REVIEW: Lipophilic Triphenylphosphonium Cations as Tools in Mitochondrial Bioenergetics and Free Radical Biology
M. F. Ross1, G. F. Kelso2, F. H. Blaikie2, A. M. James1, H. M. Cochemé1, A. Filipovska1, T. Da Ros3, T. R. Hurd1, R. A. J. Smith2, and M. P. Murphy1*
1MRC Dunn Human Nutrition Unit, Hills Road, Cambridge CB2 2XY, UK; fax: +44-1223-252905; E-mail: mpm@mrc-dunn.cam.ac.uk2Department of Chemistry, University of Otago, Box 56, Dunedin, New Zealand
3Pharmaceutical Science Department, Trieste University, 34127 Trieste, Italy
* To whom correspondence should be addressed.
Received September 24, 2004
Lipophilic phosphonium cations were first used to investigate mitochondrial biology by Vladimir Skulachev and colleagues in the late 1960s. Since then, these molecules have become important tools for exploring mitochondrial bioenergetics and free radical biology. Here we review why these molecules are useful in mitochondrial research and outline some of the ways in which they are now being utilized.
KEY WORDS: lipophilic phosphonium cations, mitochondria, membrane potential, oxidative damage, antioxidants
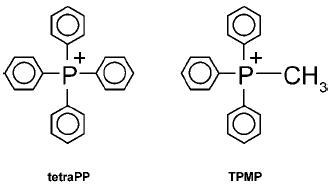
Abbreviations: epsilon) dielectric constant; GSH) glutathione; GSSG) glutathione disulfide; tetraPP) tetraphenylphosphonium cation; IBTP) (4-iodobutyl)triphenylphosphonium cation; Deltapsi) membrane potential; PBN) phenylbutylnitrone; TBTP) (4-thiobutyl)triphenylphosphonium cation; TPB) tetraphenylborate anion; TPMP) methyltriphenylphosphonium cation; TPP) triphenylphosphonium cation; TrxSH) reduced thioredoxin; TrxSS) oxidized thioredoxin; WB) Born energy.
In the late 1960s, the chemiosmotic coupling hypothesis of Peter
Mitchell was fiercely disputed as a mechanistic explanation of
mitochondrial oxidative phosphorylation [1]. Many
researchers had difficulty accepting that a protonmotive force
comprising a membrane potential (Deltapsi) and a pH
gradient across the mitochondrial inner membrane could be the
intermediate between respiration and ATP synthesis [1]. Consequently, a paper published by Vladimir
Skulachev and colleagues in 1969 had considerable impact, as it
provided strong evidence that a large Deltapsi existed
across the mitochondrial inner membrane [2]. They
demonstrated the existence of the mitochondrial Deltapsi
by showing that the lipophilic dibenzylammonium cation accumulated in
the mitochondrial matrix in response to Deltapsi [2]. The ability of lipophilic cations to permeate
artificial phospholipid bilayers had been known for some time [3], but this was the first application of these
molecules to mitochondrial biology. The mitochondrial uptake of the
dibenzylammonium cation used in the initial report was relatively poor
and required the lipophilic tetraphenylborate anion (TPB) [2]. Therefore, in subsequent studies Skulachev's group
used the methyltriphenylphosphonium (TPMP) cation (Fig. 1), which was accumulated by energized mitochondria
without the requirement for TPB [4, 5].
At the time these papers were published, their chief significance lay in showing that mitochondrial respiration generated a significant Deltapsi. However, they were also important for showing that lipophilic cations were accumulated in energized mitochondria and for introducing the use of triphenylphosphonium (TPP) cations to mitochondrial biology. Up until that point, phosphonium cations had been widely used in synthetic organic chemistry, for example in the Wittig reaction, and in exploring the electrical properties of phospholipid bilayers [3].
The finding that lipophilic cations were taken up by energized mitochondria also made sense of the fact, known since the early 20th century, that many vital stains used to visualize mitochondria in microscopy, such as Janus Green, were lipophilic cations [6]. Nowadays fluorescent lipophilic cations such as rhodamine, JC-1, and the MitoTracker--> series of compounds are routinely used to visualize mitochondria within cells.
The structures of TPMP and the closely related tetraphenylphosphonium cation (tetraPP) are shown in Fig. 1. Such lipophilic phosphonium cations are able to cross the mitochondrial inner membrane without the assistance of ionophores, and accumulate within the mitochondrial matrix, driven by Deltapsi. For this reason, the lipophilic phosphonium cations introduced by Skulachev and colleagues have proven to be among the most useful probes for the investigation of mitochondrial function. In this review we outline why lipophilic phosphonium cations are taken up by mitochondria, and discuss some of the ways in which these molecules have been used to investigate mitochondrial biology over the past 35 years.Fig. 1. Structure of tetraPP and TPMP.
WHY ARE LIPOPHILIC PHOSPHONIUM CATIONS TAKEN UP BY
MITOCHONDRIA?
