Zinc Ions as Cytochrome c Oxidase Inhibitors: Two Sites of Action
S. S. Kuznetsova, N. V. Azarkina, T. V. Vygodina, S. A. Siletsky, and A. A. Konstantinov*
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Vorobjevy Gory, 119992 Moscow, Russia; fax: (7-095) 939-3181; E-mail: konst@genebee.msu.su* To whom correspondence should be addressed.
Received September 6, 2004
Zinc ions are shown to be an efficient inhibitor of mitochondrial cytochrome c oxidase activity, both in the solubilized and the liposome-reconstituted enzyme. The effect of zinc is biphasic. First there occurs rapid interaction of zinc with the enzyme at a site exposed to the aqueous phase corresponding to the mitochondrial matrix. This interaction is fully reversed by EDTA and results in a partial inhibition of the enzyme activity (50-90%, depending on preparation) with an effective Ki of ~10 µM. The rapid effect of zinc is observed with the solubilized enzyme, it vanishes upon incorporation of cytochrome oxidase in liposomes, and it re-appears when proteoliposomes are supplied with alamethicin that makes the membrane permeable to low molecular weight substances. Zinc presumably blocks the entrance of the D-protonic channel opening into the inner aqueous phase. Second, zinc interacts slowly (tens of minutes, hours) with a site of cytochrome oxidase accessible from the outer aqueous phase bringing about complete inhibition of the enzymatic activity. The slow phase is characterized by high affinity of the inhibitor for the enzyme: full inhibition can be achieved upon incubation of the solubilized oxidase for 24 h with zinc concentration as low as 2 µM. The rate of zinc inhibitory action in the slow phase is proportional to Zn2+ concentration. The slow interaction of zinc with the outer surface of liposome-reconstituted cytochrome oxidase is observed only with the enzyme turning over or in the presence of weak reductants, whereas incubation of zinc with the fully oxidized proteoliposomes does not induce the inhibition. It is shown that zinc ions added to cytochrome oxidase proteoliposomes from the outside inhibit specifically the slow electrogenic phase of proton transfer, coupled to a transition of cytochrome oxidase from the oxo-ferryl to the oxidized state (the F --> O step corresponding to transfer of the 4th electron in the catalytic cycle).
KEY WORDS: cytochrome c oxidase, zinc ion, Zn2+, proteoliposomes
Abbreviations: DETAPA) diethylenetriaminepentaacetate; TMPD) N,N,N´,N´-tetramethyl-p-phenylene diamine; CCCP) carbonyl cyanide m-chlorophenylhydrazone; COX) cytochrome oxidase; RuBpy) Ru(II)-tris-bipyridine.
Both mitochondrial and bacterial respiratory chains supply the cell with
energy due to the conversion of energy released from the oxidation of
substrates into transmembrane proton gradient across the coupling
membrane. The electron-transfer complexes of the respiratory chain,
such as NADH:ubiquinone reductase (complex I), ubiquinol:cytochrome
c reductase (complex III, or the complex of cytochromes
bc1), and cytochrome c oxidase (COX, complex
IV) are the coupling membrane-bound multisubunit proteins, which act as
redox-driven proton pumps translocating protons from the inside of
mitochondrion to the outside [1-3].
The study of the mechanism of transmembrane proton transport by respiratory chain complexes is one of main tasks of modern bioenergetics. A traditional approach is the use of specific inhibitors for proton pumps of the respiratory chain. Site-directed mutagenesis, which allows a selective blocking of one or another stage of the catalytic mechanism of the enzymes studied, is a powerful technique playing a pivotal role in studies on the bacterial respiratory chain that is closely related to the mitochondrial one. Unfortunately, this potent method is not applicable to studies on the respiratory chains of higher organisms, so in this case the search for new inhibitors still remains important.
Already in 1967, Skulachev and coworkers [4] had found that the electron transport in the mitochondrial respiratory chain complex III is effectively inhibited by zinc ions at the level of Rieske iron-sulfur protein. Further detailed studies performed in other laboratories confirmed these data and demonstrated a selective binding of Zn2+ to three sites in the surroundings of the Rieske iron-sulfer protein [5, 6]; at least in one case [6] the inhibition caused by zinc was associated with suppression of proton-transferring activity of complex III.
Later it was found that Zn2+ also inhibits the secondary quinone reduction in reaction centers of photosynthesizing bacteria by suppressing proton transfer in the proton-transducing pathways of these enzymes [7, 8].
Finally, some recent studies report that zinc taken at micromolar concentration inhibits the activity of cytochrome c oxidase (COX) [9-13]. Although the first study devoted to the inhibitory activity of zinc on COX was published long ago [9], this publication appeared in a little-known journal and remained unfollowed. A discovery of a new effective inhibitor of mitochondrial oxidase is very important. All respiratory inhibitors acting at the level of COX affect either the oxygen-reducing center of the enzyme (cyanide, carbon dioxide, azide, or formate) or interaction of COX with the natural electron donor cytochrome c (alkaline proteins and polypeptides [14]), that is, the electron exit and entry sites of cytochrome oxidase, whereas inhibitors blocking the intramolecular electron transfer were unknown.
