
|
Methylation of the Tumor Suppressor Gene RASSF1A in Human TumorsG. P. Pfeifer1* and R. Dammann21Department of Biology, Beckman Research Institute, City of Hope Cancer Center, Duarte, California 91010, USA; fax: 626-358-7703; E-mail: gpfeifer@coh.org2AG Tumorgenetik der Medizinischen Fakultät, Martin-Luther-Universität Halle-Wittenberg, Halle/Saale 06097, Germany * To whom correspondence should be addressed.
|
Received November 10, 2004
Loss of heterozygosity of a segment at 3p21.3 is frequently observed in lung cancer and several other carcinomas. We have identified the Ras-association domain family 1A gene (RASSF1A), which is localized at 3p21.3 in a minimum deletion sequence. De novo methylation of the RASSF1A promoter is one of the most frequent epigenetic inactivation events detected in human cancer and leads to silencing of RASSF1A expression. Hypermethylation of RASSF1A was frequently found in most major types of human tumors including lung, breast, prostate, pancreas, kidney, liver, cervical, thyroid and many other cancers. The detection of RASSF1A methylation in body fluids such as serum, urine, and sputum promises to be a useful marker for early cancer detection. The functional analysis of RASSF1A reveals a potential involvement of this protein in apoptotic signaling, microtubule stabilization, and cell cycle progression.
KEY WORDS: RASSF1A, DNA methylation, tumor suppressor, cancer
Abbreviations: RASSF1) Ras-association domain family 1; NORE1) novel Ras effector 1; LOH) loss of heterozygosity; RA domain) RalGDS/AF6 Ras-association domain; aa) amino acid; HPV) human papilloma virus; ATM) ataxia telangiectasia mutated; C1) protein kinase C conserved region 1; SCLC) small cell lung cancer; NSCLC) non-small cell lung cancer.
Several regions on chromosome 3 including 3p12, 3p14, 3p21, and 3p24-25,
frequently undergo loss of heterozygosity in human solid tumors [1]. Potential tumor suppressor genes in these
chromosomal segments include the van Hippel-Lindau disease (VHL)
gene at 3p25 [2], the gene FHIT at 3p14.2
[3], and the DUTT1/ROBO1 gene at 3p12 [4]. At 3p21.3, heterozygous and homozygous deletions
have been observed in several cancer cell lines and in primary lung
tumors [1, 5-8]. A region of minimum homozygous deletion at 3p21.3
spans approximately 120 kb [9] and eight genes are
located in this region [10]. However, mutations in
these eight genes are rarely detected in tumors. Recently, we and
others have cloned and characterized the RASSF1 gene, which is
one of the eight genes residing in the common deletion area at 3p21.3
[10-12]. The C-terminus of
RASSF1 is homologous to the mammalian Ras-effector protein NORE1 [13] and encodes a Ras-association domain. Thus, the
gene has been named Ras-association domain family 1 gene [11]. The three major splice variants RASSF1A,
RASSF1C, and RASSF1F are transcribed from two different
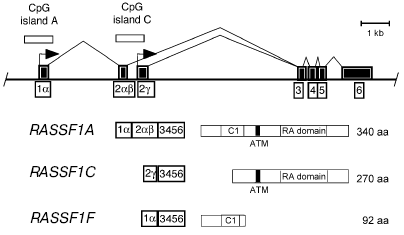
CpG islands, which are separated by approximately 3.5 kb (Fig. 1). RASSF1A and RASSF1C have the four
C-terminal exons in common (Fig. 1). The N-terminus
of RASSF1A has high homology to the protein kinase C conserved region 1
(C1), which contains a zinc finger motif [14]. The
RASSF1F transcript skips exon 2alphabeta and
encodes a truncated polypeptide [12].
RASSF1A transcripts are frequently missing in several cancer
cell lines and in primary tissues [11, 15, 16]. Silencing of
RASSF1A is due to de novo methylation of its CpG island
promoter. RASSF1A expression can be reactivated by treating
cells with inhibitors of DNA methyltransferase such as
5-aza-2´-deoxycytidine [11]. In this review,
we summarize published work on the epigenetic inactivation and presumed
biochemical function of RASSF1A in normal and malignant cells.
Fig. 1. Map of the RASSF1 locus. The two major promoters of RASSF1 (arrows) are located in two separate CpG islands (open squares). Three major isoforms (RASSF1A, RASSF1C, and RASSF1F) are made by alternative promoter usage and alternative splicing of the exons (black boxes). The cDNA of RASSF1A codes for a protein of 340 amino acids with a calculated molecular mass of 38.8 kD. Transcript RASSF1C initiates in exon 2gamma located at CpG island C. The cDNA encodes a 270 amino acid protein with a molecular mass of 31.2 kD. The RASSF1F transcript skips exon 2alphabeta and encodes a truncated peptide of 92 amino acids. The protein domains indicated are: C1, diacylglycerol/phorbol ester (protein kinase C conserved region 1) domain; RA, RalGDS/AF6 Ras-association domain; and ATM, putative ATM phosphorylation site.