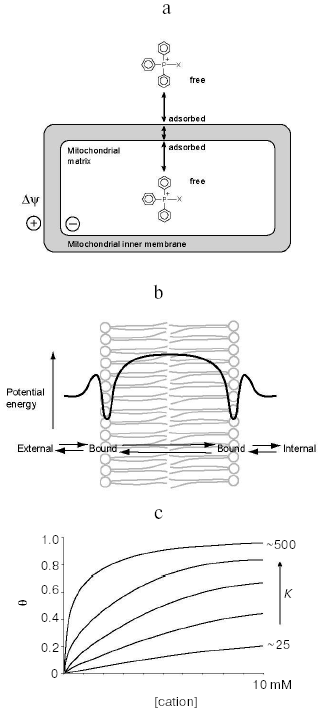
Lipophilic cations such as tetraPP and TPMP (Fig. 1) have the unusual property of being relatively lipid-soluble, despite their net positive charge. Consequently, they can pass easily through phospholipid bilayers into mitochondria [7, 8] (Fig. 2a). This is in contrast to hydrophilic cations such as Na+, which cannot cross biological membranes except when transport is facilitated by ionophores or carrier proteins. The impermeability of biological membranes to hydrophilic cations is largely due to the energy demand of moving an ion from an aqueous environment to the non-polar lipid interior of the membrane [7]. The activation energy for movement through the hydrophobic core of the membrane is therefore very high, while for lipophilic cations this activation energy is far lower, enabling their passage.
The activation energy for movement from the aqueous medium to the hydrophobic core of the membrane has contributions from electrostatic interactions and from hydrophobic forces [7, 9]. The main electrostatic energy component, the Born energy (WB), is due to the enthalpy input required to remove solvating water molecules from the cation upon transfer from the aqueous environment to the lipid core of the membrane [10, 11]. The Born energy is given by:Fig. 2. Uptake and binding of phosphonium cations by mitochondria. a) Uptake into mitochondria of a triphenylphosphonium cation conjugated to X, showing passage through the inner membrane and adsorption to the phospholipid bilayer. b) The energy profile for the movement of a triphenylphosphonium cation through a phospholipid bilayer, showing adsorption to potential energy wells close to the carbonyls of the phospholipid fatty acids. c) Adsorption of phosphonium cations to a phospholipid bilayer as described by a Langmuir adsorption isotherm, which shows Theta, the fraction of binding sites on the membrane occupied by phosphonium cation, as a function of the cation concentration at different values of K.
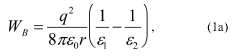
![]()
In addition to the Born energy, two other electrostatic forces contribute to the movement of a cation through a lipid bilayer. Both are significantly smaller than the Born energy. One is the image energy, which is due to the electrostatic forces at the dielectric interface [9, 10, 13]. The other is the dipole energy, which is the local electrical potential within the core of the phospholipid bilayer, caused by the orientation of the dipoles of the fatty acid carbonyl groups in phospholipids [10, 13]. While the overall contribution of the dipole force is small, it is interesting because it affects cations and anions in opposite ways, lowering the activation energy for transport of lipophilic anions such as TPB but increasing the activation energy for the lipophilic cation tetraPP, which has a similar ionic radius and hydrophobicity [13]. The dipole energy explains why lipophilic anions permeate phospholipid bilayers much more quickly than lipophilic cations [13].
The electrostatic forces described above, which are dominated by the Born energy, are all repulsive, requiring an enthalpy input to drive movement of the cation through the phospholipid bilayer. However, there is also an important component of the activation energy of uptake that is due to the hydrophobicity of the cation. This is the energy required to move an uncharged molecule identical in size and hydrophobicity from the aqueous environment into the lipid core of the membrane. This hydrophobic component of the activation energy is an attractive force for lipophilic cations, and is due to increased entropy (loss of water structure) on moving a molecule with a large hydrophobic surface area into lipid. Consequently, the greater the solvent-accessible surface area of the cation, the larger the hydrophobic effect (about 92 J/mol per Å2 of solvent-accessible surface [10]). The large hydrophobic surface area of tetraPP contributes to its decreased activation energy for transport, reducing it to about 29 kJ/mol [10].
The above analysis indicates why lipophilic cations have a far lower activation energy for movement through lipid bilayers than hydrophilic cations. The main contributors are the decrease in the repulsive Born energy due to the large ionic radii of lipophilic cations, and the attractive hydrophobic effect due to the extensive hydrophobic surface of the molecules. Considering only the Born energy, the difference in radii indicates that the activation energy should be ~4-fold greater for Na+ over tetraPP. The rate of movement of the cation through the phospholipid bilayer will follow the Arrhenius rate equation and will be proportional to e-E/RT, where E is the activation energy. This largely accounts for the 107-108-fold faster rate of permeation through phospholipid bilayers of tetraPP over small hydrophilic cations such as Na+ [10].
Numerous studies have extended these basic findings and we now have a detailed understanding of how lipophilic cations such as tetraPP and TPMP move through phospholipid bilayers [7]. The energy profile for the movement of a lipophilic cation through a phospholipid bilayer (shown in Fig. 2b) is derived by combining the attractive hydrophobic effect with the repulsive electrostatic forces. In addition to the energy barrier at the center of the membrane, the other notable feature is the potential energy wells close to the surfaces of the membrane. These wells arise because the attractive hydrophobic force, which favors the uptake of the hydrophobic compound into the membrane, becomes large very close to the surface of the membrane, while the repulsive electrostatic forces increase more gradually on moving through the membrane [7]. The binding of tetraPP within this potential energy well is on the hydrophobic side of the lipid/water interface of the membrane at about the level of the carbonyls and C2 carbons of the phospholipid fatty acyl groups [7, 9, 10, 14-16]. The enthalpy for binding is positive (i.e., repulsive) due to electrostatic forces, and the binding is in fact driven by the increase in entropy due to the hydrophobic driving force for binding [9].
