promoter and the intermittent uncoding methylated domains, after their possible association with m5C-binding proteins [107], would physically interfere with the slip of RNA polymerase (RNApol) on the transcriptional unit. The block of transcription would start from methylated promoter (previously recognized by a type A m5C-binding protein able to compete with RNApol) and then be potentiated, inside the gene, by an intermittent association of uncoding methylated domains with a type B m5C-binding protein [46].
But what could be supposed about the diversity of the methylation code at the level of the promoter and at that of the intervening uncoding sequences? An answer to this question sprung from studies on targets for methylation [76-78]. After finding that it preferentially involves CpG dinucleotides in bacteria [1], as in eukaryotes [76-78], clear-cut evidence about the existence of two classes of targets for met emerged from the analysis of HeLa cell double-helical DNA in alkaline Ag+/Cs2SO4 [77]. As mentioned, this gradient separated a heavier fraction (representing about 20% of the total) from a lighter one (representing about 80% of the total). The analytical ultracentrifugation of these fractions in CsCl showed that the heavier, banding at 1.715 g/cm3, contained 53% CG (10% of the total CG), whereas the lighter, banding at 1.703 g/cm3, contained 40% CG (32% of the total CG) [77].
In relation to the diversity of the targets, four possible triplets as sites for met recognition were suggested: GCG and CCG, in CG-rich sequences; ACA and ACG, in AT-rich sequences [78]. The hypermethylation of CG-rich sequences was not surprising, as said, because of its statistical character especially if one considered that against a larger C target the probability of methylation would be higher. Instead, the paradoxically significant methylation observed along AT-rich sequences was not to be expected because it would occur against a smaller C target. Since the statistical character of methylation could not be taken into consideration, the existence of specific recognition mechanisms for AT-rich sequences by met was assumed [77].
Another group of experiments performed in HeLa cells showed that the specific methylation of CG-rich sequences (such as those for rRNAs) was maximal in early S and that the specific methylation of AT-rich sequences was maximal in late S [78].
This, on one hand, demonstrated that DNA sequences were replicated and methylated with an order during S [78] and, on the other, suggested that met activity played the role of maintenance modification along CG-rich sequences and the role of de novo modification along AT-rich sequences [78].
Such an important suggestion was in harmony with the fact that the extra-S phase methylation (about 10% of the total) involved some AT-rich sequences along the old and the new chains, while the S phase methylation (about 90% of the total) almost exclusively involved newly replicating chains [41, 73] (in S the maintenance met activity [105] followed at a distance of about 30 min [41] the pol alpha activity [105]). The reasoning made sense because, during S, maintenance modification, involving CpG targets along newly replicating chains, would be “induced” by -CH3 of m5C present in the complementary strand as a genetically encoded signal (a CpG dinucleotide would be methylated on the new chain in the presence of a complementary already methylated GpC dinucleotide on the old chain [41, 42]); in contrast, during the extra-S time, a de novo modification occurring on an ApC dinucleotide, for instance, would not be induced by a signal coming from the complementary TpG dinucleotide which cannot be methylated at all.
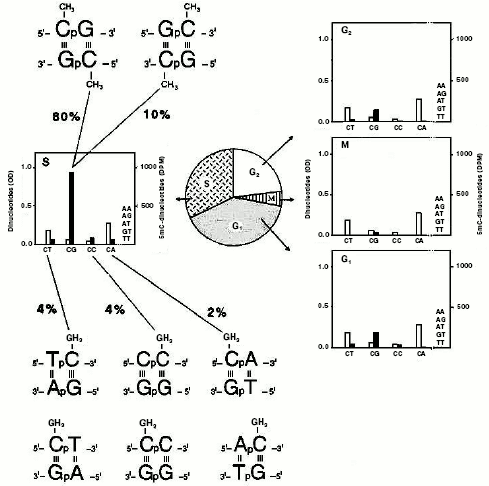
Identification of monomethylated and dimethylated dinucleotide pairs. Sedimentation of CG- and AT-rich sequences in alkaline Ag+/Cs2SO4 suggested the existence of at least four targets for methylation: GCG, CCG, ACA, and ACG [77]. In one of these trinucleotides there was a methylatable CpA; instead, in three of them, the methylatable CpG was repeated. A direct isolation of such restricted targets was required (Fig. 5). This was achieved by exploiting DNase I digests [81, 102, 108] of DNA samples methylated in isolated nuclei of previously synchronized cells with [3H-methyl]SAM [10, 41, 97].