FREQUENT METHYLATION OF THE RASSF1A GENE IN HUMAN TUMORS
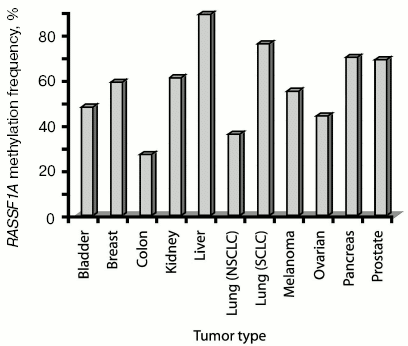
Silencing of genes by DNA methylation is a common phenomenon occurring in human cancer cells [17]. It has been reported that promoter hypermethylation plays an essential role in loss of function of certain tumor suppressor genes [18]. Aberrant promoter methylation of RASSF1A has been frequently detected in several tumor types (see Fig. 2 for examples). Alphabetically, in bladder cancer, a high frequency of RASSF1A methylation was observed and was correlated with advanced tumor stage and poor prognosis [19-22]. In brain cancer, RASSF1A methylation is common in neuroblastoma, glioblastoma, and medulloblastoma [23-30]. Methylation of RASSF1A was frequently found in breast cancer and was detected even in serum of breast cancer patients [12, 31-39]. In cervical cancer, Kuzmin et al. have reported an inverse correlation between methylation of RASSF1A and human papilloma virus infection [40]. In cholangiocarcinoma, 69% of the cases had RASSF1A promoter hypermethylation [41]. In colon cancer, RASSF1A methylation is less frequent [42-45]. In 52% of esophageal squamous cell carcinoma, RASSF1A methylation was reported [46]. In gastric cancers, RASSF1A hypermethylation was more frequently found in tumors of advanced stage and RASSF1A methylation was rarely detected in non-carcinoma tumor samples [47-52]. In head and neck cancer, RASSF1A methylation frequency is less than 20% [53-56]. There is an inverse correlation between RASSF1A methylation and human papilloma virus (HPV) infection in these tumors [55]. In hepatocellular carcinoma, RASSF1A methylation frequencies are very high, sometimes occurring in up to 100% of the cases [57-60]. In Hodgkin's lymphoma, 65% of the tumors showed RASSF1A methylation [61]. Several reports have demonstrated a high frequency of RASSF1A methylation in kidney tumors [42, 62-65]. Intensive methylation (>70%) of RASSF1A was reported in small cell lung cancer (SCLC). In about 40% of non-small cell lung cancers (NSCLCs), RASSF1A hypermethylation is found [11, 12, 30, 32, 35, 66-77]. In malignant mesothelioma, 32% of RASSF1A inactivation was found and this event correlated with the presence of SV40 DNA in these tumors [78, 79]. For melanoma, frequent RASSF1A methylation has been reported [80-82]. In nasopharyngeal carcinoma, aberrant RASSF1A methylation was frequently (>60%) observed [83-88]. In 40% of osteosarcomas, hypermethylation of RASSF1A occurred [89]. In ovarian cancer, frequent RASSF1A methylation was reported in several studies [32, 42, 90-92]. In endocrine tumors of the pancreas, the frequency of RASSF1A inactivation was higher compared to pancreatic adenocarcinoma [93, 94]. In childhood tumors, RASSF1A methylation was found in Wilms' tumor, medulloblastoma, retinoblastoma, rhabdomyosarcoma, neuroblastoma, hepatoblastoma, leukemia, pancreatoblastoma, adrenocortical carcinoma, and lymphoma [44, 95-97]. In prostate cancer, a high frequency (>70%) of RASSF1A methylation was reported in several studies. In testicular germ cell tumors, studies have shown that RASSF1A methylation occurs frequently [98-101]. In thyroid cancers, RASSF1A hypermethylation was also frequently found [102, 103].
Methylation of RASSF1A has been rarely detected in normal tissues, however methylation was also found in some non-cancerogenous tissue specimen mostly adjacent to the tumors. This methylation could represent infiltration of tumor cells into normal tissue, or could be related to a general defect leading to carcinogenesis.Fig. 2. Methylation frequency of RASSF1A in several common types of human tumors. The frequency (% of cases with methylated RASSF1A gene) of RASSF1A methylation for several of the most common human tumors is shown. Data were used when at least three different studies analyzed RASSF1A methylation in one type of tumor and the percentages calculated represent the average methylation frequency of the independent studies (see text for literature citations).
METHYLATION OF RASSF1A AS A BIOMARKER FOR TUMOR
DIAGNOSIS
The detection of cancer at early stages by noninvasive methods may be aided by the development of cancer-specific biomarkers for the detection of these biomarkers in body fluids. Methylation-specific polymerase chain reaction (PCR) (methylation-specific PCR, MSP) for detection of promoter hypermethylation has been used in several pilot studies to detect cancer cell DNA in bodily fluids [104, 105].
Several studies have analyzed the methylation of the RASSF1A promoter in bodily fluids of cancer patients. In nasopharyngeal carcinoma, Wong et al. found a RASSF1A methylation frequency of only 5% using MSP out of sera, whereas 65% of the primary tumors showed hypermethylation [87]. In four out of 14 (29%) lung cancer patients who showed hypermethylation of RASSF1A in the primary tumor, the bronchoalveolar lavages were positive for hypermethylated RASSF1A [77]. In serum of patients with NSCLC, Ramirez et al. detected a high frequency (34%) of RASSF1A methylated DNA and a correlation between methylation in tumor tissue and serum (p = 0.0001) [30].
Sputum of lung cancer patients was investigated for hypermethylated DNA of tumor suppressor genes [35, 106]. Honorio et al. found that hypermethylation of RASSF1A was detectable in 50% of SCLC and in 21% of NSCLC sputum samples [35]. In 50% of the sera and tumors of glioblastoma patients, RASSF1A methylation was detected [30]. RASSF1A methylation was investigated in the urine of patients with bladder and kidney cancer [21, 22, 64]. In 19 of 23 (82%) of patients with RASSF1A methylation in bladder cancer, Dulaimi et al. detected RASSF1A hypermethylation in the corresponding urine samples [22]. Chan et al. detected methylation of RASSF1A in seven out of 14 (50%) urine specimens and all positive probes showed also epigenetic inactivation of RASSF1A in the corresponding primary bladder tumors [21]. In urine samples of patients with kidney tumors, Battagli et al. found hypermethylated RASSF1A in 25 out of 50 (50%) of the patients [64]. Only for a single case, no RASSF1A methylation was detected in the urine despite a methylated tumor [64]. In sera of breast cancer patients, hypermethylation of RASSF1A was detected in six out of 26 (23%) cases and this was associated with a worse prognosis [37]. RASSF1A methylation was detected in bodily fluids (serum, plasma, and peritoneal fluid) of ovarian cancer patients with 100% specificity [92]. Methylation was undetectable in controls. In summary, RASSF1A hypermethylation is frequently detected in bodily fluids of cancer patients. Methylation analysis of tumor-related genes in bodily fluids is a promising new diagnostic approach to screen putative cancer patients.
THE TUMOR SUPPRESSOR FUNCTION OF RASSF1A
Given its common epigenetic inactivation in tumors, the question arises if RASSF1A is a bona fide tumor suppressor gene. This test will need to be passed by many genes that are found inactivated in tumors. In other words, the question needs to be addressed if methylation of a gene in a tumor is simply a phenomenon associated with tumorigenesis or if it can have a causative role. This assessment will prove to be difficult in many cases, in particular when germ line or somatic mutations are not observed at any significant frequency in the gene silenced by methylation.
