Folding and Stability of Chimeric Immunofusion VL-Barstar
Y. I. Tsybovsky1, D. V. Shubenok1, O. A. Stremovskiy2,3, S. M. Deyev4, and S. P. Martsev1*
1Institute of Bioorganic Chemistry, National Academy of Sciences of Belarus, ul. Akademika Kuprevicha 5/2, Minsk 220141, Belarus; fax: (+375) 172-64-87-61; E-mail: martsev@iboch.bas-net.by2Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, Moscow 117984, Russia; E-mail: ostr@mail.ru
3University of Oslo, Boks 1072 Blindern NO-0316 Oslo, Norway; fax: (+47) 22-85-50-50
4Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow 117997, Russia; E-mail: deyev@ibch.ru
* To whom correspondence should be addressed.
Received March 18, 2004; Revision received May 13, 2004
A chimeric protein VL-barstar that comprises the VL domain of anti-human ferritin monoclonal antibody F11 and barstar, the naturally occurring inhibitor of bacterial RNase barnase, has been constructed for study of structure-function characteristics of chimeric immunoglobulin fused proteins. Such chimeric constructs may be potentially employed for development of bivalent/bispecific antibodies on the basis of the high affinity interaction between barstar and barnase (the association constant is about 1014 M-1). We have developed a protocol for VL-barstar expression in E. coli and purification and refolding from inclusion bodies that yields a homogeneous and soluble form of this protein. Differential scanning calorimetry in combination with fluorescence and CD spectroscopy revealed that the VL-barstar formed well-resolved ordered secondary and compact tertiary structures. However, partial loss of tertiary interactions resulted in low stability of the recombinant protein and the lack of functional activity of the two constituent modules. These conformational features suggest that the protein might be referred to the class of native molten globules, which comprises partially unfolded conformations stabilized under physiological conditions. Since individually expressed VL domain and barstar retain completely folded conformation and stable spatial structure, the incomplete folding of the chimeric protein may be attributed to interaction between heterologous domains, which appears at the folding stage preceding formation of a system of tertiary interactions in both structural modules. The results provide evidence for non-native interactions between heterologous modules that may occur in chimeric proteins composed of taxonomically distinct fusion partners.
KEY WORDS: chimeric immunofusions, recombinant antibody, VL domain, barstar, protein folding, differential scanning calorimetry, antiferritin antibody
Abbreviations: ANS) 8-anilino-1-naphthalenesulfonate; IPTG) isopropyl beta-D-thiogalactopyranoside; VL) variable domain of immunoglobulin light chain.
Recombinant proteins fused with antibody fragments represent a new class
of chimeric proteins that are now being developed for immune therapy of
malignant tumors [1]. Various plant toxins [2, 3], bacterial exotoxin from
Pseudomonas [1, 4], or
various RNases [5-8] are
considered as the cytotoxic module of immunotoxins. Bacterial RNase
from Bacillus amyloliquefaciens, known as barnase, is one of the
most promising cytotoxic modules. This small (~12 kD) cytotoxic
enzyme is insensitive to mammalian/human RNase inhibitors, and it is
also compatible with bacterial expression systems. The availability of
a specific high affinity barnase inhibitor, barstar, provides an
additional advantage to the barnase-based immunotoxins. Recently a
principally new approach for construction of therapeutically effective
bivalent/bispecific antibodies based on tight complex formation between
chimeric fusion proteins antibody-barnase and antibody-barstar has been
proposed [9]. The use of such constructs is based
on the exceptionally high association constant (up to 1014
M-1) of the barnase-barstar couple [10], which is considered as an alternative to the
traditional biotin-(strept)avidin system [9].
Rational construction of chimeric proteins of this class requires
knowledge of the structure-function relationships within this new group
of proteins, which in its turn requires construction of a
representative set of model constructs.
