REVIEW: Structural Insights into Excitation-Contraction Coupling by Electron Cryomicroscopy
I. I. Serysheva
Department of Molecular Physiology and Biophysics, National Center for Macromolecular Imaging, Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA; fax: (713) 798-3456; E-mail: irinas@bcm.tmc.edu
Received August 2, 2004
In muscle, excitation-contraction coupling is defined as the process linking depolarization of the surface membrane with Ca2+ release from cytoplasmic stores, which activates contraction of striated muscle. This process is primarily controlled by interplay between two Ca2+ channels--the voltage-gated L-type Ca2+ channel (dihydropyridine receptor, DHPR) localized in the t-tubule membrane and the Ca2+-release channel (ryanodine receptor, RyR) of the sarcoplasmic reticulum membrane. The structures of both channels have been extensively studied by several groups using electron cryomicroscopy and single particle reconstruction techniques. The structures of RyR, determined at resolutions of 22-30 Å, reveal a characteristic mushroom shape with a bulky cytoplasmic region and the membrane-spanning stem. While the cytoplasmic region exhibits a complex structure comprising a multitude of distinctive domains with numerous intervening cavities, at this resolution no definitive statement can be made about the location of the actual pore within the transmembrane region. Conformational changes associated with functional transitions of the Ca2+ release channel from closed to open states have been characterized. Further experiments determined localization of binding sites for various channel ligands. The structural studies of the DHPR are less developed. Although four 3D maps of the DHPR were reported recently at 24-30 Å resolution from studies of frozen-hydrated and negatively stained receptors, there are some discrepancies between reported structures with respect to the overall appearance and dimensions of the channel structure. Future structural studies at higher resolution are needed to refine the structures of both channels and to substantiate a proposed molecular model for their interaction.
KEY WORDS: excitation-contraction coupling, ryanodine receptor, voltage-gated L-type Ca2+ channel, electron cryomicroscopy, single particle reconstruction
It has long been appreciated that muscle contraction is regulated by the intracellular Ca2+ level maintained via interaction between two membrane systems, the muscle fiber surface membrane and the sarcoplasmic reticulum (SR) membrane separating the internal Ca2+ stores of the SR from the cytosol. Excitation-contraction (E-C) coupling is the process that links electrical stimulation of muscle to the release of Ca2+ from the SR into the cytoplasm. It takes place at specialized junctions between invaginations of the muscle cell plasma membrane (transverse tubules, t-tubules) and SR membrane. Although E-C coupling proceeds by different mechanisms in different types of muscles, two main elements of the coupling process are present in each of them. These elements are the voltage-gated L-type Ca2+ channel (dihydropyridine receptor, DHPR) localized in the t-tubule membrane and the Ca2+-release channel (ryanodine receptor, RyR) of the junctional membrane of the SR. Both Ca2+ channels are highly essential for E-C coupling, and defects in these two key proteins or modulation of their activity produce human neuromuscular diseases such as malignant hyperthermia, central core disease, and hypokalemic periodic paralysis.
In both cardiac and skeletal muscles, DHPRs tightly control activation of the Ca2+ release channels by providing efficient coupling of depolarization of the plasma membrane with the rapid Ca2+ release from the SR. The DHPR is a voltage sensor that undergoes conformational changes in response to depolarization of the plasmalemma [1], and as one of the results of these changes, a slow L-type Ca2+ current is activated, which in turn mediates entry of extracellular Ca2+ through the DHPR. In cardiac muscle, entry of Ca2+ through DHPRs activates ryanodine receptors 2, RyR2s (cardiac isoform of RyR), causing them to release Ca2+ from the SR (Ca2+-induced Ca2+ release mechanism) [2]. In skeletal muscle, however, entry of Ca2+ through the DHPR is not required for E-C coupling [3]. Instead, voltage-dependent conformational changes in DHPR produce a signal, which has been hypothesized to allosterically activate ryanodine receptors 1, RyR1s (skeletal muscle isoform of RyR). This signal is thought to be transmitted through a physical, possibly direct link between DHPR and RyR1 (“mechanical-link” mechanism) [4]. Ca2+-induced Ca2+ release was proposed to be supplementary to the direct molecular coupling in skeletal muscle [5].
Although it has been established that protein-protein interactions are highly essential in E-C coupling, the precise molecular mechanism underlying this signaling pathway in muscle cells is largely unclear. Knowledge of the 3D architecture of the channel complex is basic to understanding the channel function. Large size and dynamic properties of these two Ca2+ channels render their structural determination by standard structural techniques like X-ray crystallography or NMR spectroscopy impossible, while electron microscopy is able to tackle both large macromolecular assemblies as well as molecules in different functional states. Our current knowledge about structure-functional relationships within DHPR and RyR is derived primarily from structural studies carried out using electron microscopy of isolated detergent-solubilized channel complexes (single particles). Image analysis of single particles embedded in vitreous ice as observed in the electron microscope is a powerful method for structural studies of large biological molecules, such as ion channels, which are difficult to crystallize. The ice-embedding technique has been proven to preserve protein structure in its native fully hydrated conformation [6, 7].
In this review, I will focus on structures of the two Ca2+ channels, RyR and DHPR, which were determined by electron microscopy and single particle reconstruction techniques, and discuss some applications of this approach to the study of conformational changes in the macromolecular complex.