Supporting evidence for a role of a methylation-silenced gene in tumorigenesis can come from mouse models or from functional characterization of the gene/protein. For RASSF1A, we have created a knockout mouse in which exon 1alpha was specifically deleted [107]. This mimics the situation in human tumors in which the isoform RASSF1A but not RASSF1C is missing. RASSF1A-targeted mice were viable and fertile. RASSF1A-/- mice were prone to spontaneous tumorigenesis at advanced age. Whereas only two tumors developed in 48 wild type mice, six tumors were found in 35 RASSF1A+/- mice (p < 0.05) and 13 tumors were found in 41 RASSF1A-/- mice (p < 0.001). The tumors in RASSF1A-targeted mice included lymphomas, lung adenomas, and one breast adenocarcinoma. RASSF1A-/- and wild type mice were treated with the chemical carcinogens, benzo[a]pyrene or urethane, to induce skin tumors and lung tumors, respectively. RASSF1A-/- and RASSF1A+/- mice showed increased tumor multiplicity and tumor size relative to control animals [107]. The data are consistent with a tumor-suppressive role of RASSF1A, which may explain its frequent epigenetic inactivation in human tumors. RASSF1A inactivation in combination with other genetic or epigenetic alterations may produce a more severe tumor susceptibility phenotype.
Several studies have begun to investigate the biochemical function of RASSF1A although we are far from understanding its true role(s). RASSF1A is apparently involved in several growth regulatory and apoptotic pathways (Fig. 3). Ectopic expression of RASSF1A in cancer cell lines, which lack endogenous RASSF1A transcripts, resulted in reduced growth of the cells in vitro and in nude mice [11, 12, 27, 62, 108-110]. Shivakumur et al. have reported that RASSF1A can induce cell-cycle arrest by engaging the Rb family cell-cycle checkpoint. RASSF1A inhibited accumulation of cyclin D1 and the RASSF1A-induced growth arrest could be relieved by ectopic expression of cyclins [111].
The exact involvement of RASSF1A in Ras signaling pathways is unclear. Activated Ras is usually associated with enhanced proliferation, transformation, and cell survival (Fig. 3). However, Ras also induces inhibitory effects on cell proliferation [112, 113] and apoptosis [114-117]. Certain Ras effectors, like RASSF1A, may be specialized to transmit inhibitory growth signals and these inhibitory signaling pathways may need to be inactivated during tumorigenesis. It was shown that RASSF1C binds Ras in a GTP-dependent manner and expression of RASSF1C induced apoptosis [118]. Recent data indicate that in colorectal and pancreatic cancer, the inactivation of RASSF1A and mutational activation of Ras may be mutually exclusive events [93, 119], but in lung cancer this correlation was not significant [67, 72, 120]. In thyroid cancer, RASSF1A methylation occurred more commonly when BRAF was not mutated [103]. The closest homolog of RASSF1, which also encodes a Ras association domain, is the NORE1 (novel Ras effector 1) protein [13, 121]. NORE1 may act as a Ras-regulated tumor suppressor in lung cancer and melanoma [122, 123]. Epigenetic inactivation of NORE1 was seen in several cancers, including lung and kidney cancer [121, 124, 125]. Biochemical experiments have shown that binding of RASSF1A to Ras may require heterodimerization with NORE1, and that RASSF1A can bind to Ras only weakly by itself [126]. RASSF1A and NORE1 may function in the same Ras-regulated pro-apoptotic pathway. Khokhlatchev et al. showed that RASSF1A and NORE1 interact with the pro-apoptotic kinase MST1, which mediates the apoptotic effect of activated Ras [127]. The NORE1/RASSF1-MST1 complex represents a novel Ras-regulated pro-apoptotic pathway [127]. However, it is currently unknown what the upstream input and downstream output signals are that regulate this cascade.Fig. 3. Summary of putative biological roles of RASSF1A. The RASSF1A tumor suppressor, a putative Ras effector, induces apoptosis through its interaction with the pro-apoptotic kinase MST1. RASSF1A regulates cellular integrity and mitotic progression through interaction with microtubules and CDC20 thus inhibiting the anaphase-promoting complex. RASSF1A inhibits the accumulation of cyclin D1 and may inhibit cell cycle progression at the G1/S transition. The question marks indicate that details of the respective pathway are not understood.
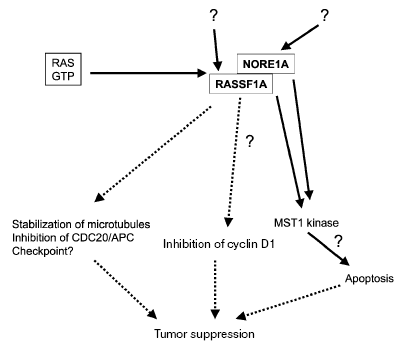
Recently, several groups have reported that RASSF1A is a microtubule-binding protein, which regulates mitotic progression [128-132]. We have shown that RASSF1A co-localizes with microtubules in interphase cells and with spindles and centrosomes during mitosis [128]. RASSF1A has a strong activity that protects cells against microtubule depolymerization induced by nocodazole or ice treatment in vivo. RASSF1A-/- cells were much more sensitive to nocodazole induced microtubule destruction than wild type cells [128]. Overexpression of RASSF1A induced mitotic arrest at metaphase with aberrant mitotic cells [128]. These results were confirmed by several other groups [129-132]. A deletion mutant of RASSF1A, which lacks the microtubule association domain is defective for the ability to promote cell cycle arrest and partially inhibits RASSF1A induced cell death [130]. Song et al. have reported that RASSF1A regulates mitosis by inhibiting the anaphase promoting complex (APC) through Cdc20 and induces G2-M arrest at pro-metaphase [129]. The function of RASSF1A is independent of the protein Emi1 (early mitotic inhibitor 1) and therefore Song et al. proposed that RASSF1A acts in early pro-metaphase, to prevent the degradation of mitotic cyclins and to delay mitotic progression [129]. RASSF1A can regulate microtubule stability and control mitotic progression, presumably by modulating centrosome and spindle function and by regulating the APC complex. Thus, RASSF1A may function as a tumor suppressor through controlling mitotic cell division [133, 134]. However, the exact mechanism of its biological function is likely to be complex and will require much additional investigation. Understanding the molecular abnormalities and the function of RASSF1A in cancer may lead to the identification of targets for new therapeutic approaches and to the development of new anticancer drugs.