Chromatographic analysis [76, 108] of the material contained in these digests yielded a number of obviously unmethylated dinucleotides (ApA, ApG, ApT, CpT, and TpT) and, in addition, four dinucleotides methylated to a different extent (m5CpT, Cpm5C, m5CpA, and m5CpG). Their methylation level was cell-cycle dependent, in harmony with [41]: in M, few CpGs were methylated; in G1 and G2, methylation of CpGs slightly increased, in comparison with that occurring in M, and there appeared some methylation on CpTs, CpCs, and CpAs; in S, again, a methylation of four dinucleotides (m5CpTs, Cpm5Cs, m5CpAs, and m5CpGs) occurred. In this phase, methylation of CpGs became intensive (90% of the total), accounting for its maintenance character along newly made chains. The occurrence of methylation on CpTs, CpCs, and CpAs accounted instead for a de novo phenomenon which characterized especially the extra-S part of the interphase [41]. Considering the two possible directions of the separated dinucleotides, due to the double helix antiparallelism (the 5´-3´ and 3´-5´ directions could not be distinguished chromatographically), the analysis actually regarded the identification of eight words written in the genome in terms of dinucleotide pairs: 5´-m5CpG-3´/3´-Gpm5C-5´ and 5´-Gpm5C-3´/3´-m5CpG-5´, 5´-Tpm5C-3´/3´-ApG-5´ and 5´-m5CpT-3´/3´-GpA-5´, 5´-Cpm5C-3´/3´-GpG-5´ and 5´-m5CpC-3´/3´-GpG-5´, 5´-m5CpA-3´/3´-GpT-5´ and 5´-Apm5C-3´/3´-TpG-5´. In sum, if the CpG targets only allowed a final dimethylation of dinucleotide pairs, as expected from semiconservative methylation [41, 42] (Fig. 2, b and d), the other combinations always led to a monomethylation of dinucleotide pairs.Fig. 5. Methylated “words” in eukaryotic genomic DNA. Experiments with synchronized HeLa cells demonstrated that, in double helix (characterized by antiparallelism of complementary chains) two symmetrically dimethylated palindromic dinucleotide pairs and six asymmetrically monomethylated non-palindromic dinucleotide pairs were detected [120]. DNA was labeled in isolated nuclei with [14C-methyl]SAM, as for Fig. 2c [120]: after extraction, in the middle of each cell-cycle phase (G1, S, G2, and M), it was digested with pancreatic DNase I to chromatographically separate various dinucleotides, as in [108]. The white columns show the dinucleotide molar concentration, expressed in OD at 256 nm; the black columns, expressed in DPM (decompositions per minute), show the radioactivity of their m5C.
Location of methylatable words in restriction gene mini-maps. In agreement with the design of the eukaryotic gene showing the intermittence of coding hypomethylated and uncoding hypermethylated regions (Fig. 4), it was observed that the calcitonin gene (representative of about thirty fully sequenced and m5C-regulated TSs [26]) was heavily methylatable upstream and slightly methylatable downstream, with at least four pairs of dinucleotides: two monomethylatable (5´-Tpm5C-3´/3´-ApG-5´; 5´-Cpm5C-3´/3´-GpG-5´) in exons and two dimethylatable (5´-m5CpG-3´/3´-Gpm5C-5´; 5´-Gpm5C-3´/3´-m5CpG-5´) in promoter and introns, respectively [46]. The average quantitative distribution of these pairs (that corresponded to those previously isolated from the DNAse I digests [81, 97]), was similar in HKs and TSs [26, 46], with the exception that in the case of TSs, from 5´ to 3´, the monomethylatable dinucleotide pairs clearly increased, while the dimethylatable clearly decreased [46].