ARCHITECTURE OF RYANODINE RECEPTOR
Ryanodine receptors are a family of largest integral membrane Ca2+ channels that mediate the gated release of Ca2+ from intracellular Ca2+ stores into the cytosol. In mammals, three different RyR isoforms are identified and characterized, each encoded by distinct genes located on different chromosomes. The three isoforms share ~70% sequence homology. RyR1 and RyR2 isoforms are the predominant Ca2+ release channels in cardiac and skeletal muscles, respectively [8-11]. RyR3 is primarily expressed in diaphragm, smooth muscle, and brain [12-14]. The functional Ca2+ release channel is a homotetramer composed of four RyR subunits, each ~560 kD, which constitute a single ion channel pore [15].
Extensive efforts have been made by several groups to study the three-dimensional structure (3D) of RyR by using electron microscopy of both single particles and two-dimensional crystals [16-20]. So far, only one group has reported ordered arrays of the RyR1, which enabled the construction of a two-dimensional projection map at 25 Å resolution [20]. Nevertheless, electron cryomicroscopy of single particles has been used at 22-30 Å resolution to define the 3D molecular architecture of the channel complex and to delineate the structural domains associated with molecular functions [16-19]. Thus, two groups have analyzed 3D structure of RyR by electron cryomicroscopy and single particle reconstruction using two different approaches: the random conical reconstruction [16] and the angular reconstitution methods [18, 21]. In the random conical method, the specimen is tilted in the microscope in order to obtain many views of the channel complex in different orientations, whose relative angles are defined by settings of the goniometer in the microscope. The other approach exploits random orientations of ice-embedded molecules imaged in the electron microscope without tilting the specimen holder. In this approach, the relative orientations of particles are computationally determined by searching their common line projections. The 3D reconstructions of RyR1 generated in these studies are quite similar. Additionally, a remarkable similarity is observed between the 3D reconstructions of RyR1 and the two other channel isoforms, RyR2 and RyR3 [22, 23]. The RyR channel structure exhibits a four-fold symmetry and has a characteristic mushroom shape, which comprises two square-shaped regions interconnected by four column densities (Fig. 1, see color insert). A large square-shaped region with overall dimensions of 270 × 270 × 120 Å represents a bulky cytoplasmic region exposed to the gap between the SR membrane and the t-tubule membrane. The cytoplasmic (CY) assembly is strikingly empty with numerous distinctive structural domains and intervening cavities that appear suitable for interaction with channel-specific ligands known to bind within the N-terminal cytoplasmic region of RyR (Fig. 2, see color insert). The clamp-shaped regions, located at the corners of the CY assembly are most likely the regions inter-digitating with neighboring molecules of RyR1 as seen in situ [24] or with modulatory auxiliary proteins [25, 26]. The clamp-shaped regions are interconnected by “handle” domains and form a continuous network with the central rim and the column domains of the CY region via several bridging densities. The small square-shaped structure with dimensions of 120 × 120 × 60 Å is rotated by ~40° with respect to the CY assembly and represents the region which spans the SR membrane. The proposed assignment of two major morphological regions within the 3D reconstruction of RyR is quite consistent with the topological arrangement that is predicted based on hydropathy analysis of the channel protein sequence. Hydropathy profiles of RyRs suggest that transmembrane domains lie within one-fifth of the C-terminal of the protein molecule while the remaining RyR sequences form cytoplasmic domains [8, 9]. Predictions for the number of transmembrane helices range from four [8] to twelve [9] segments, and recently an eight transmembrane sequence model has been derived [27]. The estimation of the volume of the putative transmembrane region from the 30 Å map of RyR1 suggests that this region could easily accommodate a bundle of ten nearly parallel alpha-helices per subunit [16, 18]. It is unlikely that this issue can be fully clarified until higher resolution 3D structure of RyR1 is available and alpha-helices can be clearly resolved.
Fig. 1. 3D reconstructions of the ice-embedded RyR1 in two functional states: closed, obtained by depletion of Ca2+ with EGTA [31], and open in the presence of 100 µM Ca2+ and 100 nM ryanodine [19]. Structures are shown in the same orientation--tilted views from the SR lumen toward the cytoplasm with the cytoplasmic side facing upward. Note the differences in the clamp-shaped domains (marked with asterisk) and the presence and absence of the channel opening in the transmembrane regions (marked with arrow). The volume of shown surfaces corresponds to a protein mass of ~2400 kD assuming protein density of 1.35 g/cm3. The scale bar represents 100 Å.