The cations adsorb to the membrane as a monolayer in the potential energy well. They then jump rapidly through the hydrophobic core of the membrane to the potential energy well on the other surface of the membrane before desorbing from the membrane (Fig. 2b). This mechanism enables lipophilic cations to pass easily through phospholipid bilayers and thus these cations can traverse the mitochondrial inner membrane and access the matrix even in the absence of Deltapsi. However, in the presence of Deltapsi the distribution of singly charged lipophilic cations such as tetraPP across the membrane will equilibrate with Deltapsi leading to the extensive accumulation of the cation in the mitochondrial matrix to an extent given by the Nernst equation:
![]()
The final factor that requires analysis is the extent of binding of the cations to the potential energy wells both outside and within mitochondria. The potential energy wells are narrow; consequently the lipophilic cations adsorb to the surface of the membrane as a monolayer. The extent to which this adsorption saturates the binding sites for cations on the membrane surface depends on the concentration of cation free in solution. A number of models have been used to describe this relationship, but the simplest one is the Langmuir adsorption isotherm [18]. This straightforward model assumes that the cations adsorb independently to the surface binding sites.
This assumption becomes invalid at high cation concentrations, and more accurate models, such as the Volmer isotherm and the Stern isotherm, give a better description of binding to membranes and account for the repulsive charges between bound cations [18, 19]. However, for our purposes the Langmuir adsorption isotherm adequately describes the main features of binding of lipophilic cations to phospholipid bilayers at relatively low concentrations [13, 20]. The Langmuir adsorption isotherm is given by:
![]()
For lipophilic cations with relatively low values of K, such as TPMP, there is negligible binding to membranes when micromolar concentrations of cation are mixed with deenergized mitochondria [14, 23]. In this case, the free concentration of the cation is the same inside and outside the mitochondria, and the Langmuir adsorption isotherm indicates that the fraction of sites occupied will be similar outside the mitochondrial matrix (i.e., binding to the inside and outside of the outer membrane and to the outer surface of the inner membrane) and inside mitochondria (i.e., binding to the inner surface of the inner membrane). As the concentration is increased to millimolar levels, binding becomes more extensive and occurs to a similar degree of binding site saturation on both the inside and outside of mitochondria [21].
For more hydrophobic TPP cation compounds, K is far larger and consequently the degree of saturation of the binding sites at a given concentration is far greater (Fig. 2c). For example, in a series of alkyltriphenylphosphonium cations where the alkyl group is increased from 1 to 5 carbons in length, the amount of adsorption to the membrane increases in line with the hydrophobicity [16, 20]. Interestingly, increasing the hydrophobicity of the side chain does not alter the nature of the interaction of the phosphonium head group with the membrane; the TPP cation is still present in the potential well and the repulsive enthalpy of binding is unaffected [16]. However, as the alkyl group increases in hydrophobicity it is the larger entropic driving force that increases the amount bound to the membrane [14, 16, 20]. For example, this leads to a 10-fold increase in K on going from TPMP to amylTPP, and a 6-fold increase in K on going from TPMP to tetraPP [20]. An implication of these findings is that for all TPP derivatives bound to phospholipid bilayers, the TPP head group will be found at the same place on the hydrophobic side of the membrane lipid/water interface, with the hydrophobic tail penetrating into the hydrophobic core of the membrane [14, 23]. Therefore, when bound to membranes the TPP moiety of even the most hydrophobic derivatives are not present within the hydrophobic core of the membrane, except transiently during transport. This is supported by measurements of the partition coefficients of TPP derivatives between organic solvents and aqueous buffers: octan-1-ol mimics the less hydrophobic outer layer of phospholipid bilayers where water is present and consequently a significant amount of hydrogen bonding occurs. The octanol/water partition coefficients for TPP derivatives are relatively high: 0.35 for TPMP and 2760 for MitoQ10 (a quinol linked to TPP by a ten-carbon alkyl chain) [23], indicating that the cations move easily to the water/phospholipid interface. In contrast, even the most hydrophobic TPP cations such as MitoQ10 have negligible partition coefficients into nonpolar solvents, such as cyclohexane, that mimic the low dielectric environment at the center of the membrane.