Research presented in this review was supported by BMBF grant FKZ 01ZZ0104, Land Sachsen-Anhalt, DFG grant DA552-1 to Reinhard Dammann, and NIH grant CA88873 to Gerd P. Pfeifer.
REFERENCES
1.Kok, K., Naylor, S. L., and Buys, C. H. (1997)
Adv. Cancer Res., 71, 27-92.
2.Kaelin, W. G., Jr., and Maher, E. R. (1998)
Trends Genet., 14, 423-426.
3.Sozzi, G., Veronese, M. L., Negrini, M., Baffa, R.,
Cotticelli, M. G., Inoue, H., Tornielli, S., Pilotti, S., De Gregorio,
L., Pastorino, U., Pierotti, M. A., Ohta, M., Huebner, K., and Croce,
C. M. (1996) Cell, 85, 17-26.
4.Xian, J., Clark, K. J., Fordham, R., Pannell, R.,
Rabbitts, T. H., and Rabbitts, P. H. (2001) Proc. Natl. Acad. Sci.
USA, 98, 15062-15066.
5.Killary, A. M., Wolf, M. E., Giambernardi, T. A.,
and Naylor, S. L. (1992) Proc. Natl. Acad. Sci. USA, 89,
10877-10881.
6.Wei, M. H., Latif, F., Bader, S., Kashuba, V.,
Chen, J. Y., Duh, F. M., Sekido, Y., Lee, C. C., Geil, L., Kuzmin, I.,
Zabarovsky, E., Klein, G., Zbar, B., Minna, J. D., and Lerman, M. I.
(1996) Cancer Res., 56, 1487-1492.
7.Todd, S., Franklin, W. A., Varella-Garcia, M.,
Kennedy, T., Hilliker, C. E., Jr., Hahner, L., Anderson, M., Wiest, J.
S., Drabkin, H. A., and Gemmill, R. M. (1997) Cancer Res.,
57, 1344-1352.
8.Wistuba, II, Behrens, C., Virmani, A. K., Mele, G.,
Milchgrub, S., Girard, L., Fondon, J. W., 3rd, Garner, H. R., McKay,
B., Latif, F., Lerman, M. I., Lam, S., Gazdar, A. F., and Minna, J. D.
(2000) Cancer Res., 60, 1949-1960.
9.Sekido, Y., Ahmadian, M., Wistuba, II, Latif, F.,
Bader, S., Wei, M. H., Duh, F. M., Gazdar, A. F., Lerman, M. I., and
Minna, J. D. (1998) Oncogene, 16, 3151-3157.
10.Lerman, M. I., and Minna, J. D. (2000) Cancer
Res., 60, 6116-6133.
11.Dammann, R., Li, C., Yoon, J. H., Chin, P. L.,
Bates, S., and Pfeifer, G. P. (2000) Nat. Genet., 25,
315-319.
12.Burbee, D. G., Forgacs, E., Zochbauer-Muller, S.,
Shivakumar, L., Fong, K., Gao, B., Randle, D., Kondo, M., Virmani, A.,
Bader, S., Sekido, Y., Latif, F., Milchgrub, S., Toyooka, S., Gazdar,
A. F., Lerman, M. I., Zabarovsky, E., White, M., and Minna, J. D.
(2001) J. Natl. Cancer Inst., 93, 691-699.
13.Vavvas, D., Li, X., Avruch, J., and Zhang, X. F.
(1998) J. Biol. Chem., 273, 5439-5442.
14.Newton, A. C. (1995) Curr. Biol.,
5, 973-976.
15.Dammann, R., Schagdarsurengin, U., Strunnikova,
M., Rastetter, M., Seidel, C., Liu, L., Tommasi, S., and Pfeifer, G. P.
(2003) Histol. Histopathol., 18, 665-677.
16.Pfeifer, G. P., Yoon, J. H., Liu, L., Tommasi,
S., Wilczynski, S. P., and Dammann, R. (2002) Biol. Chem.,
383, 907-914.
17.Jones, P. A., and Baylin, S. B. (2002) Nat.
Rev. Genet., 3, 415-428.
18.Herman, J. G., and Baylin, S. B. (2003) N.
Engl. J. Med., 349, 2042-2054.
19.Lee, M. G., Kim, H. Y., Byun, D. S., Lee, S. J.,
Lee, C. H., Kim, J. I., Chang, S. G., and Chi, S. G. (2001) Cancer
Res., 61, 6688-6692.
20.Maruyama, R., Toyooka, S., Toyooka, K. O.,
Harada, K., Virmani, A. K., Zochbauer-Muller, S., Farinas, A. J.,
Vakar-Lopez, F., Minna, J. D., Sagalowsky, A., Czerniak, B., and
Gazdar, A. F. (2001) Cancer Res., 61, 8659-8663.
21.Chan, M. W., Chan, L. W., Tang, N. L., Lo, K. W.,
Tong, J. H., Chan, A. W., Cheung, H. Y., Wong, W. S., Chan, P. S., Lai,
F. M., and To, K. F. (2003) Int. J. Cancer, 104,
611-616.
22.Dulaimi, E., Uzzo, R. G., Greenberg, R. E.,
Al-Saleem, T., and Cairns, P. (2004) Clin. Cancer Res.,
10, 1887-1893.
23.Astuti, D., Agathanggelou, A., Honorio, S.,
Dallol, A., Martinsson, T., Kogner, P., Cummins, C., Neumann, H. P.,
Voutilainen, R., Dahia, P., Eng, C., Maher, E. R., and Latif, F. (2001)
Oncogene, 20, 7573-7577.
24.Horiguchi, K., Tomizawa, Y., Tosaka, M.,
Ishiuchi, S., Kurihara, H., Mori, M., and Saito, N. (2003)
Oncogene, 22, 7862-7865.
25.Lusher, M. E., Lindsey, J. C., Latif, F.,
Pearson, A. D., Ellison, D. W., and Clifford, S. C. (2002) Cancer
Res., 62, 5906-5911.