In the present study we have constructed, expressed, and investigated the chimeric fusion protein VL-barstar. This includes the variable VL domain of the light chain of mouse F11 antibody directed to the tumor marker, human ferritin. Full-length radiolabeled antibodies against ferritin have successfully been employed for cytotoxic immune therapy of some tumors [11, 12], which explains much interest in recombinant fusion proteins containing antiferritin antibody module. Besides evident prospects for immune therapy, VL-barstar also represents an interesting model for studies of folding of recombinant proteins comprising two small heterologous domains related to phylogenetically distant and functionally unrelated proteins.
Since spatial structure, stability, and functional activity of proteins are predominantly determined by a primary structure, fusion to a new amino acid sequence may significantly change conformation and activity of the chimeric protein. However, some examples are also known when significant changes in protein primary structure did not alter its conformation and activity. For example, Collinet and coauthors demonstrated that insertion of dihydrofolate reductase or beta-lactamase into four different positions of the primary structure of phosphoglycerate kinase did not change the activity of the mutants [13]. As another essential yet poorly understood source that contributes to conformation and activity of chimeric proteins, domain interactions should be carefully analyzed. A few studies were reported where interactions between domains of chimeric proteins have been considered. Depending on the structure of chimeric proteins, both effects of an altered stability and activity of a partner domain [14] and lack of any changes [15] have been reported. Thus, at the present time, it is impossible to a priori predict correlation between structure, conformation, and activity of chimeric proteins, and these should be investigated in detail for each particular case.
Both isolated VL domain and barstar possess tertiary structure typical for globular proteins. Barstar is a small protein (~10 kD) consisting of 89 amino acid residues. Its spatial structure and conformational stability are well established [10, 16-19]. F11 antibody VL domain is a homodimer, with molecular mass of each monomer of 12 kD; it effectively binds to ferritin [20-22]. Using VL domain we have recently constructed and studied the fusion immunotoxin, VL-barnase, which constitutes a structurally minimized model of an immunotoxin. The two modules of this chimeric protein, the antigen-binding domain and RNase, preserved their functional activity and structural integrity, which suggests independent folding of these two heterologous parts [23].
In the present study, we provide a detailed description for the conformation of VL-barstar using a combination of differential scanning calorimetry, circular dichroism (CD), and fluorescence spectroscopy. The data indicate that after refolding from inclusion bodies the chimeric protein possessed sufficient solubility and compact conformation. This study also provides the first demonstration that heterologous modules in chimeric immunoconjugates may interact at a stage preceding complete folding. These early interactions between the two structural modules of VL-barstar result in incomplete folding of the whole chimeric protein, which is characterized by an intermediate structural state that possesses significant changes in secondary and tertiary structures and partial loss of stabilizing tertiary interactions.
MATERIALS AND METHODS
Expressing plasmid. The pET(VL-barstar) plasmid encoding the variable domain of the antiferritin antibody F11 light chain fused to barstar was obtained using standard gene engineering methods (Fig. 1). The KpnI-HindIII fragment of plasmid pET(VL-barnase) [23], encoding the variable domain of F11 antibody light chain fused to barnase, was ligated to the KpnI-HindIII fragment of pET(F11-barstar) plasmid (our unpublished data), encoding scFv-fragment of F11 antibody fused to barstar, which was flanked by a histidine hexapeptide at the C-end.
Expression of the recombinant fusion protein. Escherichia coli BL21(DE3) cells transformed with the pET(VL-barstar) plasmid were incubated at 37°C in 1 liter of LB medium containing 100 mg/liter of ampicillin. After 3-4 h of growth isopropyl beta-D-thiogalactopyranoside (IPTG) was added to the final concentration of 0.5 mM and the cells were cultivated for an additional 3-5 h. The cells were sedimented by centrifugation at 10,000g for 10 min and suspended in 80-100 ml of 0.1 M Tris-HCl buffer, pH 8.1. After disintegration by sonication, the cells were centrifuged at 20,000g for 25 min. The sediment was washed twice with the same buffer. Inclusion bodies usually contained VL-barstar of 50-70% purity.Fig. 1. a) Plasmid pET(VL-barstar) encoding the amino acid sequence of chimeric protein VL-barstar. Designations: T7 lac, promoter of T7 RNA polymerase; Amp, ampicillin resistance gene; L, peptide linker; KpnI and HindIII, restriction sites for the corresponding enzymes. Detailed description of the pET22 plasmid can be found at www.invitrogen.com; b) primary structure of VL-barstar.