The description above applies to the binding of TPP cations to deenergized mitochondria. However, the considerations for binding become more critical when discussing energized mitochondria. For example, for energized mitochondria incubated with micromolar concentrations of cation, the Nernst equation indicates that the concentration within the matrix will be millimolar. For molecules like TPMP, values of K for outside the mitochondrial matrix are similar to those for inside mitochondria [21]. Consequently, the dependence of degree of binding site saturation will be given by a similar Langmuir adsorption isotherm for binding to the outside and to the inside. However, in most situations involving mitochondria, the variable measured is the amount of cation associated with the mitochondria in total. Under these circumstances, the proportion of the cation within the mitochondria that is bound to the membrane is far greater than the amount bound to the outside of the mitochondria. For example, when deenergized mitochondria are incubated with micromolar TPMP, the amount of TPMP bound to the mitochondrial membranes is negligible, but once the mitochondria are energized, the internal concentration will be millimolar and a high proportion of the internal TPMP will be membrane-bound. This greater proportion of bound cation is due to the high volume of matrix-facing membrane relative to that of the aqueous phase within the mitochondria. The matrix volume is typically 0.5-1 µl/mg protein. In contrast, the volume of the outer face of the inner membrane, and the inner and outer faces of the outer membrane, is very low compared to that of the extramitochondrial aqueous phase (typically 1-5 ml).
To see why this is so, consider the binding of the cation to the surface of the membrane where the partition coefficient P is given by:
![]()
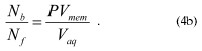
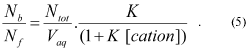
For more hydrophobic compounds, the binding to deenergized mitochondria can be extensive [23], with large amounts bound to both the outside and inside of the mitochondria. On induction of Deltapsi, there will be a several hundred-fold increase in the amount of free compound in the matrix, but as K is now higher most of the accumulated compound will be bound to the inner surface of the inner membrane. In situations where there is considerable binding to membranes of deenergized mitochondria, the induction of Deltapsi will lead to a rearrangement of the compounds, from the surfaces of the outer membrane and from the outer surface of the inner membrane to the inner surface of the inner membrane. So, while the amount of compound associated with the mitochondria may not change greatly on energizing the mitochondria, the location of the compound within the mitochondria will.
The model outlined in Fig. 2 therefore describes the uptake and distribution of all TPP cations within mitochondria, with increasing hydrophobicity leading to greater adsorption. Within mitochondria, the cations are in equilibrium between those free in solution and those bound to the membrane surface as a monolayer.
USING PHOSPHONIUM CATIONS TO MEASURE THE MITOCHONDRIAL MEMBRANE
POTENTIAL
The most widespread use for phosphonium cations such as tetraPP and TPMP in mitochondrial biology is in measuring the mitochondrial Deltapsi. Both the theoretical background and the practical aspects of these measurements have been reviewed extensively [17, 18, 25], and are only summarized here. To measure Deltapsi of isolated mitochondria the phosphonium cation, usually tetraPP or TPMP, is incubated with energized mitochondria and the distribution of the cation between the matrix and extramitochondrial environment is allowed to equilibrate in response to Deltapsi. The amounts of cation in the extramitochondrial fluid and in the mitochondrial matrix are then quantified. This can be done by using radiolabeled cation or by measuring the extramitochondrial concentration with an ion-selective electrode sensitive to TPMP [17, 18, 26]. The ion-selective electrode has the advantage that the measurements can be done in real time and can also be carried out simultaneously with other measurements such as respiration rate [17]. Once the amounts of cation in the mitochondrial matrix and in the extramitochondrial environment have been determined, the amount in the mitochondrial matrix is converted to a free concentration by the use of the mitochondrial volume and by taking into account membrane binding by using a correction factor [17, 18]. Matrix and extramitochondrial concentrations are then inserted into the Nernst equation, and Deltapsi is calculated. This approach is the standard way in which Deltapsi of isolated mitochondria is measured.
The distribution of phosphonium cations can also be used to determine the mitochondrial Deltapsi within cells, since the plasma membrane potential drives their uptake into the cytoplasm, from where they are further accumulated in mitochondria by the mitochondrial Deltapsi [17, 27]. Fluorescent dyes such as rhodamine or JC-1, in conjunction with confocal microscopy, flow cytometry, or fluorimetry can be used to give an indication of changes of the mitochondrial Deltapsi within cells; however, these approaches are difficult to quantify, and lipophilic phosphonium cations present a more quantitative alternative. This is usually done using radiolabeled tetraPP or TPMP, as the uptake is usually too slow to be measured effectively with an ion-selective electrode, the response of which tends to drift. Therefore, the experimental measurement is usually a comparison of the amount of radiolabeled cation accumulated by the cells against that remaining in the extracellular environment.
Several adjustments need to be made to derive Deltapsi from the total cell uptake: the plasma Deltapsi must be measured along with further adjustment for the cytoplasmic and extracellular binding to give the free concentration in the cytoplasm, and then the mitochondrial volume and binding correction are used to calculate the free concentration in the matrix. The mitochondrial Deltapsi is then calculated by inserting the free concentrations in the cytoplasm and mitochondrial matrix into the Nernst equation.
The rigorous application of these corrections to the uptake of radiolabeled TPMP by cells has enabled definitive measurement of Deltapsi of mitochondria in thymocytes, hepatocytes, and lymphocytes [27-29]. As lipophilic phosphonium cations pass easily through all biological membranes, they can also accumulate within the energized mitochondria of intact perfused organs. Therefore, the uptake of radiolabeled TPMP has been used to assess mitochondrial Deltapsi in perfused heart [30, 31], liver [32], and skeletal muscle [33].