It is worth noting that the works presently published on the effect of zinc on cytochrome oxidase are rather contradictory. This concerns a significant scatter in effective zinc concentrations and, and what is more important, the question on the location of the binding site of the inhibitor, which is known to not penetrate the membrane [10]. Thus, the data [10-12] suggest that zinc inhibits the enzyme binding to a site exposed to the inner aqueous phase in both mitochondria and bacteria. At the same time, the initial report of Nicholls [9] described COX inhibition by zinc via interaction of the cation with the surface exposed to the external aqueous phase. According to data of Ferguson-Miller's laboratory [13], as well as to the data obtained from another study [9], the electron transfer by COX incorporated into liposomes is only inhibited by exogenous zinc when the liposomal membrane is energized; however, no inhibition of cytochrome oxidase activity was observed in the presence of protonophore and valinomycin with potassium. In our laboratory we have found that zinc added to bovine heart cytochrome oxidase incorporated into liposomes specifically decelerates 5-6-fold the slow phase of the formation of transmembrane electric potential difference (Deltapsi) coupled to the transfer of the fourth electron in the catalytic cycle of the enzyme (the so-called F --> O transition) [15], and this effect is completely developed already during the first cytochrome oxidase turnover, when no significant membrane energization has yet arisen, which is difficult to reconcile with the results of Ferguson-Miller et al. [13].
The contradictory data on the interaction of zinc ions with COX might be partially explained by the diverse origin of the studied enzyme (in particular, COX has been isolated from cardiac mitochondria [9], Rhodobacter sphaeroides [11-13], and Paracoccus denitrificans [10]). Moreover, a possibility of zinc interaction with more than one site in COX cannot be excluded. In the present study, a comparison is made between the inhibitory effect of zinc on bovine heart mitochondrial cytochrome c oxidase in detergent solution or in liposomes. The experiments reveal at least two distinct sites of zinc action located on opposite sides of the coupling membrane.
MATERIALS AND METHODS
Materials. Commercial chemicals of high purity were purchased from Serva (Germany), Sigma (USA), Merck (Germany), Fluka (Switzerland), and Anatrace (USA); some chemicals of ultra purity grade were produced in Russia. Bovine heart mitochondrial COX was isolated according to the method of Fowler et al. [16] followed by additional purification according to MacLennan and Tsagoloff [17]. Mitochondria were isolated as described by Crane et al. [18]. The aa3 -type COX from a mutant strain of Rhodobacter sphaeroides, which contains six extra His residues on the C-terminus of the first subunit, was purified by affinity chromatography on a Ni-NTA agarose column [19]. Samples of the ba3-type COX and cytochrome c-552 from Thermus thermophilus were kindly provided by Dr. Tewfik Soulimane (Zurich). Proteoliposomes containing COX were prepared by cholate dialysis method [20, 21] using azolectin (phosphatidylcholine type IV-S, Sigma) as the phospholipid.
Zn2+-citrate buffer system. In most experiments Zn2+-citrate buffer system was used for maintenance of the required level of free Zn2+. The Kd value of Zn2+-citrate complex (1.11*10-5 M at pH 7.0) allows maintenance of stable Zn2+ concentration in the range of micromolar to tens of micromolar. Zinc ions were added as ZnCl2 on the background of 5 mM citrate. Additions providing for required concentration of free Zn2+ in a sample under given conditions (such as concentrations of citrate and other chelators, pH, ionic strength, and temperature) were calculated using the software package WinMAXC version 2.05.
Cytochrome oxidase activity assay. COX activity was monitored either amperometrically following decrease in the concentration of O2 in the medium or spectrophotometrically by oxidation of reduced cytochrome c. All measurements were conducted at 25°C under continuous stirring. Amperometric measurements of O2 consumption were performed with a covered Clark-type oxygen electrode. When the respiratory activity was monitored for more than 30 min, the following oxygen-regenerating system was used: catalase was added to the medium before the experiment, and 0.38 mM H2O2 was added repeatedly when the level of O2 decreased to ~20% of its initial value. Cytochrome c2+ oxidation was determined by the decrease of optical absorption at 550 nm relative to the 540 nm reference point using a SLM-Aminco DW-2000 spectrophotometer (SLM Instruments) in dual-wavelength kinetic mode at the monochromator slit width of 3 nm. The reaction was performed in a standard rectangular cuvette with a 10-mm light path. Reduced cytochrome c was obtained from its oxidized form by addition of sodium dithionite right before the experiment; excess dithionite was removed by gel filtration on a 5-ml Sephadex G-25 column.
Spectrophotometric measurements. Optical absorption spectra of COX were recorded using a Cary Bio 300 spectrophotometer (Varian, USA), in a split-beam mode at the monochromator slit width of 3 nm and the scan rate of 2 nm/sec in a standard cuvette with 10-mm light path at 25°C. The extinction coefficient Deltaepsilon605-630 = 27 mM-1*cm-1 was used to evaluate the concentration of COX in the sample from the “dithionite-reduced minus oxidized” difference absorption spectrum [22].
Electrometric measurements. Rapid kinetics of transmembrane electric potential difference (DeltaPsi) generation induced by liposome-reconstituted COX transition from the ferryl-oxo (F) to the oxidized (O) form was measured electrometrically as described previously [23]. Measurements were conducted at room temperature. RuBpy was used as a photoactive electron donor. It was photoexcited by 15-nsec flashes from a Quantel (France) YAG 471 laser operated at double-frequency (2nd harmonic) mode (lambda = 532 nm, 30-50 mJ/flash).