Sequencing of m5C in the promoter of the transglutaminase gene. Using the model of hTGc (human transglutaminase gene) [48] and the method based on bisulfite conversion of C to T residues along a DNA filament [47], the following analysis showed that m5C, behaving as bisulfite-independent base [109, 110], can be sequenced directly [27], although its molar proportion is low [4, 33-35]. The hTGc gene was chosen as one of those regulated by methylation (a number of m5Cs was assigned to it by methylation-sensitive restrictases [48]) and a sequence limited at both ends by a CpG dinucleotide in which at least five CpGs are present with a maximal distance of 100 bp between each other was defined as CpG-enriched domain [27]. This allowed to distinguish, in the 1665 bp long hTGc promoter [111], three CpG-rich domains [27] (Fig. 6): the first (330 bp), close to 5´, contained 12 CpGs corresponding to an average frequency of 3.63% with respect to the total number of nucleotides; the second (227 bp), roughly located in the middle of the promoter, contained eight CpGs corresponding to an average frequency of 3.52%; the third (264 bp), close to 3´ (including 70 bp of the 73 bp long 5´-UTR), contained 31 CpGs corresponding to an average frequency of 11.74%.
In leukocytes and lymphocytes, where hTGc is silent [48], out of the three CpG-rich domains, only the first two were found to be methylated, while the third, on the 3´-side, did not present any m5Cs: the 11 CpGs of domain 1 and the seven CpGs of domain 2 were methylated 100%; the 31 CpGs of domain 3 were instead unmethylated 100% (in this domain the 5´-UTR, also resulting 100% unmethylated, was almost entirely included) [27]. The lack of m5C in domain 3 was expected, also in agreement with the conventional definition of unmethylated CpG-rich island (where the intrafilament CpG/GpC ratio has to be higher than 0.6); but the occurrence of methylation in all CpGs of domains 1 and 2 was unexpected. Domain 1 contains 79 Cs and 11 CpGs out of 330 bases; domain 2 contains 44 Cs and seven CpGs out of 227 bases; domain 3 contains 116 Cs and 31 CpGs out of 264 bases.Fig. 6. Methylation language of promoter in repressed hTGc gene [27]. The dashed circles show the CpGs external to the sequenced bisulfite-converted fragments A and B; the closed circles show the methylated CpGs clustered in domains 1 and 2; the open circles show the unmethylated CpGs clustered in domain 3; the open circles marked with x show the CpGs that were present in the sequence described by [111] but not found in [27]. The dashed line in fragment A shows a 24 bp long element which may be repeated. The dotted line between fragments A and B shows the promoter part which was not investigated in [27]. Starting from site +1, fragment B (and domain 3) overlapped a 5´-UTR, also unmethylated.
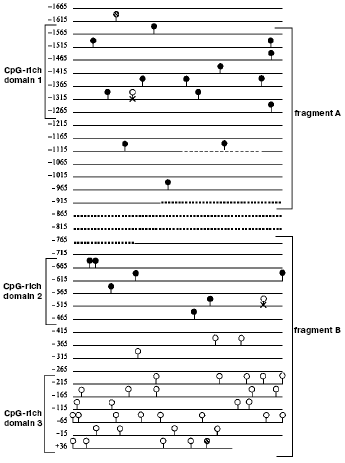
Compared with the average 5%-methylation of human genomic DNA [112], methylation of domains 1 and 2 corresponded to 15.18 and 18.18%, respectively [27]. This confirmed the idea that the promoter of an inactive gene should be characterized by hypermethylation [10, 26, 46]. In fact, the staminal HUVEC cells join their hTGc gene activity with a loss of m5C along the hTGc promoter domain 1, at least at the -1380, -1349, -1338, and -1320 CpG sites (Virgili, Cacciamani, and Volpe, unpublished). This was an appropriate example of direct base-sequence analysis showing the inverse correlation between the activity of a gene and the methylation of its promoter.