DERIVATIVES OF TRIPHENYLPHOSPHONIUM CATIONS
Targeting compounds to mitochondria. Mitochondrial oxidative phosphorylation is at the heart of cell life and death, and there are many situations where it would be of value to be able to concentrate a bioactive molecule selectively within mitochondria in order to report on or to manipulate mitochondrial function, either as a research tool or as a potential therapy [8, 34-36]. As lipophilic cations are accumulated by mitochondria, the covalent attachment of a neutral bioactive compound to a lipophilic cation should lead to its selective delivery to mitochondria [8, 35].
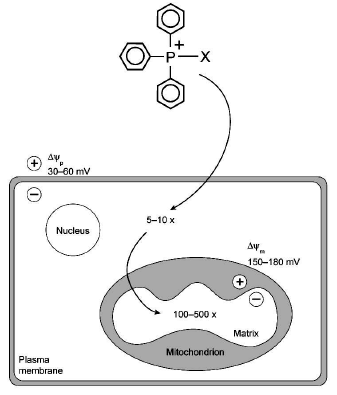
This approach was first used by Chen, who showed that attachment of the lipophilic cation rhodamine to the anti-cancer drug cisplatin facilitated its delivery to cells [37]. However, alkyltriphenylphosphonium cations are better candidates for the general delivery of compounds to mitochondria as they accumulate to a greater extent within mitochondria in cells. In addition, the chemistry required to form alkyltriphenylphosphonium cations from either the reaction of triphenylphosphine with an appropriate precursor [14, 22, 38], or by conjugation of a pre-formed alkyltriphenylphosphonium cation to a molecule is relatively straightforward [39, 40]. Therefore, it is possible to make a wide range of mitochondria-targeted lipophilic phosphonium cations. Such cations should be accumulated several hundred-fold by mitochondria within cells (Fig. 3). This approach can be used to direct a wide range of potential probe or therapeutic molecules to mitochondria, and some of these are outlined below.
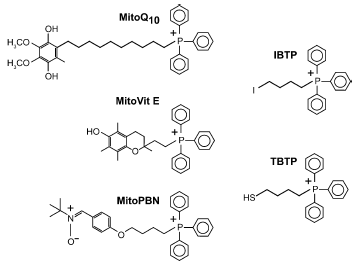
Mitochondria-targeted antioxidants. The mitochondrial respiratory chain is a major source of reactive oxygen species (ROS) within the cell, and these contribute to a number of degenerative diseases and also to redox signaling [41-45]. Therefore there is considerable interest in developing mitochondria-specific antioxidants [8, 35, 46]. If an antioxidant is attached to TPP, it should accumulate several hundred-fold within mitochondria in cells and selectively block mitochondrial oxidative damage and manipulate mitochondrial redox signaling (Fig. 3). We have synthesized a series of mitochondria-targeted antioxidants that comprise the TPP cation covalently attached to an antioxidant. Targeted antioxidants include derivatives of the endogenous antioxidants ubiquinol (MitoQ) [14, 22, 23] and alpha-tocopherol (MitoVit E) [38], and of the synthetic spin trap PBN (MitoPBN) [39] (Fig. 4).Fig. 3. Uptake of a mitochondria-targeted compound. The triphenylphosphonium cation attached to the moiety X to be delivered to mitochondria is first accumulated 5-10-fold in the cytoplasm, driven by the plasma membrane potential (Deltapsip) and then further accumulated (100-500-fold) by the mitochondrial membrane potential (Deltapsim).
These compounds have been found to block oxidative damage in isolated mitochondria and cells far more effectively than untargeted antioxidant analogs due to their concentration within mitochondria [22, 47, 48]. In addition, these molecules have been shown to block oxidative damage and prevent cell death in a model of Friedreich's ataxia [47], a disease that is thought to arise due to increased mitochondrial oxidative damage [49, 50]. This raises the possibility that mitochondria-targeted antioxidants may be of therapeutic benefit in those degenerative diseases involving mitochondrial oxidative damage. The utility of these compounds as therapies is supported by the finding that oral administration of lipophilic phosphonium cations leads to their accumulation in the brain, heart, and muscle which are all tissues that are affected by mitochondrial oxidative damage [51].Fig. 4. Mitochondria-targeted antioxidants and thiol reagents.
A further aspect of mitochondrial oxidative metabolism is the participation of mitochondria in redox signaling, by which they relay signals from the mitochondrion to the rest of the cell by the production of ROS, such as hydrogen peroxide, which then act on redox-sensitive effectors such as transcription factors [44, 45]. Mitochondria-targeted antioxidants have been shown to block many of these putative mitochondrial redox signaling and metabolic regulation pathways, including those involving p53 [52], damage to telomeres [53], and uncoupling protein (UCP) activation [39, 54]. Therefore, these compounds are also important tools in exploring the significance and mechanism of mitochondrial redox signaling.
Anti-cancer drugs. In many transformed cells, the mitochondrial and plasma membrane potentials are reported to be higher than in controls [37, 55-58]. Therefore, these cells should accumulate more of an administered lipophilic phosphonium cation than unaffected cells [37, 56], and if the compound were toxic to the cells, this could lead to a potential mechanism of differential toxicity. This approach has been tried by administering tetraPP at levels where its mitochondrial accumulation is great enough to lead to cell death in the cancer cells due to nonspecific disruption to mitochondrial function, but is sufficiently low to avoid killing the control cells [56]. This has also been extended to include the conjugation of toxic components such as protein alkylating reagents to TPP, thereby increasing the relative concentration of these compounds within cancer cells to selectively promote their death [59-62].