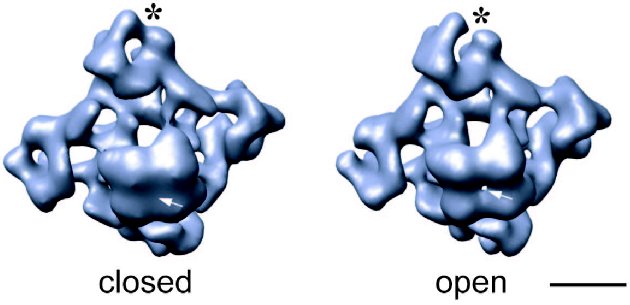
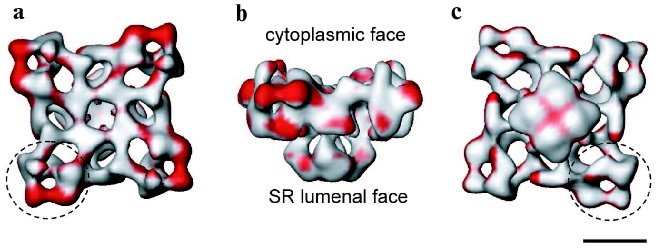
It is well established that during E-C coupling the Ca2+ release channel exists in several functional states, which can be characterized by specific channel gating parameters. The functional channel transitions are regulated by a wide variety of endogenous molecules and pharmacological agents [28-30]. One of the greatest advantages of electron cryomicroscopy of ice-embedded single particles is its direct visualization of conformational changes, since the macromolecular complex can be trapped in the solution under conditions similar to those for electrophysiological or biochemical characterization. While analyzing conformation transitions of a biological macromolecule, one should keep in mind that the population of the molecules in one functional state has to be large enough for the detection as well as to be stable enough to be frozen for electron microscope analysis. Thus, the structure of RyR has been analyzed under conditions that drive the channel population predominantly to its “open” (conducting) and “closed” (non-conducting) states (Fig. 1) [19, 31, 32]. Although the limiting resolution of these reconstructions was 25-30 Å, global conformational changes were detected in both the cytoplasmic region and the putative membrane-spanning portion. The clamp-domains at the four corners of the cytoplasmic region appear in a more open conformation in the presence of Ca2+/ryanodine or Ca2+/ATP, which are reported to induce open states of the channel. In addition, a central cavity and mass movements are detected in the transmembrane region, whereas there was no apparent hole in the closed-state channel obtained by depletion of Ca2+ with EGTA and in the absence of either ryanodine or ATP. Moreover, in the ryanodine-modified open channel the transmembrane domain is twisted by ~4° with respect to its position in the closed state. Based on these observations a mechanism for the opening of the channel was proposed to be similar to the opening-closing of the iris in a camera diaphragm [31]. Thus, structural studies of RyR in different functional states suggest that channel activation is associated with significant mass rearrangements in the channel complex, implying a highly allosteric regulation of channel gating (Fig. 2). A number of other studies also support a model of long-range rather than local conformational changes in the quaternary architecture of the Ca2+ release channel. These include fluorescent studies by Ikemoto et al. [33]. The majority of RyR mutations, which are associated with malignant hyperthermia and with central core diseases and effect the channel gating, have been mapped to two regions (amino acid residues 35-614 and 2163-2458) within the N-terminal putative cytoplasmic portion of the channel protein [34]. Topological analysis also suggests that the N-terminal portion of RyR contains most of the ligand binding sites. In addition, binding sites for regulatory proteins such as CaM and FKBP12 were mapped within the cytoplasmic assembly of the 3D channel structure using electron cryomicroscopy [25].Fig. 2. Significance map of differences between two 3D reconstructions of RyR1 in closed and open states is imaged onto the surface representation of the 3D map of the closed channel. Regions of significant differences (confidence level greater than 99.9 %) are shown in red. The channel structure is shown in (a) top view (view from the cytoplasm), (b) side view, and (c) bottom view (view from the SR lumen). The clamp-shaped domains are indicated with dashed circles. The scale bar represents 100 Å.
ARCHITECTURE OF VOLTAGE-GATED L-TYPE Ca2+
CHANNEL
L-Type (or slow) Ca2+ channels, also frequently designated as dihydropyridine receptors (DHPRs) because of their high affinity to dihydropyridine drugs, are hetero-oligomeric membrane protein complexes with a total mass ~430 kD. The DHPRs are localized in the t-tubule membrane and mediate Ca2+ influx in response to membrane depolarization. In skeletal muscle, the DHPR primarily functions as the voltage sensor. The skeletal DHPR is composed of five subunits arranged in a 1 : 1 : 1 : 1 : 1 stoichiometry: the pore-forming alpha1-subunit (190-212 kD) is associated with auxiliary alpha2 (140 kD), delta (25 kD), beta (53 kD), and gamma (33 kD) subunits [35-38]. Each subunit is encoded by a separate gene, with the exception of the delta subunit, arising as a result of proteolysis from the C-terminus of the alpha2/delta primary polypeptide. The alpha2 and delta subunits form a transmembrane disulfide-linked glycoprotein dimer [39]. A topological model of the voltage-dependent L-type Ca2+-channel has been predicted based on the primary sequence and the hydrophobicity profiles [40]. The alpha1 subunit is organized into four transmembrane domain repeats, each of which contains the canonical voltage-dependent ion channel organization--six putative transmembrane segments (S1-S6). The S5 and S6 helices together with the S5-S6 inter-linking loop from each of repeated domains is believed to form the Ca2+ channel pore. The S4 segment of each repeat contains an ordered pattern of five to six positively charged amino acids, suggesting its essential role in the channel gating as the voltage sensor. Although the alpha1 subunit is shown to carry the characteristic pharmacological and functional properties of the Ca2+-channel for voltage sensing, ion permeability, and drug binding, complete receptor function (including targeting and modulation) requires the presence of all the subunits [3, 41-43].