Remarks on mechanisms switching-on and -off transcription. What hypothesis could one deduce on the role of methylation in regulating gene activity from the picture described herewith? In harmony with the existing literature [26, 46], analysis based on the use of methylation-sensitive restriction endonucleases further suggested that, in human lymphocytes and monocytes, an inverse correlation between methylation of hTGc promoter and expression of hTGc gene takes place [48]. In addition, the study based on direct sequencing of m5C showed that in the same cell species, in the absence of hTGc activity, hTGc promoter is hypermethylated in its CpG-rich domains 1 and 2 and unmethylated in its CpG-rich domain 3 [27]. This neat division of hTGc promoter into two parts, one methylated and the other not, should deserve particular attention in correlating DNA heterochromatization with m5CpG-binding proteins [100, 113, 114]. In the case of the hTGc promoter [27], one could assume that they may transform domains 1 and 2 into heterochromatic structures and that, in turn, these structures would be sufficient to prevent transcription (methylation would be relevant to the repressor complex able to bind the promoter 5´-end).
But why, in hTGc promoter, the unmethylated domain 3, remaining in a normal relaxed state, should be unable to interfere with the basal transcription machinery at the promoter 3´-end? This question remains open. Combined with the inactivity of hTGc gene in leukocytes [27], the postulated heterochromatization of domains 1 and 2, in its promoter, could acquire great interest if considered within the framework of the repair-modification scheme (Fig. 3b). By causing an at random DNA de-methylation [82, 115], it would lead to switch-on of previously silent genes, as in the case of those for alpha and beta chains of hemoglobin (Hb) in Friend erythroleukemia cells [46, 116]. In such a case the switch-on and -off of transcription, implying conformational changes of the two Hb gene promoters, corresponded, first, to their de-methylation and, then, to their re-methylation [46, 116, 117]. In other words, following the damages caused to double helix by ionizing radiations [82, 115], excision-repair is sufficient to guarantee a complete reconstruction of previously unmethylated regions; however, to complete the reconstruction of previously methylated regions, a coupling has to occur between excision-repair (re-establishing the basic code in A, G, T, and C) and met (re-establishing the position of m5C among A, G, T, and C) [11]. The coupling, properly meaning repair-modification, takes place in S [118]: a specific DNA polymerase, crucial for excision-repair, is active throughout the whole cycle [119]; met is highly active in S and almost inactive during the major part of the extra-S time [105]. Once de-methylated through excision-repair, given genes--silent when methylated--could be expressed if their transcription is inversely correlated with their methylation.
This review was written to celebrate the 70th birthday of Prof. Boris F. Vanyushin whom I greatly admired for his pioneering and brilliant discoveries regarding DNA methylation in eukaryotes. His studies, performed without interruptions for about forty years, have always exhibited an original character. His elegant demonstrations, contributing to show that m5C is the sole modified base in DNA of animals and plants, represented a milestone in Genomics of Eukaryotes, since until them it was only known that in bacteria both m6A and m5C participate in restriction-modification reactions. Particularly relevant were the experiments performed by him on age-dependent and tissue-dependent DNA methylation, on methylation of mtDNA, and on the involvement of Okazaki fragments in maintenance methylation.
I wish to express my gratitude to Prof. Tamilla Eremenko, of the former Institute of Experimental Medicine of CNR in Rome, for fruitful discussion during the preparation of this manuscript. Financial support by the University of Rome “Tor Vergata” is also acknowledged.
REFERENCES
1.Doskocil, J., and Sormova, Z. (1965) Biochim.
Biophys. Acta, 95, 513-515.
2.Vanyushin, B. F., Belozersky, A. N., Kokurina, N.
A., and Kadirova, D. X. (1968) Nature, 218,
1066-1067.
3.Scarano, E., Iaccarino, M., Grippo, P., and
Winckelmans, D. (1965) J. Mol. Biol., 14, 603-607.
4.Vanyushin, B. F., Tkacheva, S. G., and Belozersky,
A. N. (1970) Nature, 225, 948-949.
5.Palmer, B. R., and Marinus, M. G. (1994)
Gene, 143, 1-12.