Purification of the recombinant fusion protein. The sediment containing inclusion bodies was dissolved at room temperature in 10 ml of 6 M guanidine hydrochloride for 2 h under stirring. After centrifugation at 40,000g, the supernatant was dialyzed against buffer 1, containing 7 M urea, 25 mM Tris-HCl, pH 7.0. Insoluble sediment was removed by centrifugation and the sample was then applied onto a Ni-NTA-Sepharose (Novagen, USA) column (80 × 10 mm) equilibrated with buffer 1. The column was washed with five volumes of the same buffer. Bound VL-barstar was eluted by 0.1 M imidazole in buffer 1. The fraction containing VL-barstar was dialyzed against 5 M urea in 0.1 M Tris-HCl-buffer, pH 8.1.
Refolding of the recombinant chimeric protein. Refolding was carried out by stepwise dialysis against buffers with gradually reducing urea concentrations. At various stages of dialysis the following buffers were used (depending on the refolding scheme): 50-100 mM sodium phosphate-HCl, pH 2.0-2.5 and 7.4; 50-100 mM sodium acetate, pH 3.0-5.0; 0.1 M Tris-HCl, pH 8.0-9.0; 50 mM sodium borate, pH 8.5-10.0. In some cases, refolding was carried out in the presence of 0.1-0.4 M sodium chloride. At the final stage, the VL-barstar samples were dialyzed against 0.1 M sodium phosphate buffer, pH 7.4.
Molecular weight was determined by gel chromatography using a Sephacryl S-200 column (0.8 × 106 cm) with the following molecular weight markers: immunoglobulin G (150 kD), BSA (66 kD), ovalbumin (45 kD), and lysozyme (14.4 kD).
Thiol-disulfide analysis. Disulfide number in purified and folded protein was determined by the method of Thannhauser [24] in the presence of 3 M guanidine thiocyanate. Free thiols were determined according to Ellman [25], also in the presence of the denaturant.
Binding to antibodies against the VL domain. The isolated VL domain and VL-barstar (2 µg in 0.25 ml of 50 mM sodium borate buffer, pH 8.5) were immobilized by adsorption on the surface of polystyrene tubes during overnight incubation. The tubes were then washed twice with the same buffer and free protein binding sites were blocked by incubating with 1% BSA in 0.1 M sodium phosphate buffer, pH 7.4. Coated tubes were incubated with increasing concentrations of polyclonal rabbit antibodies against the isolated recombinant VL domain. The binding reaction was carried out in 0.25 ml of 50 mM sodium borate buffer, pH 8.5. After 2 h and two washings, 200 ng of anti-rabbit IgG sheep antibodies conjugated with horseradish peroxidase were added in 0.25 ml of 50 mM sodium borate buffer, pH 8.5. After 1.5 h of incubation, the tubes were washed and 0.6 ml of 0.02 M o-phenylenediamine and 0.02 M H2O2 in 0.1 M sodium citrate, pH 5.0, were added, and the mixture was incubated for 10 min under stirring. The reaction was stopped by adding 0.4 ml of 10% H2SO4, and the absorbance was determined at 492 nm.
Analysis of antigen binding. The ferritin binding affinity of VL-barstar was determined in an enzyme-linked immunosorbent assay (ELISA). All experiments were performed in triplicates and the mean values were used for calculations.