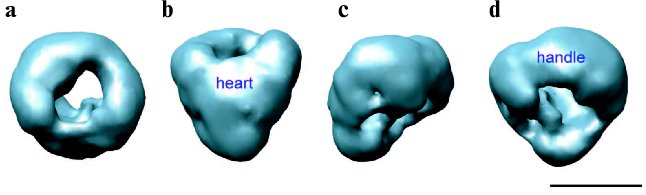
Despite extensive current research into the biochemical, structural, pharmacological, and electrophysiological properties of the L-type Ca2+ channels, little experimental data is available concerning the quaternary arrangement of the channel complex. The structural studies of DHPR are hampered by difficulties in isolation, purification, and expression of the channel protein complex. A number of groups have been pursuing structural studies of the DHPR by various types of electron microscopy [44-48]. Electron microscopy of the freeze-dried, rotary shadowed DHPR revealed ovoid particles with dimensions of 160 × 220 Å [44]. The 2D image analysis of negatively stained images of the L-type Ca2+ channel showed an asymmetric structure of a rod-like domain decorated with a small protrusion on one end, which was suggested to be formed by alpha2/delta [45]. Four 3D maps of the DHPR were reported recently based on single-particle reconstruction from specimens prepared by ice embedding [46, 48] and negative staining [47, 49]. Our group has reported the first 30 Å resolution 3D structure of the skeletal muscle L-type Ca2+ channel determined from frozen-hydrated protein [46]. The asymmetrical channel structure measures about 130 × 115 × 120 Å and consists of two major regions: a heart-shaped region connected at its widest end with a handle-shaped region (Fig. 3, see color insert). Our 3D structure of DHPR corresponds more closely with the structure reported by Wolf et al. [48] when compared at similar resolution and contour levels. However, there is a poor agreement between these two studies with respect to assignment of extracellular and transmembrane regions within 3D maps. Due to low resolution in both structures, the molecular boundaries of individual subunits were not determined and electron densities were assigned primarily based on molecular mass and proposed topological arrangement of the channel subunits in the t-tubule membrane. Thus, in our model the heart-shaped region accounts for the main pore-forming alpha1 subunit associated with the gamma- and beta-subunits, and the handle-shaped region comprises the alpha2/delta complex. Therefore, the heart-shaped region spans the membrane and its narrow part is exposed to the cytoplasm, while the major protein density comprising the handle-shaped region and the upper lobes of the heart-shaped structure is located on the extracellular side. This topology is quite consistent with the model proposed by Murata and coworkers based on antibody labeling [45]. In contrast, the DHPR model reported by Wolf et al. [48] suggests that the major protein densities comprising the alpha1, gamma, delta, and beta subunits are embedded within the membrane, placing the extracellular alpha2 subunit within the smaller decorating region (“leg” region). However, we argue that the beta-subunit is a hydrophilic, peripheral protein located in the cytoplasm as was well established by many functional, biochemical, and structural studies [50-53]. Moreover, recently reported crystal structures of the conserved core region of the beta-subunit alone as well as in complex with the AID domain (alpha1-interaction domain) [54-56] give us insights into the molecular mechanism of the alpha1-beta interaction, conferring high affinity binding between the intracellular domain of alpha1 subunit and the beta subunit functional core, which is most likely located intracellularly. Notably, although the transmembrane region of the alpha1 subunit is predicted to be comprised of four homologous domains and, therefore, to be oriented in a pseudo-fourfold symmetric fashion in the membrane, no symmetry was revealed in the reported 3D structures of DHPR, probably, due to the presence of auxiliary subunits within the L-type Ca2+ channel complex.
Another 27 Å resolution map of the DHPR was reported based on negative-stain electron microscopy and single-particle reconstruction [47]. This 3D map is strikingly different from structures determined from frozen-hydrated channel protein [46, 48]. It exhibits a ring shape with a diameter of ~230 Å and is ~80 Å thick. The central cavity formed by the main body of protein has a diameter of ~70 Å. Two finger-like protrusions of protein density extend over the surface of the central cavity on both sides of the ring-shaped structure. It was estimated that the volume of 3D reconstruction of DHPR at the chosen threshold level could accommodate the mass corresponding to a DHPR dimer. Thus, it was suggested that studied particles presumably correspond to a dimeric form of DHPR. A similar structure for the cardiac voltage-gated L-type Ca2+ channel in its dimer form has also been recently reported by the same group [49]. It seems unlikely that discrepancies between reported 3D structures of DHPR would arise from image processing since the reconstruction of the DHPR dimer was initially performed using the random conical tilt approach and was recently refined with the EMAN software suite [57]. The formation of channel dimers may represent consequences of different detergents used in these studies for solubilization of the channel complex. Wang and coworkers used CHAPS/asolecithin [47], while both structural studies of frozen-hydrated protein utilized digitonin in their channel purification procedures [46, 48]. Clearly more experimentation is required to determine the cause of these discrepancies as well as to establish functional and physiological relevance of the DHPR dimer. Further structural analysis at higher resolution combined with specific antibody-labeling technique is expected in the nearest feature.Fig. 3. 30 Å resolution 3D structure of DHPR obtained by electron cryomicroscopy and single particle reconstruction [46]. The structure is shown in four different views: (a) top view; (b) front view obtained by 90° rotation along the horizontal axis of the top view in (a); (c) and (d) are views obtained by stepwise rotation of the view in (b) along the vertical axis by 90°. The handle-shaped structure and the upper lobes of the heart-shaped region were proposed to account for the extracellular channel region and to include the alpha2 subunit. Thus, the heart-shaped region includes the voltage-sensitive transmembrane region of the L-type Ca2+ channel and the cytoplasmically located beta subunit. The scale bar represents 100 Å.