26.Lindsey, J. C., Lusher, M. E., Anderton, J. A.,
Bailey, S., Gilbertson, R. J., Pearson, A. D., Ellison, D. W., and
Clifford, S. C. (2004) Carcinogenesis, 25, 661-668.
27.Hesson, L., Bieche, I., Krex, D., Criniere, E.,
Hoang-Xuan, K., Maher, E. R., and Latif, F. (2004) Oncogene,
23, 2408-2419.
28.Balana, C., Ramirez, J. L., Taron, M., Roussos,
Y., Ariza, A., Ballester, R., Sarries, C., Mendez, P., Sanchez, J. J.,
and Rosell, R. (2003) Clin. Cancer Res., 9,
1461-1468.
29.Astuti, D., Da Silva, N. F., Dallol, A., Gentle,
D., Martinsson, T., Kogner, P., Grundy, R., Kishida, T., Yao, M.,
Latif, F., and Maher, E. R. (2004) Br. J. Cancer, 90,
515-521.
30.Ramirez, J. L., Taron, M., Balana, C., Sarries,
C., Mendez, P., de Aguirre, I., Nunez, L., Roig, B., Queralt, C.,
Botia, M., and Rosell, R. (2003) Rocz. Akad. Med. Bialymst.,
48, 34-41.
31.Dammann, R., Yang, G., and Pfeifer, G. P. (2001)
Cancer Res., 61, 3105-3109.
32.Agathanggelou, A., Honorio, S., Macartney, D. P.,
Martinez, A., Dallol, A., Rader, J., Fullwood, P., Chauhan, A., Walker,
R., Shaw, J. A., Hosoe, S., Lerman, M. I., Minna, J. D., Maher, E. R.,
and Latif, F. (2001) Oncogene, 20, 1509-1518.
33.Lehmann, U., Langer, F., Feist, H., Glockner, S.,
Hasemeier, B., and Kreipe, H. (2002) Am. J. Pathol., 160,
605-612.
34.Chen, C. M., Chen, H. L., Hsiau, T. H., Hsiau, A.
H., Shi, H., Brock, G. J., Wei, S. H., Caldwell, C. W., Yan, P. S., and
Huang, T. H. (2003) Am. J. Pathol., 163, 37-45.
35.Honorio, S., Agathanggelou, A., Schuermann, M.,
Pankow, W., Viacava, P., Maher, E. R., and Latif, F. (2003)
Oncogene, 22, 147-150.
36.Fackler, M. J., McVeigh, M., Evron, E., Garrett,
E., Mehrotra, J., Polyak, K., Sukumar, S., and Argani, P. (2003)
Int. J. Cancer, 107, 970-975.
37.Muller, H. M., Widschwendter, A., Fiegl, H.,
Ivarsson, L., Goebel, G., Perkmann, E., Marth, C., and Widschwendter,
M. (2003) Cancer Res., 63, 7641-7645.
38.Mehrotra, J., Vali, M., McVeigh, M., Kominsky, S.
L., Fackler, M. J., Lahti-Domenici, J., Polyak, K., Sacchi, N.,
Garrett-Mayer, E., Argani, P., and Sukumar, S. (2004) Clin. Cancer
Res., 10, 3104-3109.
39.Krassenstein, R., Sauter, E., Dulaimi, E.,
Battagli, C., Ehya, H., Klein-Szanto, A., and Cairns, P. (2004)
Clin. Cancer Res., 10, 28-32.
40.Kuzmin, I., Liu, L., Dammann, R., Geil, L.,
Stanbridge, E. J., Wilczynski, S. P., Lerman, M. I., and Pfeifer, G. P.
(2003) Cancer Res., 63, 1888-1893.
41.Wong, N., Li, L., Tsang, K., Lai, P. B., To, K.
F., and Johnson, P. J. (2002) J. Hepatol., 37,
633-639.
42.Yoon, J. H., Dammann, R., and Pfeifer, G. P.
(2001) Int. J. Cancer, 94, 212-217.
43.Van Engeland, M., Weijenberg, M. P., Roemen, G.
M., Brink, M., de Bruine, A. P., Goldbohm, R. A., van den Brandt, P.
A., Baylin, S. B., de Goeij, A. F., and Herman, J. G. (2003) Cancer
Res., 63, 3133-3137.
44.Wagner, K. J., Cooper, W. N., Grundy, R. G.,
Caldwell, G., Jones, C., Wadey, R. B., Morton, D., Schofield, P. N.,
Reik, W., Latif, F., and Maher, E. R. (2002) Oncogene,
21, 7277-7282.
45.Lee, S., Hwang, K. S., Lee, H. J., Kim, J. S.,
and Kang, G. H. (2004) Lab. Invest., 84, 884-893.
46.Kuroki, T., Trapasso, F., Yendamuri, S.,
Matsuyama, A., Alder, H., Mori, M., and Croce, C. M. (2003) Clin.
Cancer Res., 9, 1441-1445.
47.Byun, D. S., Lee, M. G., Chae, K. S., Ryu, B. G.,
and Chi, S. G. (2001) Cancer Res., 61, 7034-7038.
48.Kang, G. H., Lee, S., Kim, W. H., Lee, H. W.,
Kim, J. C., Rhyu, M. G., and Ro, J. Y. (2002) Am. J. Pathol.,
160, 787-794.
49.To, K. F., Leung, W. K., Lee, T. L., Yu, J.,
Tong, J. H., Chan, M. W., Ng, E. K., Chung, S. C., and Sung, J. J.
(2002) Int. J. Cancer, 102, 623-628.
50.Kang, G. H., Lee, H. J., Hwang, K. S., Lee, S.,
Kim, J. H., and Kim, J. S. (2003) Am. J. Pathol., 163,
1551-1556.
51.Kang, G. H., Lee, S., Kim, J. S., and Jung, H. Y.
(2003) Lab. Invest., 83, 519-526.
52.House, M. G., Guo, M., Efron, D. T., Lillemoe, K.
D., Cameron, J. L., Syphard, J. E., Hooker, C. M., Abraham, S. C.,
Montgomery, E. A., Herman, J. G., and Brock, M. V. (2003) J.
Gastrointest. Surg., 7, 1004-1014.
53.Hasegawa, M., Nelson, H. H., Peters, E.,
Ringstrom, E., Posner, M., and Kelsey, K. T. (2002) Oncogene,
21, 4231-4236.