6.Raleigh, E. A., and Wilson, G. (1986) Proc.
Natl. Acad. Sci. USA, 83, 9070-9074.
7.Heitman, J., and Model, P. (1987) J.
Bacteriol., 169, 3243-3250.
8.Raleigh, E. A., Murray, N. E., Revel, H.,
Blumenthal, R. M., Wastaway, D., Reith, A. D., Rigby, P. W. J., Elhai,
J., and Hanahan, D. (1988) Nucleic Acids Res., 16,
1563-1575.
9.Marinus, M. G. (1987) Ann. Rev. Biochem.,
21, 113-131.
10.Volpe, P., and Eremenko, T. (1974) FEBS
Lett., 44, 121-126.
11.Volpe, P., and Eremenko, T. (1989) Cell
Biophys., 15, 41-60.
12.Feng, T. Y., and Chang, K. S. (1984) Proc.
Natl. Acad. Sci. USA, 81, 3438-3442.
13.Volpe, P., and Cascio, O. (1993) Phys. Proc.
Acad. Lincei, 4, 345-357.
14.Volpe, P., and Cascio, O. (1994) Phys. Proc.
Acad. Lincei, 5, 79-87.
15.Franchina, M., Hooper, J., and Kay, P. H. (2001)
Int. J. Biochem. Cell Biol., 33, 1104-1115.
16.Hattman, S., Brooks, J. E., and Masurekar, M.
(1978) J. Mol. Biol., 126, 367-380.
17.Messer, W., and Noyer-Weidnetr, M. (1988)
Cell, 54, 735-737.
18.Marinus, M. G., and Morris, N. R. (1974) J.
Mol. Biol., 85, 309-322.
19.Marinus, M. G., and Morris, N. R. (1973) J.
Bacteriol., 114, 1143-1150.
20.May, M. S., and Hattman, S. (1975) J.
Bacteriol., 123, 768-770.
21.Arber, W. (1965) J. Mol. Biol., 11,
247-256.
22.Arber, W., and Linn, S. (1969) Ann. Rev.
Biochem., 38, 467-500.
23.Arber, W. (1974) Progr. Nucl. Acids Res. Mol.
Biol., 14, 1-37.
24.Jacob, F., and Monod, J. (1961) J. Mol.
Biol., 3, 318-356.
25.Kruger, D., Schroeder, C., Reuter, M., Bogdarina,
I., Buryanov, Y., and Bickle, T. (1985) Eur. J. Biochem.,
150, 323-330.
26.Volpe, P., Iacovacci, P., Butler, R. H., and
Eremenko, T. (1993) FEBS Lett., 329, 233-237.
27.Cacciamani, T., Virgili, S., Centurelli, M.,
Bertoli, E., Eremenko, T., and Volpe, P. (2002) Gene,
297, 103-112.
28.Holliday, R., and Pugh, J. E. (1975)
Science, 187, 226-232.
29.Sutter, D., and Doerfler, W. (1980) Proc.
Natl. Acad. Sci. USA, 77, 253-256.
30.Razin, A., and Riggs, A. D. (1980)
Science, 210, 604-610.
31.Tentravahi, V., Guntaka, R. V., Erlanger, B. F.,
and Miller, O. J. (1981) Proc. Natl. Acad. Sci. USA, 78,
489-493.
32.Liau, M. C., Chang, C. F., Saunders, G. F., and
Tsai, Y. H. (1981) Arch. Biochem. Biophys., 208,
261-272.
33.Vanyushin, B. F., Mazin, A. L., Vasilyev, V. K.,
and Belozersky, A. N. (1973) Biochim. Biophys. Acta, 299,
397-403.
34.Vanyushin, B. F., Nemirovsky, L. E., Klimenko, V.
V., Vasilyev, V. K., and Belozersky, A. N. (1973) Gerontologia,
19, 138-152.
35.Romanov, G. A., and Vanyushin, B. F. (1981)
Biochim. Biophys. Acta, 653, 204-218.