VL-barstar (2 µg in 0.25 ml of 50 mM sodium borate buffer, pH 8.5) was adsorbed overnight in the wells of a polystyrene plate. The wells were then washed with the same buffer, and the remaining binding sites on the polystyrene surface were blocked by incubating with BSA in sodium phosphate buffer for 40 min. The coated wells were incubated with increasing amounts of human spleen ferritin dissolved in 0.1 M sodium phosphate buffer containing 1% BSA. After incubation for 2 h and two washings, 200 ng of monoclonal antiferritin antibody G-10 conjugated with horseradish peroxidase were added, and peroxidase activity was determined as above.
Analysis of inhibition of barnase RNase activity. Inhibition of barnase RNase activity by barstar within the recombinant protein was analyzed by assaying RNase activity of samples containing barnase and VL-barstar according to Kunitz [26] by spectrophotometrically following the decrease in RNA absorbance at 300 nm in a 1-cm cuvette containing 1 mg/ml yeast RNA dissolved in 0.1 M sodium phosphate, pH 7.4. The molar ratio of barnase to VL-barstar varied from 1 : 1 to 1 : 50 at 0.5 µM barnase and 0.5-25 µM VL-barstar (calculated per mole of monomer).
CD spectra were recorded using a J-20 spectropolarimeter (Jasco, Japan) in a thermostatted cuvette using pathlength of 1 and 10 mm in far and near ultraviolet regions, respectively. Protein concentration was 0.3-0.5 mg/ml, and the scanning rate was 5 nm/min with time constant of 16 sec. Each spectrum represents the mean of three scanning. The mean amino acid molecular weight used for calculation of molar ellipticity was 111.2 daltons.
Fluorescent studies. Intrinsic fluorescence spectra of the proteins and fluorescence of protein-bound 8-anilino-1-naphthalenesulfonate (ANS) were recorded at 25°C in a 1-cm pathlength quartz cuvette using an SFL-1211 fluorimeter (Solar, Belarus) connected to a PC. Samples (protein concentration 0.05 mg/ml) were subjected to overnight incubation at 4°C. Protein fluorescent spectra were corrected for fluorescence of buffered solutions. Fluorescence was excited at 280 and 295 nm and recorded from 290 to 390 nm (excitation at 280 nm) or from 305 to 405 nm (excitation at 295 nm). ANS fluorescence was recorded from 400 to 600 nm with excitation at 360 nm. Protein-bound ANS spectra were obtained by subtracting ANS spectra recorded in corresponding buffer solutions. The molar ANS-to-protein ratio was 10 : 1 per protein domain. The following buffer systems were used: 0.1 M sodium phosphate-HCl, pH 2.0-2.5 and 6.0-7.4; 0.1 M citrate-phosphate, pH 2.5-6.0; 0.1 M Tris-HCl, pH 7.5-9.0; 0.1 M sodium borate, pH 9.0-10.0.
Differential scanning calorimetry (DSC). Measurements were carried out using a DASM-4M scanning calorimeter (Biopribor, Russia) equipped with a computer interface in a temperature range of 10-100°C at a scan rate of 60 K/h. In all cases, the reference cell was filled with the buffer used for dialysis of the protein sample. The resultant curves were corrected for baseline obtained when both cells were filled with the buffer. The protein concentrations varied between 0.8 and 2.0 mg/ml. Each sample was scanned from 3 to 8 times; the error of enthalpy measurements did not exceed 6%. Enthalpy of heat denaturation was calculated using TERMCALC software (Biopribor).
Other methods. Native electrophoresis was carried out using 6-10% separating polyacrylamide gel without stacking gel using the following buffer system: 0.35% sodium acetate, pH 5.0, for separating gel; 35 mM beta-alanine-acetic acid, pH 5.0, and 0.1 M sodium acetate, pH 4.0, as upper and lower electrode buffers, respectively.
The protein purity was evaluated by electrophoresis in polyacrylamide gel in the presence of sodium dodecyl sulfate (SDS-PAGE), using 5% stacking and 15% separating gels, respectively. Samples were reduced with 2% beta-mercaptoethanol and heated on a boiling water bath for 2 min. Gels were stained using Coomassie brilliant blue R-250.