54.Hogg, R. P., Honorio, S., Martinez, A.,
Agathanggelou, A., Dallol, A., Fullwood, P., Weichselbaum, R., Kuo, M.
J., Maher, E. R., and Latif, F. (2002) Eur. J. Cancer,
38, 1585-1592.
55.Dong, S. M., Sun, D. I., Benoit, N. E., Kuzmin,
I., Lerman, M. I., and Sidransky, D. (2003) Clin. Cancer Res.,
9, 3635-3640.
56.Maruya, S., Issa, J. P., Weber, R. S., Rosenthal,
D. I., Haviland, J. C., Lotan, R., and El-Naggar, A. K. (2004) Clin.
Cancer Res., 10, 3825-3830.
57.Yu, M. Y., Tong, J. H., Chan, P. K., Lee, T. L.,
Chan, M. W., Chan, A. W., Lo, K. W., and To, K. F. (2003) Int. J.
Cancer, 105, 204-209.
58.Schagdarsurengin, U., Wilkens, L., Steinemann,
D., Flemming, P., Kreipe, H. H., Pfeifer, G. P., Schlegelberger, B.,
and Dammann, R. (2003) Oncogene, 22, 1866-1871.
59.Zhong, S., Yeo, W., Tang, M. W., Wong, N., Lai,
P. B., and Johnson, P. J. (2003) Clin. Cancer Res., 9,
3376-3382.
60.Lee, S., Lee, H. J., Kim, J. H., Lee, H. S.,
Jang, J. J., and Kang, G. H. (2003) Am. J. Pathol., 163,
1371-1378.
61.Murray, P. G., Qiu, G. H., Fu, L., Waites, E. R.,
Srivastava, G., Heys, D., Agathanggelou, A., Latif, F., Grundy, R. G.,
Mann, J. R., Starczynski, J., Crocker, J., Parkes, S. E., Ambinder, R.
F., Young, L. S., and Tao, Q. (2004) Oncogene, 23,
1326-1331.
62.Dreijerink, K., Braga, E., Kuzmin, I., Geil, L.,
Duh, F. M., Angeloni, D., Zbar, B., Lerman, M. I., Stanbridge, E. J.,
Minna, J. D., Protopopov, A., Li, J., Kashuba, V., Klein, G., and
Zabarovsky, E. R. (2001) Proc. Natl. Acad. Sci. USA, 98,
7504-7509.
63.Morrissey, C., Martinez, A., Zatyka, M.,
Agathanggelou, A., Honorio, S., Astuti, D., Morgan, N. V., Moch, H.,
Richards, F. M., Kishida, T., Yao, M., Schraml, P., Latif, F., and
Maher, E. R. (2001) Cancer Res., 61, 7277-7281.
64.Battagli, C., Uzzo, R. G., Dulaimi, E., Ibanez de
Caceres, I., Krassenstein, R., Al-Saleem, T., Greenberg, R. E., and
Cairns, P. (2003) Cancer Res., 63, 8695-8699.
65.Dulaimi, E., De, C., II, Uzzo, R. G., Al-Saleem,
T., Greenberg, R. E., Polascik, T. J., Babb, J. S., Grizzle, W. E., and
Cairns, P. (2004) Clin. Cancer Res., 10, 3972-3979.
66.Dammann, R., Takahashi, T., and Pfeifer, G. P.
(2001) Oncogene, 20, 3563-3567.
67.Tomizawa, Y., Kohno, T., Kondo, H., Otsuka, A.,
Nishioka, M., Niki, T., Yamada, T., Maeshima, A., Yoshimura, K., Saito,
R., Minna, J. D., and Yokota, J. (2002) Clin. Cancer Res.,
8, 2362-2368.
68.Toyooka, S., Toyooka, K. O., Maruyama, R.,
Virmani, A. K., Girard, L., Miyajima, K., Harada, K., Ariyoshi, Y.,
Takahashi, T., Sugio, K., Brambilla, E., Gilcrease, M., Minna, J. D.,
and Gazdar, A. F. (2001) Mol. Cancer Ther., 1, 61-67.
69.Toyooka, S., Maruyama, R., Toyooka, K. O.,
McLerran, D., Feng, Z., Fukuyama, Y., Virmani, A. K., Zochbauer-Muller,
S., Tsukuda, K., Sugio, K., Shimizu, N., Shimizu, K., Lee, H., Chen, C.
Y., Fong, K. M., Gilcrease, M., Roth, J. A., Minna, J. D., and Gazdar,
A. F. (2003) Int. J. Cancer, 103, 153-160.
70.Endoh, H., Yatabe, Y., Shimizu, S., Tajima, K.,
Kuwano, H., Takahashi, T., and Mitsudomi, T. (2003) Int. J.
Cancer, 106, 45-51.
71.Kim, D. H., Kim, J. S., Ji, Y. I., Shim, Y. M.,
Kim, H., Han, J., and Park, J. (2003) Cancer Res., 63,
3743-3746.
72.Kim, D. H., Kim, J. S., Park, J. H., Lee, S. K.,
Ji, Y. I., Kwon, Y. M., Shim, Y. M., Han, J., and Park, J. (2003)
Cancer Res., 63, 6206-6211.
73.Yanagawa, N., Tamura, G., Oizumi, H., Takahashi,
N., Shimazaki, Y., and Motoyama, T. (2003) Cancer Sci.,
94, 589-592.
74.Li, J., Zhang, Z., Dai, Z., Popkie, A. P., Plass,
C., Morrison, C., Wang, Y., and You, M. (2003) Neoplasia,
5, 362-366.
75.Zochbauer-Muller, S., Lam, S., Toyooka, S.,
Virmani, A. K., Toyooka, K. O., Seidl, S., Minna, J. D., and Gazdar, A.
F. (2003) Int. J. Cancer, 107, 612-616.
76.Maruyama, R., Sugio, K., Yoshino, I., Maehara,
Y., and Gazdar, A. F. (2004) Cancer, 100, 1472-1477.
77.Topaloglu, O., Hoque, M. O., Tokumaru, Y., Lee,
J., Ratovitski, E., Sidransky, D., and Moon, C. S. (2004) Clin.
Cancer Res., 10, 2284-2288.