SPATIAL ARRANGEMENT OF DHPRs AND RyRs IN MEMBRANE
JUNCTIONS
The groundwork for understanding ultrastructural and spatial organization of both the DHPRs and RyRs was laid with studies by Franzini-Armstrong and coworkers using freeze-fracture methods [24, 58-60]. These elegant studies have demonstrated that the physiological difference in the E-C coupling mechanism between skeletal and cardiac muscles is reflected in the striking ultrastructural difference in the relative arrangement of DHPR and RyR within junctional domains. Electron microscopy of thin sections through the junctional regions between the plasma membrane and the SR membrane showed that RyRs and DHPRs are facing each other but are organized into different geometries in two muscle types. The RyRs, which appear as rectangular dense structures (“junctional feet”) in freeze-fracture replicas, form a two-rowed regular array on the junctional membrane of the SR in both skeletal and cardiac muscles. Freeze-fracture of t-tubules reveals rectangular dense particles that were identified as DHPRs. In skeletal muscle, DHPRs are clustered into groups of four DHPRs (tetrads) overlaying every other RyR1 [24]. In cardiac muscle, the DHPRs are not organized in regular arrays of tetrads. These observations provide the direct structural evidence supporting the mechanical nature of the Ca2+ release triggering mechanism of skeletal muscle E-C coupling. Other current evidence in favor of physical coupling includes co-immunoprecipitation of solubilized voltage-gated Ca2+ channel and RyR1 [61], as well as identification of sequences of the DHPR alpha1 subunit (the II-III loop, the III-IV loop, and the C-terminal) and the RyR1 (residues 1076-1112) that are involved in interactions [62-66]. Nevertheless, attempts to show direct binding between the two channels have failed [67, 68].
It is noteworthy that the interaction between the voltage-gated L-type Ca2+ channel and RyR1 is bidirectional: not only does L-type Ca2+ channel activate RyR1 (“orthograde” signaling from DHPR to RyR1), but RyR1 also strongly effects L-type Ca2+ gating by transmitting a signal that enhances L-type Ca2+ current (“retrograde” signaling from RyR1 to DHPR) [69, 70]. The precise mechanism for the retrograde signaling in skeletal muscle is still not clear. Recent observation that RyR1 also effects expression of DHPR, its activation kinetics, modulation by DHP agonists, and divalent conductance suggests that RyR1 is an allosteric modulator of the L-type Ca2+ channel, which influences the activity of the DHPR by the same mechanism as the “orthograde” signaling, i.e., a direct RyR1-DHPR physical interaction [70]. Nevertheless, other mechanisms have yet to be excluded.
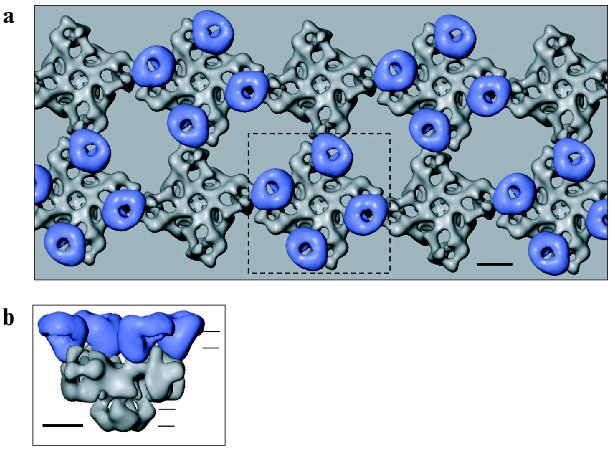
Thus, specific regions within both the DHPR and RyR proteins have been reported to mediate DHPR-RyR1 coupling and should be located in close opposition. Putting together observations from freeze-fracture studies and the molecular shapes of DHPR and Ca2+ release channel determined by electron cryomicroscopy, we put forward a model of their interactions as shown in Fig. 4 (see color insert) [46]. The handedness of the RyR array was inferred from a recent study by electron tomography of the frozen-hydrated triad junction isolated from skeletal muscle [71]. The site of inter-oligomeric contact within the RyR array corresponds to the clamp-shaped domain in single-particle 3D reconstruction of RyR (Fig. 2) [31]. These regions in the RyR structure undergo significant conformational changes upon opening of the Ca2+ release channel [19, 31]. Also, divergent regions 2 (residues 1342-1403 of RyR1) and 3 (residues 1872-1923 of RyR1) which were implicated to mediate interaction of RyR with DHPR, were localized to the clamp-shaped region [72, 73]. Given the location of FKBP12 and CaM binding sites within the 3D structure of RyR [25], it should be pointed out that both binding sites are most likely not involved in the interaction between two adjacent RyRs.