RESULTS AND DISCUSSION
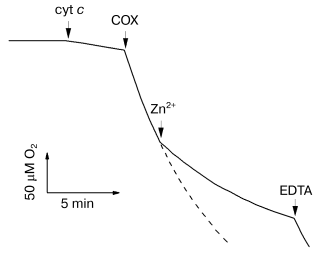
Inhibition of solubilized cytochrome oxidase by Zn2+. Biphasic character of the inhibitory effect of zinc. Figure 1 shows the effect of zinc ions on the rate of oxygen consumption by solubilized cytochrome oxidase isolated from bovine heart mitochondria. Addition of 200 µM Zn2+ results in strong inhibition of COX activity. We found that the inhibition develops on two time scales: fast (<30 sec) and slow (tens of minutes). The fast phase can be completely reversed, whereas the slow phase is only partially reversed by excess EDTA.
The activity of R. sphaeroides aa3-type cytochrome c oxidase is also inhibited by zinc ions (data not shown), and the inhibition parameters are similar to those of the mitochondrial enzyme (see below). However, the T. thermophilus ba3-type COX is totally resistant to Zn2+ taken at concentration up to several millimolar (data not shown).Fig. 1. Inhibition of solubilized COX by Zn2+. The reaction was initiated by 0.05 µM bovine heart mitochondrial COX added into the polarographic cell. The slow decline of O2 between the additions of the respiration substrate “cyt c” (5 mM ascorbate, 0.1 mM TMPD, and 4 µM cytochrome c) and COX is due to nonenzymatic oxidation of ascorbate. The dashed curve shows the oxidation in absence of Zn2+. Additions: Zn2+ (as ZnCl2), 200 µM; EDTA, 250 µM. Experimental medium: 50 mM HEPES/KOH, pH 7.8, 50 mM K2SO4, and 0.05% dodecylmaltoside.
To our knowledge, the possibility of an additional slow effect of zinc ions on COX activity has not yet been considered. In the study [13], rapid-mixing experiments revealed the inhibition on the second and subsecond time scales. Other publications devoted to the interaction of COX with Zn2+ report no data on the enzyme-inhibitor incubation time [9-12]. It is likely that, in some cases, the fast effect of zinc ions was partially superimposed by a slower process, which could be one of the reasons for the scatter in estimations of Ki (from 2.6 µM [11] to 75 µM [10]). In this work, we aimed at characterizing separately the fast and slow components of the inhibitor effect.
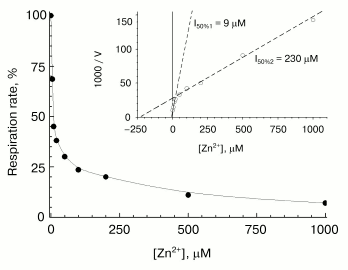
Fast phase of zinc action. A typical concentration-dependent inhibitory effect of zinc on COX activity is shown in Fig. 2. Activity was assayed within 15 sec after mixing of the enzyme with the inhibitor. Part of the curve at free zinc concentrations up to 100-200 µM corresponds essentially to contribution of the fast inhibition component. At [Zn2+] > 100-200 µM, the contribution of the slow phase of inhibitor action shows up (because the rate of the slow phase increases proportionally to zinc concentration--see below). One can see that the oxygen consumption rate measured right after mixing of the working enzyme with the inhibitor does not fall to zero (decreases by ~70%). The part of the activity sensitive to the fast reversible inhibition by zinc varied from 50 to 90% depending on the preparation. The linear Dixon plot (see the inset in Fig. 2) allows to estimate Ki = 9 µM for the fast phase, which exactly agrees with the value for the solubilized R. sphaeroides COX [13]. As in [13], the residual COX activity is inhibited by high zinc concentrations, which, as mentioned above, is due to the contribution of the slow inhibition phase. This phase is characterized by the apparent value of [I]50 = 230 µM (inset in Fig. 2), which, however, is a purely formal characteristic, because the slow binding of zinc is virtually irreversible, and the inhibition level depends on the incubation time.
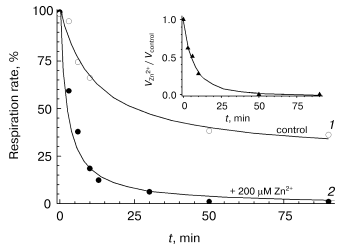
Slow phase of zinc action. The studies on the slow-phase of inhibition of COX activity by zinc are complicated by several circumstances. In particular, the turning over enzyme undergoes slow spontaneous inactivation (see for instance Fig. 1, dashed line; Fig. 3, curve 1). This inactivation seems to be partially caused by the presence of contaminating heavy-metal cations, because chelators, such as EDTA and DETAPA, stabilize COX activity significantly (but not completely, data not shown). To minimize zinc-independent inactivation of COX, subsequent experiments were carried out in the presence of 50 µM EDTA. We also used the Zn2+-citrate buffer system (see “Materials and Methods”) to provide the required concentration of free zinc ions. Nonetheless, the COX when turning over underwent slow inactivation even in the presence of chelators, so that it lost about a half of the activity in 1-1.5 h (see Fig. 3, curve 1). The resting oxidized enzyme virtually does not lose its activity (data not shown). This observation is consistent with the relevant data on the bacterial cytochrome c oxidase [24, 25] and, according to inferences from those studies, is explained by irreversible escape of one of the catalytic intermediates into inactive form of the enzyme; this process is particularly pronounced upon dissociation of the third subunit of COX.Fig. 2. Inhibitory effect of Zn2+ on solubilized COX. The cytochrome oxidase reaction was initiated by addition of 0.05 µM of mitochondrial COX and ZnCl2 up to the specified concentration into the medium containing 50 mM HEPES/KOH, pH 7.8, 50 mM K2SO4, 0.05% dodecylmaltoside, and the respiration substrate (5 mM ascorbate, 0.1 mM TMPD, and 4 µM cytochrome c). Activity was determined within 15 sec after the beginning of respiration. Initial activity of the control sample without Zn2+ is taken as 100%. Inset, the data linearized in Dixon's coordinates.