36.Vanyushin, B. F., and Belozersky, A. N. (1959)
Dokl. Akad. Nauk USSR, 129, 944-946.
37.Urieli-Shove, S., Gruenbaum, Y., Sedat, J., and
Razin, A. (1982) FEBS Lett., 146, 148-152.
38.Achwal, C. W., Ganguly, P., and Chandra, H. S.
(1984) EMBO J., 3, 263-266.
39.Gowher, H., Leismann, O., and Jeftsch, A. (2000)
EMBO J., 19, 6918-6923.
40.Lyko, F., Ramsahoye, B. H., and Jaenisch, R.
(2000) Nature, 408, 538-540.
41.Geraci, D., Eremenko, T., Cocchiara, R.,
Granieri, A., Scarano, E., and Volpe, P. (1974) Biochem. Biophys.
Res. Commun., 57, 353-361.
42.Bird, A. P. (1978) J. Mol. Biol.,
118, 49-60.
43.Geraci, D., Eremenko, T., Granieri, A., Scarano,
E., and Volpe, P. (1973) Congr. Ital. Biochem. Soc., Trieste,
Abstr., p. 291.
44.Volpe, P., Menna, T., and Eremenko, T. (1976)
Bull. Mol. Biol. Med., 1, 18-28.
45.Volpe, P. (1976) Horizons Biochem.
Biophys., 2, 285-340.
46.Volpe, P., Esposito, C., Iacovacci, P., Butler,
R. H., and Eremenko, T. (1993) Macromol. Funct. Cell, 7,
59-71.
47.Clark, S. J., Harrison, J., Paul, C. L., and
Frommer, M. (1994) Nucleic Acids Res., 22, 2990-2997.
48.Lu, S., and Davies, P. J. A. (1997) Proc.
Natl. Acad. Sci. USA, 94, 4692-4697.
49.Kudryashova, I. B., and Vanyushin, B. F. (1976)
Biokhimiya, 41, 1106-1115.
50.Vanyushin, B. F., Kadirova, D. K., Karimov, K.
K., and Belozersky, A. N. (1971) Biokhimiya, 36,
1251-1258.
51.Buryanov, Y. I., Eroshina, N. V., Vagabova, L.
M., and Ilyin, A. V. (1972) Dokl. Akad. Nauk USSR,
205, 700-703.
52.Gorodsky, M. A., Hattman, S., and Plager, G. L.
(1973) J. Cell Biol., 56, 697-701.
53.Cummings, D. J., Tait, A., and Goddard, J. M.
(1974) Biochim. Biophys. Acta, 374, 1-11.
54.Kirnos, M. D., Merkulova, N. A., Borkhsenius, S.
N., and Vanyushin, B. F. (1980) Dokl. Akad. Nauk USSR,
255, 225-227.
55.Buryanov, Y. I., Ilyin, A. V., and Skryabin, G.
K. (1970) Dokl. Akad. Nauk USSR, 195, 728-730.
56.Pakhmova, M. V., Zaitseva, G. N., and Belozersky,
A. N. (1968) Dokl. Akad. Nauk USSR, 182, 712-715.
57.Rae, P. M. M. (1976) Science, 194,
1062-1064.
58.Wyatt, G. R. (1951) Biochem. J.,
48, 584-590.
59.Adams, R. L. P., MacKay, E. L., Graig, L. M., and
Burdon, R. H. (1979) Biochim. Biophys. Acta, 563,
72-81.
60.Lyko, F., Ramsahoye, B. H., Kashevsky, H., Tudor,
M., Mastrangelo, M. A., Orr-Weaver, T. L., and Jaenish, R. (1999)
Nat. Genet., 23, 363-366.
61.Bestor, T. H., and Ingram, V. M. (1983) Proc.
Natl. Acad. Sci. USA, 80, 5559-5563.
62.Borek, E. (1975) Science, 190,
591-593.
63.Evans, H. H., Evans, T. E., and Littman, S.
(1973) J. Mol Biol., 74, 563-574.