Obviously, in order to substantiate this model, it is necessary to perform further experiments to prove the sidedness of the observed structure of DHPR in the membrane and to correlate the linear sequences of RyR to the 3D structure.Fig. 4. Model of physical coupling between RyR1 and DHPR in skeletal muscle based on freeze-fracture studies and using 3D reconstructions of two Ca2+ channels, generated by electron cryomicroscopy and single particle reconstruction. a) Two arrays of RyRs are overlaid by arrays of DHPRs grouped into tetrads. b) Side view of RyR1 coupled with the tetrad indicated with the dashed line in (a). Horizontal lines indicate the approximate position of the surface and SR membranes. The scale bar represents 100 Å.
OUTLOOK
In conclusion, while crystal structure determination of both Ca2+ channel complexes has been in high demand for a long time, no high-resolution structures are yet available. To date, low resolution structures of DHPR and RyR generated using electron microscopy and single particle reconstruction techniques have been providing the basis for interpreting structure-function relations for these proteins. Although our understanding of the molecular mechanism of E-C coupling is expanding rapidly, many essential questions on DHPR-RyR coupling cannot be currently solved due to the absence of information about the arrangement of the primary sequences of DHPR and RyR in three-dimensions. Through improvements in cryospecimen preparation and image processing algorithms in the EMAN software suite, we have now achieved a substantial improvement to 14 Å resolution in the 3D structure of RyR1 [74]. Encouragingly, recent studies from several groups have demonstrated that 6-10 Å resolution which is sufficient to detect secondary structure is possible to achieve using electron cryomicroscopy and single-particle approach for isolated macromolecular complexes [75-77]. Although the resolution reached by the single particle approach is still far from quasi-atomic resolution, many important biological questions can be clarified at this level. Moreover, for solving some biological questions, such as physiological conformations of molecules or the interaction between molecules, crystallization of molecules may not be desired. In addition, direct determination of the tertiary and quaternary structures of the entire macromolecular complex provides a three-dimensional framework for assembling the crystal structures of individual channel subunits or their sub-domains once they become available. Another way to increase the information attainable from structural studies by single particle approach is to combine these studies with bioinformatics [21]. As we continue to make progress in this exciting field of electron cryomicroscopy and single particle reconstruction, we can enjoy the great challenges of elucidation of the structural organization of membrane proteins and their structure-function mechanisms.
This review is dedicated to the memory of my teacher, Professor Boris F. Poglazov.
Original research described in this review has been supported by NIH grants (P41RR02250, P01GM99116, R01AR44864, and RO1AR41729) and an MDA grant.
REFERENCES
1.Rios, E., and Brum, G. (1987) Nature,
325, 717-720.
2.Nabauer, M., Callewaert, G., Cleemann, L., and
Morad, M. (1989) Science, 244, 800-803.
3.Tanabe, T., Beam, K. G., Powell, J. A., and Numa,
S. (1988) Nature, 336, 134-139.
4.Schneider, M. F., and Chandler, W. K. (1973)
Nature, 242, 244-246.
5.Pizarro, G., Csernoch, L., Uribe, I., Rodriguez,
M., and Rios, E. (1991) J. Gen. Physiol., 97,
913-947.
6.Chiu, W., Downing, K. H., Dubochet, J., Glaeser, R.
M., Heide, H. G., Knapek, E., Kopf, D. A., Lamvik, M. K., Lepault, J.,
Robertson, J. D., Zeitler, E., and Zemlin, F. (1986) J.
Microsc., 141, 385-391.
7.Dubochet, J., Adrian, M., Chang, J. J., Homo, J.
C., Lepault, J., McDowall, A. W., and Schultz, P. (1988) Q. Rev.
Biophys., 21, 129-228.
8.Takeshima, H., Nishimura, S., Matsumoto, T.,
Ishida, H., Kangawa, K., Minamino, N., Matsuo, H., Ueda, M., Hanaoka,
M., Hirose, T., et al. (1989) Nature, 339, 439-445.
9.Zorzato, F., Fujii, J., Otsu, K., Phillips, M.,
Green, N. M., Lai, F. A., Meissner, G., and MacLennan, D. H. (1990)
J. Biol. Chem., 265, 2244-2256.
10.Hakamata, Y., Nakai, J., Takeshima, H., and
Imoto, K. (1992) FEBS Lett., 312, 229-235.
11.Tunwell, R. E., Wickenden, C., Bertrand, B. M.,
Shevchenko, V. I., Walsh, M. B., Allen, P. D., and Lai, F. A. (1996)
Biochem. J., 318, 477-487.
12.Sutko, J. L., and Airey, J. A. (1996) Physiol.
Rev., 76, 1027-1071.
13.Sorrentino, V., and Volpe, P. (1993) Trends
Pharmacol. Sci., 14, 98-103.
14.Sorrentino, V., and Reggiani, C. (1999) Trends
Cardiovasc. Med., 9, 54-61.
15.Lai, F. A., Misra, M., Xu, L., Smith, H. A., and
Meissner, G. (1989) J. Biol. Chem., 264, 16776-16785.