78.Toyooka, S., Pass, H. I., Shivapurkar, N.,
Fukuyama, Y., Maruyama, R., Toyooka, K. O., Gilcrease, M., Farinas, A.,
Minna, J. D., and Gazdar, A. F. (2001) Cancer Res., 61,
5727-5730.
79.Toyooka, S., Carbone, M., Toyooka, K. O.,
Bocchetta, M., Shivapurkar, N., Minna, J. D., and Gazdar, A. F. (2002)
Oncogene, 21, 4340-4344.
80.Spugnardi, M., Tommasi, S., Dammann, R., Pfeifer,
G. P., and Hoon, D. S. (2003) Cancer Res., 63,
1639-1643.
81.Hoon, D. S., Spugnardi, M., Kuo, C., Huang, S.
K., Morton, D. L., and Taback, B. (2004) Oncogene, 23,
4014-4022.
82.Reifenberger, J., Knobbe, C. B., Sterzinger, A.
A., Blaschke, B., Schulte, K. W., Ruzicka, T., and Reifenberger, G.
(2004) Int. J. Cancer, 109, 377-384.
83.Lo, K. W., Kwong, J., Hui, A. B., Chan, S. Y.,
To, K. F., Chan, A. S., Chow, L. S., Teo, P. M., Johnson, P. J., and
Huang, D. P. (2001) Cancer Res., 61, 3877-3881.
84.Tong, J. H., Tsang, R. K., Lo, K. W., Woo, J. K.,
Kwong, J., Chan, M. W., Chang, A. R., van Hasselt, C. A., Huang, D. P.,
and To, K. F. (2002) Clin. Cancer Res., 8, 2612-2619.
85.Kwong, J., Lo, K. W., To, K. F., Teo, P. M.,
Johnson, P. J., and Huang, D. P. (2002) Clin. Cancer Res.,
8, 131-137.
86.Chang, H. W., Chan, A., Kwong, D. L., Wei, W. I.,
Sham, J. S., and Yuen, A. P. (2003) Int. J. Cancer, 105,
851-855.
87.Wong, T. S., Tang, K. C., Kwong, D. L., Sham, J.
S., Wei, W. I., Kwong, Y. L., and Yuen, A. P. (2003) Int. J.
Oncol., 22, 869-874.
88.Wong, T. S., Kwong, D. L., Sham, J. S., Wei, W.
I., Kwong, Y. L., and Yuen, A. P. (2004) Clin. Cancer Res.,
10, 2401-2406.
89.Lim, S., Yang, M. H., Park, J. H., Nojima, T.,
Hashimoto, H., Unni, K. K., and Park, Y. K. (2003) Oncol. Rep.,
10, 897-901.
90.Rathi, A., Virmani, A. K., Schorge, J. O., Elias,
K. J., Maruyama, R., Minna, J. D., Mok, S. C., Girard, L., Fishman, D.
A., and Gazdar, A. F. (2002) Clin. Cancer Res., 8,
3324-3331.
91.Dhillon, V. S., Aslam, M., and Husain, S. A.
(2004) Clin. Cancer Res., 10, 5537-5545.
92.De Caceres, I., Battagli, C., Esteller, M.,
Herman, J. G., Dulaimi, E., Edelson, M. I., Bergman, C., Ehya, H.,
Eisenberg, B. L., and Cairns, P. (2004) Cancer Res., 64,
6476-6481.
93.Dammann, R., Schagdarsurengin, U., Liu, L., Otto,
N., Gimm, O., Dralle, H., Boehm, B. O., Pfeifer, G. P., and Hoang-Vu,
C. (2003) Oncogene, 22, 3806-3812.
94.House, M. G., Herman, J. G., Guo, M. Z., Hooker,
C. M., Schulick, R. D., Lillemoe, K. D., Cameron, J. L., Hruban, R. H.,
Maitra, A., and Yeo, C. J. (2003) Ann. Surg., 238,
423-431.
95.Harada, K., Toyooka, S., Maitra, A., Maruyama,
R., Toyooka, K. O., Timmons, C. F., Tomlinson, G. E., Mastrangelo, D.,
Hay, R. J., Minna, J. D., and Gazdar, A. F. (2002) Oncogene,
21, 4345-4349.
96.Ehrlich, M., Jiang, G., Fiala, E., Dome, J. S.,
Yu, M. C., Long, T. I., Youn, B., Sohn, O. S., Widschwendter, M.,
Tomlinson, G. E., Chintagumpala, M., Champagne, M., Parham, D., Liang,
G., Malik, K., and Laird, P. W. (2002) Oncogene, 21,
6694-6702.
97.Wong, I. H., Chan, J., Wong, J., and Tam, P. K.
(2004) Clin. Cancer Res., 10, 994-1002.
98.Koul, S., Houldsworth, J., Mansukhani, M. M.,
Donadio, A., McKiernan, J. M., Reuter, V. E., Bosl, G. J., Chaganti, R.
S., and Murty, V. V. (2002) Mol. Cancer, 1, 8.
99.Honorio, S., Agathanggelou, A., Wernert, N.,
Rothe, M., Maher, E. R., and Latif, F. (2003) Oncogene,
22, 461-466.
100.Kawakami, T., Okamoto, K., Kataoka, A.,
Koizumi, S., Iwaki, H., Sugihara, H., Reeve, A. E., Ogawa, O., and
Okada, Y. (2003) Genes Chromosomes Cancer, 38,
97-101.
101.Koul, S., McKiernan, J. M., Narayan, G.,
Houldsworth, J., Bacik, J., Dobrzynski, D. L., Assaad, A. M.,
Mansukhani, M., Reuter, V. E., Bosl, G. J., Chaganti, R. S., and Murty,
V. V. (2004) Mol. Cancer, 3, 16.
102.Schagdarsurengin, U., Gimm, O., Hoang-Vu, C.,
Dralle, H., Pfeifer, G. P., and Dammann, R. (2002) Cancer Res.,
62, 3698-3701.
103.Xing, M., Cohen, Y., Mambo, E., Tallini, G.,
Udelsman, R., Ladenson, P. W., and Sidransky, D. (2004) Cancer
Res., 64, 1664-1668.
104.Tsou, J. A., Hagen, J. A., Carpenter, C. L.,
and Laird-Offringa, I. A. (2002) Oncogene, 21,
5450-5461.