Mitochondrial protein thiol reagents. Mitochondrial thiol metabolism is involved in the response of mitochondria to oxidative damage and redox signaling [63]. An important aspect of this response is the reversible change to the redox state of critical protein thiols that are thought to be important in both sensing and responding to oxidative damage and redox signals [63, 64]. To identify those protein thiols that are particularly sensitive to these changes, we designed the mitochondria-targeted thiol reagents (4-iodobutyl)triphenylphosphonium (IBTP) and (4-thiobutyl)triphenylphosphonium (TBTP) [65-67] (Fig. 4). TBTP contains a free thiol that equilibrates with protein-protein disulfides formed under conditions of oxidative stress, enabling reactive protein thiols to be identified [66, 67].
In contrast, IBTP reacts selectively with exposed protein thiols to label them covalently; the labeled proteins can then be identified on Western blots using antiserum specific for the TPP moiety [65, 68]. The loss of this IBTP-labeling signal enables those proteins that are affected by mild oxidative stress to be identified. This approach has been used to identify some proteins in mitochondria, such as complex I, that have reactive thiols [68] and other thiol proteins affected by ethanol toxicity in the liver [69].
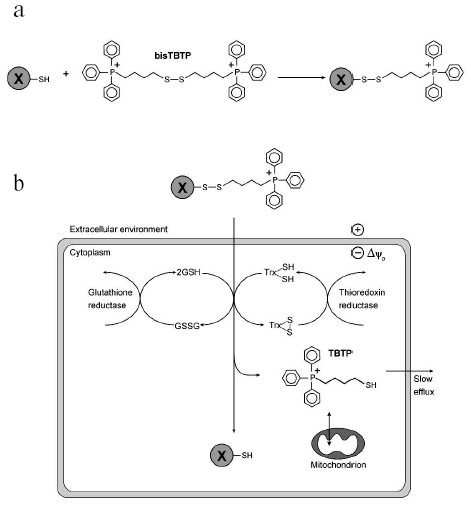
Cytoplasmic delivery vectors. Conjugation of molecules to TPP cations generally results in their delivery to mitochondria. However, TPP cations also facilitate the movement of compounds across the plasma membrane, with the plasma membrane potential (30-60 mV, negative inside) causing a 5-10-fold accumulation of TPP or TPMP cations relative to the extracellular environment. To prevent the conjugated molecule from going on to accumulate within mitochondria the TPP cation can be attached by a disulfide linkage, which will be reduced by the intracellular reductants glutathione and thioredoxin upon cellular uptake, releasing the passenger molecule into the cytosol while the TPP cation carries on to the mitochondrion (Fig. 5) [70]. As the disulfide bond should not be broken in the oxidizing extracellular environment this approach offers the potential to deliver a range of molecules selectively to the cytosol of cells in culture and also in vivo [70].
The early work of Skulachev and his collaborators introduced the use of triphenylphosphonium cations to mitochondrial biology [2, 4, 5]. Since then these compounds have been widely used to measure mitochondrial Deltapsi. More recently, their use has expanded to form the basis of a series of mitochondria-targeted probes and antioxidants. Future work will probably lead to the development of increasingly specific TPP derivatives for use as sensitive probes of mitochondrial function in cells and in vivo, and also to the development of therapies specifically targeted to mitochondria.Fig. 5. Cytoplasmic delivery of a compound by a disulfide-linked phosphonium cation. a) The molecule to be delivered, X, contains a free thiol group that is reacted with excess bisTBTP so that thiol-disulfide exchange generates X conjugated to a phosphonium cation by a disulfide bond. b) The cation leads to uptake into the cell, where the disulfide bond is reduced by intracellular reductants such as glutathione (GSH) or thioredoxin (TrxSH) to generate the free X in the cytosol. The glutathione disulfide (GSSG) or oxidized thioredoxin (TrxSS) generated are re-reduced by reductases, while the cation will be taken up by mitochondria and slowly lost from the cell.
We are grateful to Martin Brand for helpful discussions on these topics. MFR and GFK are Top Achiever Doctoral Scholars of the Tertiary Education Commission of New Zealand. AF is a New Zealand Science and Technology Postdoctoral Fellow of the Foundation for Research, Science and Technology of New Zealand. FHB holds an Otago University Doctoral Scholarship. HMC holds a PhD studentship from Research Into Ageing, UK.
REFERENCES
1.Nicholls, D. G., and Ferguson, S. J. (2002)
Bioenergetics 3, Academic Press, London.
2.Liberman, E. A., Topali, V. P., Tsofina, L. M.,
Jasaitis, A. A., and Skulachev, V. P. (1969) Nature, 222,
1076-1078.
3.Liberman, E. A., and Topaly, V. P. (1969)
Biofizika, 14, 452-461.
4.Bakeeva, L. E., Grinius, L. L., Jasaitis, A. A.,
Kuliene, V. V., Levitsky, D. O., Liberman, E. A., Severina, I. I., and
Skulachev, V. P. (1970) Biochim. Biophys. Acta, 216,
13-21.
5.Grinius, L. L., Jasaitis, A. A., Kadziauskas, Y.