64.Kappler, J. A. (1970) J. Cell Physiol.,
75, 21-34.
65.Adams, R. L. P., and Hogarth, C. (1973)
Biochim. Biophys. Acta, 331, 214-220.
66.Adams, R. L. P. (1974) Biochim. Biophys.
Acta, 335, 365-373.
67.Volpe, P., Granieri, A., and Eremenko, T. (1975)
12th Meet. Ital. Soc. Biophys. Mol. Biol., Pavia, Abstr., p.
49.
68.Demidkina, N. P., Kiryanov, G. I., and Vanyushin,
B. F. (1979) Biokhimyia, 44, 1416-1426.
69.Bashkite, E. A., Kirnos, M. D., Kiryanov, G. I.,
Alexandrushkina, N. I., and Vanyushin, B. F. (1980) Biokhimyia,
45, 1448-1456.
70.Kiryanov, G. I., Kirnos, M. D., Demidkina, N. P.,
Alexandrushkina, N. I., and Vanyushin, B. F. (1980) FEBS Lett.,
112, 225-228.
71.Vanyushin, B. F. (1984) Curr. Top. Microbiol.
Immunol., 108, 99-114.
72.Eremenko, T., and Volpe, P. (1976) 13th Meet.
Ital. Soc. Biophys. Mol. Biol., Albano laziale, Abstr., 23.
73.Eremenko, T., Palitti, F., Morelli, F.,
Whitehead, E. P., and Volpe, P. (1985) Mol. Biol. Rep.,
10, 177-182.
74.Lima de Faria, A. (1969) in Handbook of
Molecular Cytology (Lima de Faria, A., ed.) Wiley, New York, pp.
277-283.
75.Comings, D. I. (1972) Exp. Cell Res.,
74, 383-391.
76.Grippo, P., Iaccarino, M., Parisi, E., and
Scarano, E. (1968) J. Mol. Biol., 36, 195-208.
77.Eremenko, T., Granieri, A, and Volpe, P. (1978)
Mol. Biol. Rep., 4, 163-170.
78.Eremenko, T., Granieri, A., and Volpe, P. (1978)
Mol. Biol. Rep., 4, 237-240.
79.Eremenko, T., Timofeeva, M. Y., and Volpe, P.
(1980) Mol. Biol. Rep., 6, 131-136.
80.Eremenko, T., Granieri, A., Scarano, E., and
Volpe, P. (1975) 10th FEBS Meet., Paris, Abstr., 130.
81.Scarano, E. (1969) Ann. Embryol. Morphol.,
Suppl. 1, pp. 55-61.
82.Volpe, P., and Eremenko, T. (1985) in Proc.
16th FEBS Congr. (Ovcinnikov, Y. A., ed.) Vol. 3, Science Press,
Utrecht, pp. 123-129.
83.Battacharya, S. K., Ramchandani, S., Cervoni, N.,
and Szyf, M. (1999) Nature, 397, 579-583.
84.Volpe, P., and Eremenko, T. (1994) UNESCO
Techn. Rep., 19, 27-43.
85.Georgyev, G. P., and Mantieva, V. I. (1962)
Biochim. Biophys. Acta, 61, 153-162.
86.Volpe, P., and Giuditta, A. (1967) Nature,
216, 154-155.
87.Penman, S., Vesco, C., and Penman, M. (1968)
J. Mol. Biol., 34, 49-62.
88.Eremenko, T., Benedetto, A., and Volpe, P. (1972)
Nature New Biol., 237, 114-116.
89.Georgyev, G. P. (1969) J. Theor. Biol.,
25, 227-231.
90.Volpe, P., and Eremenko, T. (1973) Meth. Cell.
Biol., 6, 113-126.
91.Maclean, M., and Hilder, V. A. (1977) Int.
Rev. Cytol., 48, 54-97.
92.Darnell, J. E., Jelenek, W. R., and Molloy, G. R.
(1974) Science, 181, 1215-1221.
93.Eremenko, T., and Volpe, P. (1975) Eur. J.
Biochem., 52, 203-210.