Concentration of VL-barstar was determined spectrophotometrically using molar extinction coefficient A1%1 cm = 15.2 at 280 nm. The coefficient was calculated from the amino acid sequence using the algorithm described in [27] and the special software. The molecular mass of the VL-barstar monomer was 24,125 daltons.
RESULTS
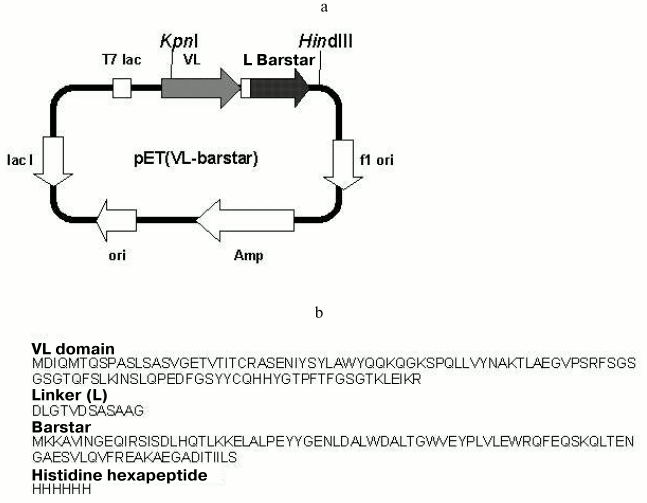
Purification and refolding. After E. coli transformation with plasmid, carrying VL-barstar gene (Fig. 1), the chimeric protein was expressed as insoluble inclusion bodies. The chimeric protein solubilized from inclusion bodies effectively bound to Ni-NTA-Sepharose (Fig. 2). The protein eluted by 0.1 M imidazole was identified as VL-barstar according to: 1) highly specific binding to polyclonal antibodies raised against the isolated VL domain (data not shown), and 2) close agreement between calculated and electrophoretically determined molecular weights (Fig. 2). Furthermore, addition of IPTG to the cell culture selectively induced expression of the protein with molecular weight close to the value calculated for VL-barstar.
Changes in dialysis scheme and duration, as well as variations of pH in the range of 2-10 at different stages of refolding did not alter the physicochemical and functional characteristics of VL-barstar. Addition of 0.1-0.4 M NaCl, an agent that is known to stabilize individual barstar [18], to the refolding buffers had also no effect on VL-barstar conformation. The most effective refolding protocol (in terms of quantitative yield of the fusion protein) included metal-chelation chromatography followed by a single step dialysis against 0.1 M Tris-HCl, pH 8.1, in the absence of detergents. The protocol typically yielded 10 mg of VL-barstar per 1 liter of the cell culture with a purity of not less than 95% as determined by SDS-PAGE (Fig. 2). The recombinant protein was stored at 4°C. Within the range of concentrations 0.1-5.5 mg/ml, no precipitation was detected during at least seven days of storage.Fig. 2. Expression and purification of VL-barstar: 1, 2) E. coli cells transformed by the pET(VL-barstar) plasmid before and 4 h after addition of IPTG, respectively; 3) inclusion bodies; 4) the protein purified by metal-chelation chromatography; 5) molecular mass markers: rabbit muscle phosphorylase b, BSA, ovalbumin, carbonic anhydrase, soybean trypsin inhibitor, and alpha-lactalbumin (on the right, their molecular masses in kD).
Lack of refolding conformers for VL-barstar. In some cases, refolding of recombinant proteins can result in formation of non-native conformations (folding conformers) or several molecular forms based on the same primary structure [28]. To rule out this possibility in the case of VL-barstar, we employed gel permeation chromatography, native electrophoresis, and chemical thiol-disulfide analysis of the fusion protein.