The oxygen-reducing center of cytochrome c oxidase is known to interact with a variety of low molecular weight anions including chloride ions [26-28]. So, we have performed control experiments showing that incubation of COX with 10 mM KCl instead of 5 mM ZnCl2 (maximal concentration in experiments with the Zn2+-citrate buffer) does not result in a decrease in COX activity (data not shown). Thus, the slow inhibition of COX in our experiments is not caused by Cl-.Fig. 3. Effect of Zn2+ on the activity of solubilized COX in the course of respiration. Activity of mitochondrial COX (0.05 µM) was determined over the given time intervals after initiation of the cytochrome oxidase reaction in the presence of 200 µM free Zn2+ (filled symbols, curve 2) or in the absence of the inhibitor (open symbols, curve 1). The inset displays the dynamics of the ratio of these values. Concentration of O2 was maintained in the range of 50-250 µM with serial addition of H2O2 aliquots in the presence of catalase (see “Materials and Methods”). The reaction mixture contained 50 mM HEPES/KOH, pH 7.8, 50 mM K2SO4, 0.05% dodecylmaltoside, 50 µM EDTA, and 5 mM citrate. Hereinafter the free Zn2+ concentration was preset using the Zn2+-citrate buffer system (see “Materials and Methods”).
Evaluation of Ki for the slow phase of COX inhibition by zinc is difficult experimentally and probably is not correct because the inhibition is a lengthy and incompletely reversible process. Nevertheless, we have made some empirical estimations for the tightness of binding. For instance, 10 µM aerobically oxidized COX completely loses its activity when incubated for 24 h in the presence of 2 µM Zn2+ (buffered concentration of free ions), whereas 30% of the activity is lost by the control sample during the same time. Hence, the effective Ki value is at least one order of magnitude less than 2 µM.
The experiments have revealed a direct proportionality between the rate of COX inactivation in the slow phase and concentration of free zinc ions ranging from 2 to 200 µM (data not shown). If a bimolecular mechanism of COX-Zn2+ interaction is assumed, the value of the second-order reaction rate constant would be 60 ± 15 M-1*sec-1 (four measurements).
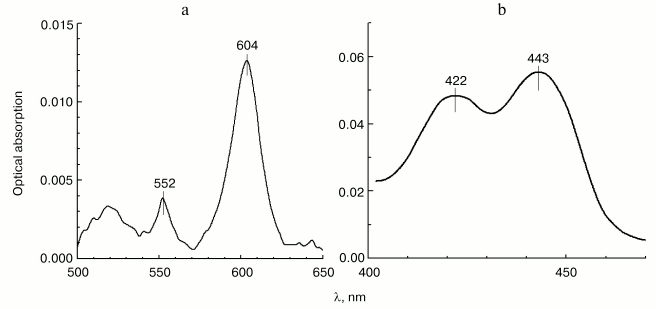
Localization of the site of zinc effect on the electron transfer in cytochrome c oxidase. In order to determine which step of intramolecular electron transfer in cytochrome c oxidase is sensitive to inhibition by zinc ions, we have studied the effect of Zn2+ on the reduction level of hemes in COX oxidizing substrates under the conditions of steady-state turnover. Figure 4 shows the spectral changes developed in 30 min after addition of 200 µM of Zn2+ to the enzyme oxidizing ascorbate aerobically in the presence of 0.1 µM cytochrome c and 10 µM TMPD. In the visible absorption spectrum (panel (a)), one can see a significant (15-20%) increase in the reduction level of low-spin heme a, as well as a small, but well-reproducible blue shift (from 605 to 604 nm) of the peak corresponding to the alpha-band of heme a. Both effects are in agreement with the observations of the effect of Zn2+ on cytochrome oxidase, which were reported originally in [9], but were not further examined in the studies of other researchers. An accompanying increase in the steady-state reduction level of cytochrome c (maximum at 552 nm in Fig. 4a) results from general deceleration of the cytochrome c oxidase reaction. In the Soret region (panel (b)), the spectral contribution of reduced cytochrome c has been subtracted allowing separate determination of the effect of zinc on the reduction levels of both COX hemes, a and a3, whose gamma-bands are comparable in amplitude. Along with the gamma-band of the additionally reduced heme a (443 nm), a distinct peak at 422 nm is also visible, whose position differs somewhat from the gamma-band of the oxidized high-spin heme a3 in the resting enzyme (usually at 414-418 nm). This absorption band might belong to one of the catalytic intermediates of heme a3, such as low-spin hydroxide complex of the oxidized heme. Formally, we cannot exclude a possibility of a Zn2+-dependent bathochromic shift of the gamma-band of the oxidized heme a3, but this interpretation is less likely. In general, our data allow to conclude that prolonged effect of Zn2+ on COX in detergent solution leads to deceleration of electron transfer between hemes a and a3, which is in agreement with the conclusions of study [9].