P., Liberman, E. A., Skulachev, V. P., Topali, V. P., Tsofina, L. M.,
and Vladimirova, M. A. (1970) Biochim. Biophys. Acta,
216, 1-12.
6.Lewis, M. R., and Lewis, W. H. (1915) Am. J.
Anat., 17, 339-401.
7.Ketterer, B., Neumcke, B., and Laeuger, P. (1971)
J. Membr. Biol., 5, 225-245.
8.Murphy, M. P. (1997) Trends Biotechnol.,
15, 326-330.
9.Flewelling, R. F., and Hubbell, W. L. (1986)
Biophys. J., 49, 531-540.
10.Honig, B. H., Hubbell, W. L., and Flewelling, R.
F. (1986) Annu. Rev. Biophys. Biophys. Chem., 15,
163-193.
11.Gennis, R. B. (1989) in Biomembranes:
Molecular Structure and Function, Springer, New York, pp.
235-269.
12.Grunwald, E., Baughman, G., and Kohnstam, G.
(1960) JACS, 82, 5801-5811.
13.Flewelling, R. F., and Hubbell, W. L. (1986)
Biophys. J., 49, 541-552.
14.Smith, R. A., Kelso, G. F., James, A. M., and
Murphy, M. P. (2004) Meth. Enzymol., 382, 45-67.
15.Cafiso, D. S., and Hubbell, W. L. (1981) Annu.
Rev. Biophys. Bioeng., 10, 217-244.
16.Ono, A., Miyauchi, S., Demura, M., Asakura, T.,
and Kamo, N. (1994) Biochemistry, 33, 4312-4318.
17.Brand, M. D. (1995) in Bioenergetics - a
Practical Approach (Brown, G. C., and Cooper, C. E., eds.) IRL,
Oxford, pp. 39-62.
18.Azzone, G. F., Pietrobon, D., and Zoratti, M.
(1984) Curr. Top. Bioenerg., 13, 1-77.
19.McLaughlin, S., and Harary, H. (1976)
Biochemistry, 15, 1941-1948.
20.Demura, M., Kamo, N., and Kobatake, Y. (1985)
Biochim. Biophys. Acta, 820, 207-215.
21.Rottenberg, H. (1984) J. Membr. Biol.,
81, 127-138.
22.Kelso, G. F., Porteous, C. M., Coulter, C. V.,
Hughes, G., Porteous, W. K., Ledgerwood, E. C., Smith, R. A. J., and
Murphy, M. P. (2001) J. Biol. Chem., 276, 4588-4596.
23.Asin-Cayuela, J., Manas, A. R., James, A. M.,
Smith, R. A., and Murphy, M. P. (2004) FEBS Lett., 571,
9-16.
24.Brown, G. C., and Brand, M. D. (1985) Biochem.
J., 225, 399-405.
25.Rottenberg, H. (1979) Meth. Enzymol.,
55, 547-569.
26.Kamo, N., Muratsugu, M., Hongoh, R., and
Kobatake, Y. (1979) J. Membr. Biol., 49, 105-121.
27.Brand, M. D., and Felber, S. M. (1984)
Biochem. J., 217, 453-459.
28.Nobes, C. D., Brown, G. C., Olive, P. N., and
Brand, M. D. (1990) J. Biol. Chem., 265, 12903-12909.
29.Scarlett, J. L., Sheard, P. W., Hughes, G.,
Ledgerwood, E. C., Ku, H.-H., and Murphy, M. P. (2000) FEBS
Lett., 475, 267-272.
30.Kauppinen, R. (1983) Biochim. Biophys.
Acta, 725, 131-137.
31.Wan, B., Doumen, C., Duszynski, J., Salama, G.,
and LaNoue, K. F. (1993) Am. J. Physiol., 265,
H445-H452.
32.Steen, H., Maring, J. G., and Meijer, D. K.
(1993) Biochem. Pharmacol., 45, 809-818.
33.Rolfe, D. F. S., and Brand, M. D. (1996)
Biochim. Biophys. Acta, 1276, 45-50.
34.Weissig, V., and Torchilin, V. P. (2001) Adv.
Drug Deliv. Rev., 49, 1-2.
35.Murphy, M. P., and Smith, R. A. J. (2000) Adv.
Drug Deliv. Rev., 41, 235-250.
36.Green, K., Brand, M. D., and Murphy, M. P. (2004)
Diabetes, 53 (Suppl. 1), S110-118.
37.Chen, L. B. (1988) Annu. Rev. Cell Biol.,
4, 155-181.
38.Smith, R. A. J., Porteous, C. M., Coulter, C. V.,
and Murphy, M. P. (1999) Eur. J. Biochem., 263,
709-716.
39.Murphy, M. P., Echtay, K. S., Blaikie, F. H.,
Asin-Cayuela, J., Cocheme, H. M., Green, K., Buckingham, J. A., Taylor,
E. R., Hurrell, F., Hughes, G., Miwa, S., Cooper, C. E., Svistunenko,
D. A., Smith, R. A., and Brand, M. D. (2003) J. Biol. Chem.,
278, 48534-48545.
40.James, A. M., Blaikie, F. H., Smith, R. A.,
Lightowlers, R. N., Smith, P. M., and Murphy, M. P. (2003) Eur. J.