Gel permeation chromatography of VL-barstar showed a single symmetric peak corresponding to an apparent molecular mass of ~54 kD (data not shown). Native polyacrylamide gel electrophoresis of the fusion protein demonstrated a single clearly defined band (data not shown). Quantitative analysis of thiol and disulfide groups of the chimeric protein comprising two Cys residues in the VL domain moiety revealed the presence of one disulfide bond per monomer of VL-barstar with no free thiol groups. This is consistent with the structure of the chimeric protein, which is composed of the cysteine-free barstar mutant (the two Cys residues are substituted by Ala) and the VL domain containing two Cys residues that form a single disulfide bond. Thus, we conclude that the refolding procedure yields a single molecular form of the recombinant protein.
Functional activity. Ferritin-binding propensity of VL-barstar was investigated by ELISA in the range of ferritin concentrations from 10 to 400 nM, the maximal ferritin concentration being one order of magnitude higher than the dissociation constant of ferritin-VL domain and ferritin-VL-barnase complexes [23]. In the case of VL-barstar, there was non-saturated behavior in dose-response curves, and at upper concentrations of ferritin, binding did not exceed 2-3% of binding of isolated VL domain or VL-barnase. Taken together these data indicate lack of antigen binding activity of the VL domain within VL-barstar. In its turn, the barstar module of the chimeric protein was unable to inhibit RNase activity of barnase within the range of VL-barstar concentrations from 0.5 to 25 µM at fixed barnase concentration of 0.5 µM. In contrast, the two isolated domains constituting the chimeric protein readily exhibit their biological activity: the VL domain binds ferritin [20-22] while barstar inhibits RNA degradation by barnase [10, 16]. Lack of functional activity of both modules within the fusion protein suggests significant rearrangements of the two functional centers and, therefore, a non-native conformation of VL-barstar.
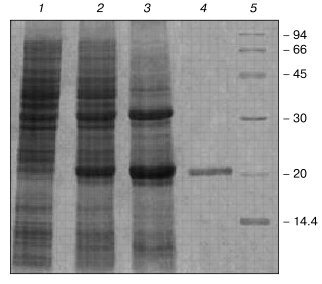
CD spectroscopy. The far-UV CD spectrum of VL-barstar at pH 7.4 displays a peak of negative ellipticity with a minimum at around 218 nm (Fig. 3). Denaturation by 8 M urea resulted in changes in the shape of the spectrum, bringing evidence for significant amount of secondary structure in the fusion protein under physiological conditions.
Barstar mutants with substitution of one or both Cys residues for alanine are reported to have similar with the wild type barstar conformational stability and inhibitory activity [19, 29]. This suggests insignificant influence of these mutations on the conformation of the barstar molecule. For example, NMR studies revealed that the double mutation did not significantly alter the spatial structure of the protein [30, 31]. Direct experimental evidence also exists that such double substitution did not affect secondary structure and, consequently, the CD spectrum [32]. This allowed us to generate the calculated CD spectrum of VL-barstar on the basis of individual curves of the VL domain and native barstar (Fig. 3). The calculation was made using the assumption that secondary structure of both constituting modules remained unchanged in the fusion protein. Comparison of the calculated and experimental spectra revealed significant differences. In particular, the calculated spectrum exhibits the negative maximum at 210 nm, which corresponds to contribution from alpha-helices. Lack of this peak in the experimental spectrum suggests that alpha-helices that are typical for individual barstar [33] are presumably not formed in the chimeric molecule.Fig. 3. Far-UV CD spectra of VL-barstar: 1) VL-barstar in 0.1 M sodium phosphate buffer, pH 7.4; 2) the same in the presence of 8 M urea; 3) the calculated curve obtained from individual spectra of the VL domain and barstar in 0.1 M sodium phosphate buffer, pH 7.4 (see “Results” section).
Near-UV CD spectra of the chimeric protein recorded with or without 8 M urea coincided with solvent spectra (data not shown). This suggests lack of asymmetric environment of aromatic groups that are known as major contributors to optical activity at 250-300 nm. Lack of asymmetric environment of aromatic residues indicates (partial) loss of tertiary interactions and formation of partially disordered tertiary structure.