94.Mandel, J. L., and Chambon, P. (1979) Nucleic
Acids Res., 7, 2081-2092.
95.Chambon, P. (1981) Sci. Amer., 244,
60-71.
96.Crick, F. (1979) Science, 204,
264-271.
97.Volpe, P., and Eremenko, T. (1973) Eur. J.
Biochem., 32, 227-232; 9th Int. Congr. Biochem.,
Stockholm, Abstr., p. 195.
98.Eden, S., and Cedar, H. (1994) Curr. Opin.
Gen. Devel., 4, 255-259.
99.Razin, A. (1998) EMBO J., 17,
4905-4908.
100.Bird, A. P., and Wolffe, A. P. (1999)
Cell, 99, 451-454.
101.Volpe, P., Menna, T., and Eremenko, T. (1978)
11th Eur. Tumor Virus Meet., Balatonfured, Abstr., 4,
194.
102.Volpe, P., and Eremenko, T. (1982) Macromol.
Funct. Cell, 2, 179-195.
103.Eremenko, T., and Volpe, P. (1984) FEBS
Lett., 169, 211-214; 173, 233-237.
104.Doerfler, W., Toth, M., Kochanek, S., Achten,
S., Freisem-Rabien, U., Behn-Krappa, A., and Orend, G. (1990) FEBS
Lett., 268, 329-333.
105.Delfini, C., Crema, A. L., Alfani, E.,
Eremenko, T., and Volpe, P. (1987) FEBS Lett., 210,
17-21; (1988) 227, 85.
106.Adams, R. L. P., Hill, J., McGarvey, M., and
Rinaldi, A. (1989) Cell Biophys., 15, 113-126.
107.Meehan, R., Antequera, F., Lewis, J., McLeod,
D., McKay, S., Kleiner, E., and Bird, A. P. (19990) Phil. Trans.
Royal Soc. London, B326, 199-205.
108.Augusti-Tocco, G., Carestia, C., Grippo, P.,
Parisi, E., and Scarano, E. (1968) Biochim. Biophys. Acta,
155, 8-18.
109.Frommer, M., McDonald, L. E., Millar, D. S.,
Collins, C., Watt, F., Grigg, G. W., Molloy, P. L., and Paul, C. L.
(1992) Proc. Natl. Acad. Sci. USA, 89, 1827-1831.
110.Clark, S. J., Harrison, J., and Frommer, M.
(1995) Nat. Genet., 10, 20-27.
111.Lu, S., Saydak, M., Gentile, V., Steins, J. P.,
and Davies, J. A. (1995) J. Biol. Chem., 270,
9748-9756.
112.Razin, A., and Cedar, H. (1977) Proc. Natl.
Acad. Sci. USA, 74, 2725-2728.
113.Nan, X., Ng, H. H., Johnson, C. A., Laherty, C.
D., Turner, B. M., Eisenman, R. N., and Bird, A. P. (1998)
Nature, 393, 386-389.
114.Wade, P. A., Gegonne, A., Jones, P. L.,
Ballestar, E., Aubry, F., and Wolffe, A. P. (1999) Nat. Genet.,
23, 62-66.
115.Volpe, P., and Eremenko, T. (1995) Rad.
Prot. Dosim., 62, 19-22.
116.Volpe, P., Parasassi, T., Sapora, O., Ravagnan,
G., and Eremenko, T. (1999) Int. J. Rad. Med., 1,
78-89.
117.Volpe, P. (2001) Int. J. Rad. Med.,
3, 123-137.
118.Eremenko, T., Delfini, C., Crema, A. L.,
Alfani, E., and Volpe, P. (1988) Macromol. Funct. Cell,
5, 37-41.
119.Delfini, C., Alfani, E., De Venezia, V.,
Oberholtzer, G., Tomasello, C., Eremenko, T., and Volpe, P. (1985)
Proc. Natl. Acad. Sci. USA, 82, 2220-2224.
120.Volpe, P., and Eremenko, T. (1978) 14th Int.
Congr. Gen., Moscow, Abstr., 1, 194.