Biochem., 270, 2827-2836.
41.Raha, S., and Robinson, B. H. (2000) Trends
Biochem. Sci., 25, 502-508.
42.Beckman, K. B., and Ames, B. N. (1998)
Physiol. Rev., 78, 547-581.
43.Wallace, D. C. (1999) Science, 283,
1482-1488.
44.Finkel, T. (1998) Curr. Opin. Cell Biol.,
10, 248-253.
45.Finkel, T., and Holbrook, N. J. (2000)
Nature, 408, 239-247.
46.Murphy, M. P. (2001) Exp. Opin. Biol.
Ther., 1, 753-764.
47.Jauslin, M. L., Meier, T., Smith, R. A. J., and
Murphy, M. P. (2003) FASEB J., 17, 1972-1974.
48.Dhanasekaran, A., Kotamraju, S., Kalivendi, S.
V., Matsunaga, T., Shang, T., Keszler, A., Joseph, J., and
Kalyanaraman, B. (2004) J. Biol. Chem., 279,
37575-37587.
49.Campuzano, V., Montermini, L., Molto, M. D.,
Pianese, L., Cossee, M., Cavalcanti, F., Monros, E., Rodius, F.,
Duclos, F., Monticelli, A., et al. (1996) Science, 271,
1423-1427.
50.Kaplan, J. (1999) Proc. Natl. Acad. Sci.
USA, 96, 10948-10949.
51.Smith, R. A. J., Porteous, C. M., Gane, A. M.,
and Murphy, M. P. (2003) Proc. Natl. Acad. Sci. USA, 100,
5407-5412.
52.Hwang, P. M., Bunz, F., Yu, J., Rago, C., Chan,
T. A., Murphy, M. P., Kelso, G. F., Smith, R. A. J., Kinzler, K. W.,
and Vogelstein, B. (2001) Nat. Med., 7, 1111-1117.
53.Saretzki, G., Murphy, M. P., and von Zglinicki,
T. (2003) Aging Cell, 2, 141-143.
54.Echtay, K. S., Murphy, M. P., Smith, R. A.,
Talbot, D. A., and Brand, M. D. (2002) J. Biol. Chem.,
277, 47129-47135.
55.Weiss, M. J., Wong, J. R., Ha, C. S., Bleday, R.,
Salem, R. R., Steele, G. D., Jr., and Chen, L. B. (1987) Proc. Natl.
Acad. Sci. USA, 84, 5444-5448.
56.Rideout, D. C., Calogeropoulou, T., Jaworski, J.
S., Dagnino, R., and McCarthy, M. R. (1989) Anti-Cancer Drug
Design., 4, 265-280.
57.Modica-Napolitano, J. S., and Singh, K. (2002)
Expert Rev. Mol. Med., 2002, 1-19.
58.Modica-Napolitano, J. S., and Aprille, J. R.
(2001) Adv. Drug Deliv. Rev., 49, 63-70.
59.Manetta, A., Gamboa, G., Nasseri, A., Podnos, Y.
D., Ema, D., Dorion, G., Rawlings, L., Carpenter, P. M., Bustamante,
A., Patel, J., and Rideout, D. (1996) Gynecol. Oncol.,
60, 203-212.
60.Patel, J., Rideout, D., McCarthy, M. R.,
Calogeropoulou, T., Wadwa, K. S., and Oseroff, A. R. (1994)
Anticancer Res., 14, 21-28.
61.Rideout, D., Bustamante, A., and Patel, J. (1994)
Int. J. Cancer, 57, 247-253.
62.Rideout, D. (1994) Cancer Invest.,
12, 189-202.
63.Costa, N. J., Dahm, C. C., Hurrell, F., Taylor,
E. R., and Murphy, M. P. (2003) Antiox. Redox Signal, 5,
291-305.
64.Beer, S. M., Taylor, E. R., Brown, S. E., Dahm,
C. C., Costa, N. J., Runswick, M. J., and Murphy, M. P. (2004) J.
Biol. Chem., [Aug 30; epub ahead of print].
65.Lin, T. K., Hughes, G., Muratovska, A., Blaikie,
F. H., Brookes, P. S., Darley-Usmar, V., Smith, R. A. J., and Murphy,
M. P. (2002) J. Biol. Chem., 277, 17048-17056.
66.Burns, R. J., and Murphy, M. P. (1997) Arch.
Biochem. Biophys., 339, 33-39.
67.Burns, R. J., Smith, R. A. J., and Murphy, M. P.
(1995) Arch. Biochem. Biophys., 322, 60-68.
68.Taylor, E. R., Hurrell, F., Shannon, R. J., Lin,
T. K., Hirst, J., and Murphy, M. P. (2003) J. Biol. Chem.,
278, 19603-19610.
69.Venkatraman, A., Landar, A., Davis, A. J.,
Ulasova, E., Page, G., Murphy, M. P., Darley-Usmar, V., and Bailey, S.
M. (2004) Am. J. Physiol. Gastrointest. Liver Physiol.,
286, G521-527.
70.Filipovska, A., Eccles, M. R., Smith, R. A., and
Murphy, M. P. (2004) FEBS Lett., 556, 180-186.