16.Radermacher, M., Rao, V., Grassucci, R., Frank,
J., Timerman, A. P., Fleischer, S., and Wagenknecht, T. (1994) J.
Cell. Biol., 127, 411-423.
17.Wagenknecht, T., and Radermacher, M. (1995)
FEBS Lett., 369, 43-46.
18.Serysheva, I. I., Orlova, E. V., Chiu, W.,
Sherman, M. B., Hamilton, S. L., and van Heel, M. (1995) Nat.
Struct. Biol., 2, 18-24.
19.Orlova, E. V., Serysheva, I. I., van Heel, M.,
Hamilton, S. L., and Chiu, W. (1996) Nat. Struct. Biol.,
3, 547-552.
20.Yin, C. C., and Lai, F. A. (2000) Nat. Cell.
Biol., 2, 669-671.
21.Baker, M. L., Serysheva, I. I., Sencer, S., Wu,
Y., Ludtke, S. J., Jiang, W., Hamilton, S. L., and Chiu, W. (2002)
Proc. Natl. Acad. Sci. USA, 99, 12155-12160.
22.Sharma, M. R., Penczek, P., Grassucci, R., Xin,
H. B., Fleischer, S., and Wagenknecht, T. (1998) J. Biol. Chem.,
273, 18429-18434.
23.Jeyakumar, L. H., Copello, J. A., O'Malley, A.
M., Wu, G. M., Grassucci, R., Wagenknecht, T., and Fleischer, S. (1998)
J. Biol. Chem., 273, 16011-16020.
24.Block, B. A., Imagawa, T., Campbell, K. P., and
Franzini-Armstrong, C. (1988) J. Cell. Biol., 107,
2587-2600.
25.Wagenknecht, T., Radermacher, M., Grassucci, R.,
Berkowitz, J., Xin, H. B., and Fleischer, S. (1997) J. Biol.
Chem., 272, 32463-32471.
26.Samso, M., and Wagenknecht, T. (2002) J. Biol.
Chem., 277, 1349-1353.
27.Du, G. G., Sandhu, B., Khanna, V. K., Guo, X. H.,
and MacLennan, D. H. (2002) Proc. Natl. Acad. Sci. USA,
99, 16725-16730.
28.Coronado, R., Morrissette, J., Sukhareva, M., and
Vaughan, D. M. (1994) Am. J. Physiol., 266,
C1485-C1504.
29.Meissner, G. (1994) Annu. Rev. Physiol.,
56, 485-508.
30.Ogawa, Y. (1994) Crit. Rev. Biochem. Mol.
Biol., 29, 229-274.
31.Serysheva, I. I., Schatz, M., van Heel, M., Chiu,
W., and Hamilton, S. L. (1999) Biophys. J., 77,
1936-1944.
32.Sharma, M. R., Jeyakumar, L. H., Fleischer, S.,
and Wagenknecht, T. (2000) J. Biol. Chem., 275,
9485-9491.
33.Ikemoto, N., Antoniu, B., and Meszaros, L. G.
(1985) J. Biol. Chem., 260, 14096-14100.
34.McCarthy, T. V., Quane, K. A., and Lynch, P. J.
(2000) Hum. Mutat., 15, 410-417.
35.Catterall, W. A., Seagar, M. J., and
Takahashi, M. (1988) J. Biol. Chem., 263,
3535-3538.
36.Ellis, S. B., Williams, M. E., Ways, N. R.,
Brenner, R., Sharp, A. H., Leung, A. T., Campbell, K. P., McKenna, E.,
Koch, W. J., Hui, A., et al. (1988) Science, 241,
1661-1664.
37.Glossmann, H., and Striessnig, J. (1988)
Vitam. Horm., 44, 155-328.
38.Jay, S. D., Ellis, S. B., McCue, A. F., Williams,
M. E., Vedvick, T. S., Harpold, M. M., and Campbell, K. P. (1990)
Science, 248, 490-492.
39.Catterall, W. A. (1995) Annu. Rev.
Biochem., 64, 493-531.
40.Isom, L. L., De Jongh, K. S., and Catterall, W.
A. (1994) Neuron, 12, 1183-1194.
41.Melzer, W., Herrmann-Frank, A., and Luttgau, H.
C. (1995) Biochim. Biophys. Acta, 1241, 59-116.
42.Catterall, W. A. (2000) Annu. Rev. Cell. Dev.
Biol., 16, 521-555.
43.McDonald, T. F., Pelzer, S., Trautwein, W., and
Pelzer, D. J. (1994) Physiol. Rev., 74, 365-507.
44.Leung, A. T., Imagawa, T., Block, B.,
Franzini-Armstrong, C., and Campbell, K. P. (1988) J. Biol.
Chem., 263, 994-1001.
45.Murata, K., Odahara, N., Kuniyasu, A., Sato, Y.,
Nakayama, H., and Nagayama, K. (2001) Biochem. Biophys. Res.
Commun., 282, 284-291.
46.Serysheva, I. I., Ludtke, S. J., Baker, M. R.,
Chiu, W., and Hamilton, S. L. (2002) Proc. Natl. Acad. Sci. USA,
99, 10370-10375.
47.Wang, M. C., Velarde, G., Ford, R. C., Berrow, N.