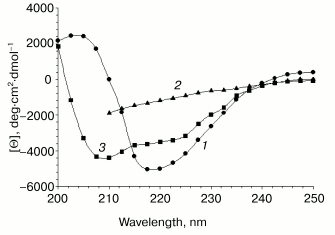
Intrinsic fluorescence. The maximum of the intrinsic fluorescence spectrum of VL-barstar located at 338 nm and the red shift to 352 nm induced by denaturation (Fig. 4a) suggest compact tertiary structure. Under physiological conditions, the amount of tertiary structure is enough for protecting of at least some of the four Trp residues against quenching by solvent. Denaturation by 7 M urea makes tryptophan residues accessible to the polar aqueous environment, which results in a large red shift of the maximal emission wavelength.
Fluorescence of bound ANS. 1-Anilino-8-naphthalenesulfonate (ANS) is widely used as a conformational probe. Due to its hydrophobic nature, ANS binds to so-called partially unfolded protein states where destabilization of the spatial structure results in accessibility of hydrophobic clusters to the probe. ANS does not bind to native (fully compact) or unfolded proteins.Fig. 4. Unfolding transitions and stability of VL-barstar monitored by fluorescent spectroscopy. a) Spectra of intrinsic fluorescence of the chimeric protein: 1) VL-barstar in 0.1 M sodium phosphate buffer, pH 7.4; 2) the same in the presence of 7 M urea. b) Unfolding of VL-barstar induced by increasing urea concentrations. The excitation wavelength was 280 nm.
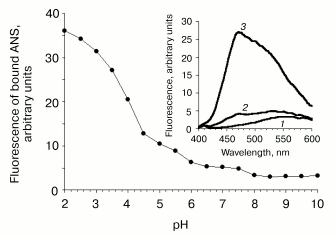
At neutral pH values, the recombinant protein demonstrated small but reliable ANS binding (Fig. 5), bringing evidence for partially disordered tertiary structure. Lowering pH was accompanied by the increase in binding of the hydrophobic probe due to subsequent loss of tertiary interactions and exposure of hydrophobic clusters. It should be noted that the total loss of tertiary structure was not observed down to pH 2.0 as indicated by no decrease in emission intensity of bound ANS. Thus, the tertiary structure of VL-barstar is intermediate between fully folded proteins and random coils.
Stability against chemical denaturation. Absence of a cooperative transition during protein unfolding induced by various denaturing factors is an indicative of a partially disordered conformation. Raising urea concentrations induce the increase in the fluorescence intensity of VL-barstar due to spatial distancing of Trp residues from their internal quencher, presumably the disulfide bond (Fig. 4b). However, the increase in emission observed at 5.6 M urea is less than 30% of the initial fluorescence intensity in the absence of the denaturant. The transition curve lacks a sigmoidal transition region that is typical for cooperative unfolding of proteins with native-like tertiary structure. Non-cooperative behavior of the transition curve suggests formation of a non-native conformation with partial loss of tertiary interactions.Fig. 5. pH stability of VL-barstar monitored by changes in protein-bound ANS fluorescence at 470 nm. Inset: fluorescence spectra of free ANS (1) and its complex with VL-barstar at pH 7.5 (2) and 2.0 (3). Excitation wavelength: 360 nm.
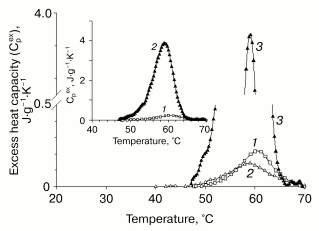
Differential scanning calorimetry (DSC). The thermal unfolding profile of VL-barstar shows a small peak of heat absorption at about 60°C (Fig. 6). The abnormally low value of the denaturation enthalpy (0.5 cal/g) suggests only marginal stability of tertiary structure resulting from significant loss of tertiary interactions (comparison of the excess heat capacity profiles of VL-barstar and isolated VL and VH domains of antibody F11, the latter domain adopting a partially structured conformation [34], is shown in Fig. 7). The second scanning of the sample in the calorimetric cell revealed an increase in heat capacity (compared with the first heating) in the temperature region below the heat absorption peak. The increase in heat capacity was induced by partial reversibility of the thermal unfolding after the first heating that had lead to exposure of hydrophobic residues on the surface of the protein molecule. The increase in heat capacity revealed by the second scanning brings strong evidence for the compact tertiary structure of the entire protein. In spite of loss of a significant part of structural interactions, it is still sufficient for shielding hydrophobic clusters against contacts with solvent. Noteworthy, the partial (about 20%) reversibility of the thermal transition of VL-barstar indicates the presence of small amount of tertiary structure even after the first heating-cooling cycle.