Effect of Zn2+ on the activity of cytochrome oxidase incorporated into liposomes. The study on the effect of zinc ions on cytochrome oxidase reconstituted in liposomes was of special interest. As we mentioned above, the data and conclusions of different authors concerning this issue are contradictory. The study [10] performed on zinc-loaded proteoliposomes, led to a conclusion that there exists only one cation-binding site localized on the surface of COX exposed on the inner side of liposomal membrane, and it was claimed that the inhibition only affects the proton-translocating function of the enzyme with no effect on the rate of electron transfer. However, the earlier publication of Nicholls and Singh [9] describes the inhibition of COX activity by zinc ions added to proteoliposomes from the outside. The group of Ferguson-Miller has published data indicating that Zn2+ added from the outside only results in fast inhibitory effect on liposome-reconstituted COX, when DeltaµH+ is present across the proteoliposome membrane [13]. Finally, our data from electrometric experiments (see below) indicate facile interaction of Zn2+ with the outer surface of liposome-reconstituted enzyme, when the membrane is not yet energized.Fig. 4. Effect of Zn2+ on the heme reduction level in turning over COX. The difference spectrum characterizing the effect (Zn2+ minus control) was obtained by subtraction of the absorption spectrum of 3 µM mitochondrial COX recorded 30 min after addition of 200 µM free Zn2+ from the spectrum recorded before the addition. The incubation medium contained 50 mM HEPES/KOH, pH 7.8, 50 mM K2SO4, 0.05% dodecylmaltoside, 50 µM EDTA, 5 mM citrate, 1 mM ascorbate, 10 µM TMPD, and 0.1 µM cytochrome c. a) Visible part of the spectrum; b) Soret-band spectrum. The spectrum of cytochrome c, which was additionally reduced in the presence of Zn2+, has been subtracted.
Since phospholipid membrane is virtually impermeable to divalent cations, a comparison of Zn2+ effects on proteoliposomes and solubilized enzyme could help to identify separate zinc-binding sites of COX and establish their localization with respect to the outer and inner aqueous phases separated by the coupling membrane.
Fast phase of zinc action. The effects of increasing concentrations of Zn2+ on the activities of solubilized and liposome-reconstituted COX are shown in Fig. 5. Experimental conditions enable preferential recording of the fast inhibition phase. One can see that the cation taken in concentration inhibiting the respiration of solubilized enzyme (curve 1), has no effect on the respiration of uncoupled proteoliposomes (curve 2). These data are consistent with the observations [13] in which the initial rate of oxygen consumption by liposome-reconstituted COX remains virtually non-inhibited by zinc when DeltaµH+ across the membrane is absent. Thus, the fast phase of the inhibitory effect of zinc on the activity of solubilized cytochrome oxidase (curve 1) disappears upon incorporation of the enzyme into liposomes. We can propose that in proteoliposomes, the binding site responsible for the fast inhibition phase is inaccessible for the externally added cation, because it is localized inside the membrane vesicles.
This suggestion is supported by the data presented in Fig. 6a. In this case, the reaction was initiated by addition of ferrocytochrome c to aerobic reaction mixture containing COX, and kinetics of cytochrome c oxidation was assayed by the decrease in optical absorption at 550 nm versus the 540 nm reference point. Under conditions of the given experiment, the cytochrome oxidase reaction is fast (complete in about 1-1.5 min). In accordance with the data presented in Fig. 5, the rate of ferrocytochrome c oxidation by COX reconstituted into liposomes remains virtually unchanged in the presence of externally added zinc ions (compare curves 1 and 2).Fig. 5. Resistance of COX incorporated into liposomes to the rapid inhibition by Zn2+. Initial cytochrome oxidase activity of solubilized (open symbols, curve 1) and incorporated in liposomes (filled symbols, curve 2) mitochondrial COX was determined in presence of 5 mM ascorbate, 0.1 mM TMPD, 4 µM cytochrome c, and specified concentrations of free Zn2+. The incubation medium for liposomes contained 50 mM MOPS/KOH, pH 7.0, 50 mM K2SO4, 5 mM citrate, 50 µM EDTA, 2 µM CCCP, and 1 µM valinomycin; the final COX concentration was 0.25 µM. Activity of the solubilized enzyme (0.05 µM) was determined in the medium containing 50 mM HEPES/KOH, pH 7.8, 50 mM K2SO4, 0.05% dodecylmaltoside, 50 µM EDTA, and 5 mM citrate.