S., Dolphin, A. C., and Kitmitto, A. (2002) J. Mol. Biol.,
323, 85-98.
48.Wolf, M., Eberhart, A., Glossmann, H.,
Striessnig, J., and Grigorieff, N. (2003) J. Mol. Biol.,
332, 171-182.
49.Wang, M. C., Collins, R. F., Ford, R. C., Berrow,
N. S., Dolphin, A. C., and Kitmitto, A. (2004) J. Biol. Chem.,
279, 7159-7168.
50.Pragnell, M., Sakamoto, J., Jay, S. D., and
Campbell, K. P. (1991) FEBS Lett., 291, 253-258.
51.Pragnell, M., de Waard, M., Mori, Y., Tanabe, T.,
Snutch, T. P., and Campbell, K. P. (1994) Nature, 368,
67-70.
52.Walker, D., Bichet, D., Campbell, K. P., and de
Waard, M. (1998) J. Biol. Chem., 273, 2361-2367.
53.Opatowsky, Y., Chomsky-Hecht, O., Kang, M. G.,
Campbell, K. P., and Hirsch, J. A. (2003) J. Biol. Chem.,
278, 52323-52332.
54.Opatowsky, Y., Chen, C. C., Campbell, K. P., and
Hirsch, J. A. (2004) Neuron, 42, 387-399.
55.Van Petegem, F., Clark, K. A., Chatelain, F. C.,
and Minor, D. L., Jr. (2004) Nature, 429, 671-675.
56.Chen, Y. H., Li, M. H., Zhang, Y., He, L. L.,
Yamada, Y., Fitzmaurice, A., Shen, Y., Zhang, H., Tong, L., and Yang,
J. (2004) Nature, 429, 675-680.
57.Ludtke, S. J., Baldwin, P. R., and Chiu, W.
(1999) J. Struct. Biol., 128, 82-97.
58.Ferguson, D. G., Schwartz, H. W., and
Franzini-Armstrong, C. (1984) J. Cell. Biol., 99,
1735-1742.
59.Franzini-Armstrong, C., and Kish, J. W. (1995)
J. Muscle Res. Cell. Motil., 16, 319-324.
60.Protasi, F., Franzini-Armstrong, C., and Flucher,
B. E. (1997) J. Cell. Biol., 137, 859-870.
61.Marty, I., Robert, M., Villaz, M., DeJohngh,
K. S., Lai, Y., Catterall, W. A., and Ronjat, M. (1994) Proc. Natl.
Acad. Sci. USA, 91, 2270-2274.
62.Lu, X., Xu, L., and Meissner, G. (1994) J.
Biol. Chem., 269, 6511-6516.
63.El-Hayek, R., Yano, M., Antoniu, B., Mickelson,
J. R., Louis, C. F., and Ikemoto, N. (1995) Am. J. Physiol.,
268, C1381-C1386.
64.Tanabe, T., Mikami, A., Numa, S., and Beam, K. G.
(1990) Nature, 344, 451-453.
65.Grabner, M., Dirksen, R. T., Suda, N., and Beam,
K. G. (1999) J. Biol. Chem., 274, 21913-21919.
66.Leong, P., and MacLennan, D. H. (1998) J.
Biol. Chem., 273, 29958-29964.
67.Kim, K. C., Caswell, A. H., Talvenheimo, J. A.,
and Brandt, N. R. (1990) Biochemistry, 29, 9281-9289.
68.Corbett, A. M., Caswell, A. H., Brandt, N. R.,
and Brunschwig, J. P. (1985) J. Membr. Biol., 86,
267-276.
69.Nakai, J., Dirksen, R. T., Nguyen, H. T., Pessah,
I. N., Beam, K. G., and Allen, P. D. (1996) Nature, 380,
72-75.
70.Avila, G., and Dirksen, R. T. (2000) J. Gen.
Physiol., 115, 467-480.
71.Wagenknecht, T., Hsieh, C. E., Rath, B. K.,
Fleischer, S., and Marko, M. (2002) Biophys. J.,
83, 2491-2501.
72.Zhang, J., Liu, Z., Masumiya, H., Wang, R.,
Jiang, D., Li, F., Wagenknecht, T., and Chen, S. R. (2003) J. Biol.
Chem., 278, 14211-14218.
73.Liu, Z., Zhang, J., Wang, R., Wayne Chen, S. R.,
and Wagenknecht, T. (2004) J. Mol. Biol., 338,
533-545.
74.Serysheva, I. I., Hamilton, S. L., Ludtke, S. J.,
and Chiu, W. (2004) Biophys. J., 86, 242.
75.Matadeen, R., Patwardhan, A., Gowen, B., Orlova,
E. V., Pape, T., Cuff, M., Mueller, F., Brimacombe, R., and van
Heel, M. (1999) Structure Fold. Des., 7,
1575-1583.
76.Gabashvili, I. S., Agrawal, R. K., Sahn,
C. M., Grassucci, R. A., Svergun, D. I., Frank, J., and Penczek,
P. (2000) Cell, 100, 537-549.
77.Ludtke, S. J., Chen, D. H., Song, J. L., Chuang,
D. T., and Chiu, W. (2004) Structure (Camb.), 12,
1129-1136.