105.Laird, P. W. (2003) Nat. Rev. Cancer,
3, 253-266.
106.Belinsky, S. A., Palmisano, W. A., Gilliland,
F. D., Crooks, L. A., Divine, K. K., Winters, S. A., Grimes, M. J.,
Harms, H. J., Tellez, C. S., Smith, T. M., Moots, P. P., Lechner, J.
F., Stidley, C. A., and Crowell, R. E. (2002) Cancer Res.,
62, 2370-2377.
107.Tommasi, S., Dammann, R., Zhang, Z., Wang, Y.,
Liu, L., Tsark, W. M., Wilczynski, S. P., Li, J., You, M., and Pfeifer,
G. P. (2005) Cancer Res., 65, 92-98.
108.Kuzmin, I., Gillespie, J. W., Protopopov, A.,
Geil, L., Dreijerink, K., Yang, Y., Vocke, C. D., Duh, F. M.,
Zabarovsky, E., Minna, J. D., Rhim, J. S., Emmert-Buck, M. R., Linehan,
W. M., and Lerman, M. I. (2002) Cancer Res., 62,
3498-3502.
109.Chow, L. S., Lo, K. W., Kwong, J., To, K. F.,
Tsang, K. S., Lam, C. W., Dammann, R., and Huang, D. P. (2004) Int.
J. Cancer, 109, 839-847.
110.Li, J., Wang, F., Protopopov, A., Malyukova,
A., Kashuba, V., Minna, J. D., Lerman, M. I., Klein, G., and
Zabarovsky, E. (2004) Oncogene, 23, 5941-5949.
111.Shivakumar, L., Minna, J., Sakamaki, T.,
Pestell, R., and White, M. A. (2002) Mol. Cell Biol., 22,
4309-4318.
112.Bar-Sagi, D., and Feramisco, J. R. (1985)
Cell, 42, 841-848.
113.Serrano, M., Lin, A. W., McCurrach, M. E.,
Beach, D., and Lowe, S. W. (1997) Cell, 88, 593-602.
114.Mayo, M. W., Wang, C. Y., Cogswell, P. C.,
Rogers-Graham, K. S., Lowe, S. W., Der, C. J., and Baldwin, A. S., Jr.
(1997) Science, 278, 1812-1815.
115.Downward, J. (1998) Curr. Opin. Genet.
Dev., 8, 49-54.
116.Chen, C. Y., Liou, J., Forman, L. W., and
Faller, D. V. (1998) J. Biol. Chem., 273,
16700-16709.
117.Shao, J., Sheng, H., DuBois, R. N., and
Beauchamp, R. D. (2000) J. Biol. Chem., 275,
22916-22924.
118.Vos, M. D., Ellis, C. A., Bell, A., Birrer, M.
J., and Clark, G. J. (2000) J. Biol. Chem., 275,
35669-35672.
119.Van Engeland, M., Roemen, G. M., Brink, M.,
Pachen, M. M., Weijenberg, M. P., de Bruine, A. P., Arends, J. W., van
den Brandt, P. A., de Goeij, A. F., and Herman, J. G. (2002)
Oncogene, 21, 3792-3795.
120.Ramirez, J. L., Sarries, C., de Castro, P. L.,
Roig, B., Queralt, C., Escuin, D., de Aguirre, I., Sanchez, J. M.,
Manzano, J. L., Margeli, M., Sanchez, J. J., Astudillo, J., Taron, M.,
and Rosell, R. (2003) Cancer Lett., 193, 207-216.
121.Tommasi, S., Dammann, R., Jin, S. G., Zhang Xf,
X. F., Avruch, J., and Pfeifer, G. P. (2002) Oncogene,
21, 2713-2720.
122.Vos, M. D., Martinez, A., Ellis, C. A.,
Vallecorsa, T., and Clark, G. J. (2003) J. Biol. Chem.,
278, 21938-21943.
123.Aoyama, Y., Avruch, J., and Zhang, X. F. (2004)
Oncogene, 23, 3426-3433.
124.Chen, J., Lui, W. O., Vos, M. D., Clark, G. J.,
Takahashi, M., Schoumans, J., Khoo, S. K., Petillo, D., Lavery, T.,
Sugimura, J., Astuti, D., Zhang, C., Kagawa, S., Maher, E. R., Larsson,
C., Alberts, A. S., Kanayama, H. O., and Teh, B. T. (2003) Cancer
Cell, 4, 405-413.
125.Hesson, L., Dallol, A., Minna, J. D., Maher, E.
R., and Latif, F. (2003) Oncogene, 22, 947-954.
126.Ortiz-Vega, S., Khokhlatchev, A., Nedwidek, M.,
Zhang, X. F., Dammann, R., Pfeifer, G. P., and Avruch, J. (2002)
Oncogene, 21, 1381-1390.
127.Khokhlatchev, A., Rabizadeh, S., Xavier, R.,
Nedwidek, M., Chen, T., Zhang, X. F., Seed, B., and Avruch, J. (2002)
Curr. Biol., 12, 253-265.
128.Liu, L., Tommasi, S., Lee, D. H., Dammann, R.,
and Pfeifer, G. P. (2003) Oncogene, 22, 8125-8136.
129.Song, M. S., Song, S. J., Ayad, N. G., Chang,
J. S., Lee, J. H., Hong, H. K., Lee, H., Choi, N., Kim, J., Kim, H.,
Kim, J. W., Choi, E. J., Kirschner, M. W., and Lim, D. S. (2004)
Nat. Cell Biol., 6, 129-137.
130.Vos, M. D., Martinez, A., Elam, C., Dallol, A.,
Taylor, B. J., Latif, F., and Clark, G. J. (2004) Cancer Res.,
64, 4244-4250.
131.Dallol, A., Agathanggelou, A., Fenton, S. L.,
Ahmed-Choudhury, J., Hesson, L., Vos, M. D., Clark, G. J., Downward,
J., Maher, E. R., and Latif, F. (2004) Cancer Res., 64,
4112-4116.
132.Rong, R., Jin, W., Zhang, J., Saeed Sheikh, M.,
and Huang, Y. (2004) Oncogene, 23, 8216-8230.
133.Jackson, P. K. (2004) Trends Cell Biol.,
14, 331-334.
134.Mathe, E. (2004) Nat. Genet., 36,
117-118.