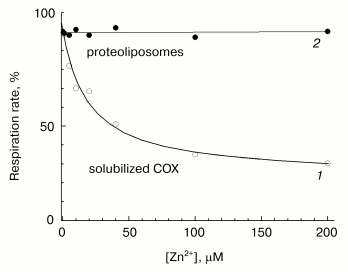
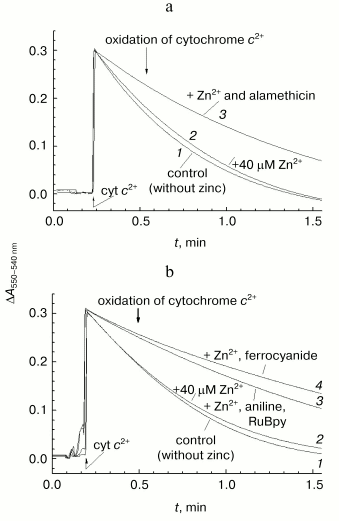
When the same experiment is performed in the presence of the channel-forming agent alamethicin, which makes proteoliposome membrane permeable for low molecular weight substances, zinc causes appreciable deceleration of the reaction (curve 3). Efficiency of inhibition observed at given concentration (40 µM) of Zn2+ corresponds to the fast effect of zinc on the solubilized enzyme. So, the fast inhibition by zinc ions is likely due to cation binding with a COX site localized on the protein surface exposed to the inner aqueous phase of the liposomes.Fig. 6. Difference in the effects of Zn2+ added from the outer and inner sides of membrane on liposome-reconstituted COX. Cytochrome oxidase activity of COX incorporated into liposomes was determined by following oxidation of ferrocytochrome c monitored as decrease in optical absorption at 550 nm relative to the 540 nm reference point. The incubation medium contained 25 mM HEPES/KOH, pH 7.5, 100 mM KCl, 5 mM citrate, and 1 µM CCCP. An upward jump of absorption at the zero time is caused by addition of 20 µM of reduced cytochrome c. a) COX from mitochondria (final concentration, 3 nM). Curves: 1) the experiment in the absence of the inhibitor; 2) in the presence of 40 µM free Zn2+; 3) in the presence of 40 µM Zn2+ and 16 µg/ml of alamethicin. b) COX from R. sphaeroides (final concentration, 0.7 nM). Curves: 1) the experiment in the absence of the inhibitor; 2-4) in the presence of 40 µM free Zn2+ and also 3) 10 mM aniline and 20 µM RuBpy or 4) 100 µM potassium ferrocyanide.
It is quite likely that the rapid effect of zinc on COX is caused by blocking proton uptake from the inner aqueous phase into the D-channel [11, 12]. Heme-copper terminal oxidases of A-class [29] (in particular, the aa3-type enzymes from mitochondria and R. sphaeroides) are characterized by the presence of a cluster of dicarboxylate and histidine residues localized near the entry of the D-channel, which might bind Zn2+ [11]. This hypothesis is consistent with the above-mentioned zinc-resistance of T. thermophilus cytochrome oxidase of ba3 type in which such a cluster is absent.
Slow phase of zinc action. As mentioned above, the resistance of respiration of the uncoupled COX-containing proteoliposomes to zinc (Fig. 5, [13]) contradicts the data of both the study of Nicholls [9], who reported inhibition of liposome-reconstituted oxidase activity, and the data obtained in our laboratory, which indicate that Zn2+ added from the outside selectively inhibits the slow phase of membrane potential generation coupled to transfer of the fourth electron in the catalytic cycle of COX [15]. A typical experiment illustrating the latter observation is presented in Fig. 7.
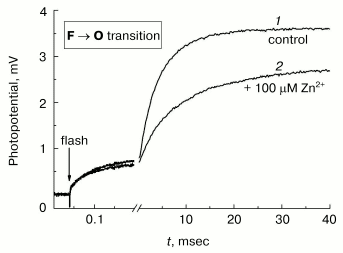
Photoinduced single-electron reduction of the input copper center CuA in COX converted to the ferryl-oxo state using hydrogen peroxide is followed by transfer of the electron from CuA to the heme a and subsequently to the oxygen-reducing center, in which the ferryl-oxo intermediate of heme a3 (Fe4+=O2-) is reduced to form the complex of oxidized heme a3 with hydroxide (Fe3+-OH-) [30-32]. This process is electrogenic and reveals three phases of membrane potential increase (see the control curve 1 in Fig. 7): rapid (tau ~ 50 µsec), intermediate (tau about 1 msec), and slow (tau ~ 4-5 msec at pH 8). The intermediate and slow phases are due to the vectorial transport of protons in the inside-to-outside direction, and the rapid phase corresponds to the vectorial transport of the electron in the opposite direction from CuA to the heme a [31, 33]. One can see that the protonic part of membrane potential generation is hindered dramatically in the presence of 100 µM Zn2+. Decomposition of kinetic curves into individual exponents indicates that Zn2+ decelerates 6- to 7-fold the slow phase with but a slight effect on the intermediate phase. It is worth noting that the slow electrogenic phase of the F --> O transition of COX corresponds to the limiting step of the enzyme oxidation; hence, its inhibition by zinc should be accompanied by inhibition of the enzyme activity under steady-state turnover conditions.Fig. 7. Inhibitory effect of Zn2+ on the electrogenic activity of COX. Generation of Deltapsi across the membrane of COX-containing proteoliposomes coupled to transition of the enzyme from F to O state, was assayed electrometrically (see “Materials and Methods”). Proteoliposomes were preincubated for 30 min in the medium containing 5 mM Tris-acetate, pH 8.0, 10 mM aniline, 40 µM RuBpy, and 4 mM H2O2. When the control curve 1 was recorded, 100 µM of Zn2+ was added into the cuvette, and curve 2 was recorded after 5 min incubation. Curves 1 and 2 were normalized to the amplitude of the rapid electrogenic phase [23, 31].
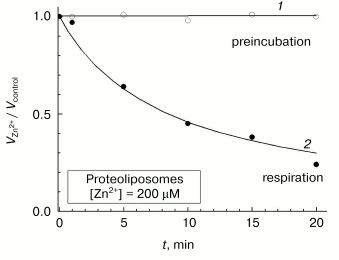
Further experiments have shown that the above-described contradictions among the reports concerned with the effect of zinc ions on liposome-reconstituted COX can be explained by peculiarities of the slow inhibition phase. Although prolonged incubation of aerobically oxidized COX-containing proteoliposomes with zinc does not result in a decrease in the respiratory activity (Fig. 8, curve 1), the enzyme undergoes slow and practically complete inactivation in the presence of the inhibitor under turnover conditions (curve 2). The rate of COX inactivation in proteoliposomes is close to that observed for the slow phase of inhibition of the solubilized sample (cf. Fig. 8 and inset in Fig. 3). The inhibition of proteoliposome respiration in the presence on Zn2+ added from the outside described in [9], apparently was developing during the enzyme turnover. On the other hand, the slow phase of the inhibitory effect of Zn2+ could not be detected in experiments of Ferguson-Miller and coworkers [13], in which COX activity in uncoupled liposomes was assayed just for a few seconds after initiation of respiration in the presence of zinc cations. We can conclude that the slow phase of the inhibitory effect of Zn2+ corresponds to its binding with the site localized on the surface of COX exposed to the outer aqueous phase, and turnover of the enzyme is pre-requisite for its inhibition by zinc in the slow phase.
The fact that zinc can only exhibit its slow inhibitory effect on COX under the enzyme turnover conditions is an unusual phenomenon, which is of significant interest. Note that the experiments were carried out under uncoupling conditions; therefore, the possibility of energy-dependent transport of Zn2+ cations into proteoliposomes down the electric field is excluded. One can suggest that the slow phase of Zn2+ binding with COX at the site exposed to external aqueous phase is redox-dependent and is absent in the case of completely oxidized enzyme. The effect could be due to a selective interaction of the cation with one or several partially reduced intermediates of the catalytic cycle or with intermediate COX states arising for a short period during the transition of one relatively stable intermediate into another.Fig. 8. Effect of Zn2+ on the activity of COX in proteoliposomes. Cytochrome oxidase activity of mitochondrial COX reconstituted in liposomes was assayed as the initial respiration rate either after incubation of the aerobically oxidized sample with Zn2+ for a given time (open symbols, curve 1) or at given time intervals after initiation of cytochrome oxidase turnover in Zn2+-containing medium (filled symbols, curve 2). Respiration substrate: 5 mM ascorbate, 0.1 mM TMPD, and 4 µM cytochrome c. The values obtained in the presence of 200 µM free Zn2+ are normalized to the activity of the control sample. Incubation medium: 50 mM MOPS/KOH, pH 7.0, 50 mM K2SO4, 2 µM CCCP, and 1 µM valinomycin. Proteoliposomes were added to the final COX concentration of 0.25 µM.
These considerations might explain yet another contradiction revealed in our work: in electrometric experiments (Fig. 7) the total inhibitory effect of 100 µM zinc is observed in 3-5 min after addition of the cation to liposome-reconstituted COX, even if the oxidation substrates are absent, whereas in the respiring proteoliposomes the effect of 200 µM zinc develops in 10-20 min.
Initially we thought that it is the ferryl-oxo state of the enzyme in the experiment presented in the Fig. 7 that reveals the selective sensitivity to zinc; however, the hypothesis was not verified. Then we realized that in the electrometric studies, a photoactive electron donor RuBpy is present in the reaction medium, as well as relatively high concentrations (10 mM) of aniline, which is an electron donor for photooxidized RuBpy and regenerates a reduced form of the latter. Aniline itself is a potential weak electron donor for the COX ferryl-oxo complex (the enzyme possesses a weak peroxidase activity).
It was found that the exposure of liposome-reconstituted COX to dim light for 1-2 min in the presence of RuBpy and aniline is sufficient for development of the inhibitory effect of zinc on oxidation of ferrocytochrome c (Fig. 6b, curve 3). The same result can be achieved when aniline and RuBpy are replaced by 100 µM ferrocyanide, a typical weak electron donor for COX (Fig. 6b, curve 4).
The steady-state rate of COX turnover during its aerobic incubation either with aniline and RuBpy or with 100 µM ferrocyanide is so low that it cannot be measured; however, some redox centers of the enzyme are apparently reduced. Careful spectrophotometric studies did not reveal any noticeable reduction of heme a or a3 or any formation of heme a3 oxygen intermediates during the incubation of COX with ferrocyanide or aniline and RuBpy (data not shown). One can anticipate that the so-called “invisible” copper (CuB) in the oxygen-reducing center of COX is the component reduction of which is responsible for zinc binding on the outer surface. Indeed, as pointed out in [34, 35], of all the redox centers of COX CuB may have the highest affinity for an electron due to the extremely high stability of the planar structure of the coordination sphere of Cu1+ formed by three histidines. To verify the hypothesis on the influence of the CuB redox state on the COX susceptibility to “slow” zinc action, further experiments are necessary with the use of ESR spectroscopy.
This study was supported by the Howard Hughes Medical Institute (Grant 55000320) and the Russian Foundation for Basic Research (Grant Nos. 03-04-48203 and 04-04-48856).
REFERENCES
1.Mitchell, P. (1966) Chemiosmotic Coupling in
Oxidative and Photosynthetic Phosphorylation, Glynn Research Ltd.,
Bodmin, UK.
2.Skulachev, V. P. (1988) Membrane
Bioenergetics, Springer, Berlin.
3.Rich, P. R. (1991) Biosci. Rep., 11,
539-571.
4.Skulachev, V. P., Chistyakov, V. V., Jasaitis, A.
A., and Smirnova, E. G. (1967) Biochem. Biophys. Res. Commun.,
26, 1-6.
5.Lorusso, M., Cocco, T., Sardanelli, A. M., Minuto,
M., Bonomi, F., and Papa, S. (1991) Eur. J. Biochem.,
197, 555-561.
6.Berry, E. A., Zhang, Z., Bellamy, H. D., and Huang,
L. (2000) Biochim. Biophys. Acta, 1459, 440-448.
7.Paddock, M. L., Graige, M. S., Feher, G., and
Okamura, M. Y. (1999) Proc. Natl. Acad. Sci. USA, 96,
6183-6188.
8.Axelrod, H. L., Abresch, C., Paddock, M. L.,
Okamura, M. Y., and Feher, G. (1999) Proc. Natl. Acad. Sci. USA,
97, 1542-1547.
9.Nicholls, P., and Singh, A. P. (1988) Life Sci.
Adv. (Biochemistry, Delhi), 7, 321-326.
10.Kannt, A., Ostermann, T., Muller, H., and
Ruitenberg, M. (2001) FEBS Lett., 503, 142-146.
11.Aagaard, A., and Brzezinski, P. (2001) FEBS
Lett., 494, 157-160.
12.Aagaard, A., Namslauer, A., and Brzezinski, P.
(2002) Biochim. Biophys. Acta, 1555, 133-139.
13.Mills, D. A., Schmidt, B., Hiser, C., Westley,
E., and Ferguson-Miller, S. (2002) J. Biol. Chem., 277,
14894-14901.
14.Konstantinov, A. A., Maslov, S. P., Severina, I.
I., and Skulachev, V. P. (1975) Biokhimiya, 40,
401-407.
15.Konstantinov, A. A. (2001) FEBS Annual
Abst., p. 37 (W08-2).
16.Fowler, L. R., Richardson, S. H., and Hatefi, Y.
(1962) Biochim. Biophys. Acta, 64, 170-173.
17.MacLennan, D. H., and Tzagoloff, A. (1965)
Biochim. Biophys. Acta, 96, 166-168.
18.Crane, F. L., Gleen, J. L., and Green, D. E.
(1956) Biochim. Biophys. Acta, 22, 475-487.
19.Mitchell, D. M., and Gennis, R. B. (1995) FEBS
Lett., 368, 148-150.
20.Racker, E. (1972) J. Membr. Biol.,
10, 221-235.
21.Hinkle, P. C. (1979) Meth. Enzymol.,
55, 748-751.
22.Nicholls, P., Petersen, L. C., Miller, M., and
Hansen, F. B. (1976) Biochim. Biophys. Acta, 449,
188-196.
23.Konstantinov, A. A., Siletsky, S., Mitchell, D.,
Kaulen, A., and Gennis, R. B. (1997) Proc. Natl. Acad. Sci. USA,
94, 9085-9090.
24.Bratton, M. R., Pressler, M. A., and Hosler, J.
P. (1999) Biochemistry, 38, 16236-16245.
25.Hosler, J. P. (2004) Biochim. Biophys.
Acta, 1655, 332-339.
26.Moody, A. J., Cooper, C. E., and Rich, P. R.
(1991) Biochim. Biophys. Acta, 1059, 189-207.
27.Giuffre, A., Stubauer, G., Brunori, M., Sarti,
P., Torres, J., and Wilson, M. T. (1998) J. Biol. Chem.,
273, 32475-32478.
28.Fabian, M., Skultety, L., Brunel, C., and Palmer,
G. (2001) Biochemistry, 40, 6061-6069.
29.Pereira, M. M., Santana, M., and Teixeira, M.
(2001) Biochim. Biophys. Acta, 1505, 185-208.
30.Nilsson, T. (1992) Proc. Natl. Acad. Sci.
USA, 89, 6497-6501.
31.Zaslavsky, D., Kaulen, A., Smirnova, I. A.,
Vygodina, T. V., and Konstantinov, A. A. (1993) FEBS Lett.,
336, 389-393.
32.Zaslavsky, D., Sadoski, R. C., Wang, K., Durham,
B., Gennis, R. B., and Millett, F. (1998) Biochemistry,
37, 14910-14916.
33.Siletsky, S., Kaulen, A. D., and Konstantinov, A.
A. (1999) Biochemistry, 38, 4853-4861.
34.Tsukihara, T., Aoyama, H., Yamashita, E.,
Takashi, T., Yamaguichi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono,
R., and Yoshikawa, S. (1996) Science, 272, 1136-1144.
35.Yoshikawa, S., Shinzawa-Itoh, K., Nakashima, R.,
Yaono, R., Inoue, N., Yao, M., Fei, M. J., Libeu, C. P., Mizushima, T.,
Yamaguchi, H., Tomizaki, T., and Tsukihara, T. (1998) Science,
280, 1723-1729.