Fig. 6. Experimental calorimetric curves of VL-barstar in 0.1 M sodium phosphate buffer, pH 7.4 (protein concentration is 1.6 mg/ml): 1) first heating; 2) second heating. Calorimetric enthalpy of denaturation was 0.5 cal/g.
The small, but reliably detected heat absorption peak on the heat capacity profile, as well as the increase in heat capacity revealed by the second scanning of the sample, suggest compact tertiary structure for VL-barstar. However, calorimetrically determined tertiary structure is only marginally stable due to the loss of a significant part of structural interactions. Compact, but significantly destabilized tertiary structure is typical for partially disordered protein molecules.Fig. 7. Comparison of the excess heat capacity profiles of VL-barstar and proteins with varying amount of structure: 1) VL-barstar; 2) the recombinant isolated VH domain of antibody F11, which belongs to the class of “native molten globules”; 3) completely folded recombinant VL domain of antibody F11. The inset shows the excess heat capacity of the VL domain and VL-barstar.
DISCUSSION
During expression of VL-barstar in E. coli cells, this chimeric protein is accumulated in inclusion bodies as insoluble aggregated species. Overexpression of a recombinant protein exceeding chaperone capacity of the host cell is one of the most typical reasons for insoluble aggregate formation in inclusion bodies. To prevent incorrect disulfide bond formation during folding, in the present study we have used the double barstar mutant (C40/82A) in which both cysteine residues (Cys40 and Cys82) have been mutated for alanine residues. Analysis of folded VL-barstar for the presence of thiols and disulfides revealed the absence of aberrant molecules with free thiol groups, or their oxygen derivatives or molecules with intermolecular disulfide bridges. The presence of only one molecular form of the resulting protein was confirmed by the gel chromatography and native electrophoresis data. The apparent molecule mass was about 54 kD. This value suggests dimerization, which is a characteristic feature of the VL domain [22] and a prerequisite for binding site formation of the antibody F11 VL domain [35]. We previously demonstrated dimerization of both the individual VL domain [22] and the fusion protein VL-barnase, a potential immunotoxin [23]. It is also possible (but less probable) that the increase in apparent molecular mass of VL-barstar may be attributed to the increase in Stokes radius, which might result from less compact spatial conformation generally found for intermediate states of some proteins.
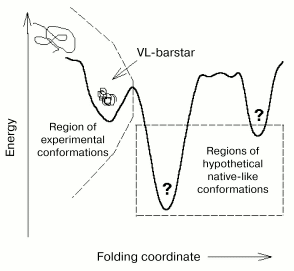
The conformation of VL-barstar obtained during refolding and purification is obviously the only achievable conformation within a wide range of conditions, because the use of various protocols for protein preparation did not yield other conformations. Long-term storage of the chimeric protein at various concentrations did not result in changes in calorimetry-revealed thermodynamic parameters. This suggests either lack of transition to more ordered conformation (provided that such conformations are attainable under other conditions, e.g., under employment of chaperones in vitro) or the lack of more compact conformations. The second suggestion implies a thermodynamic mechanism of stabilization (also known as a thermodynamic trap) due to inability of formation of a more organized chimeric protein with the given amino acid sequence (Fig. 8). The first suggestion means that the conformation of the chimeric protein is kinetically stabilized (kinetically trapped) in the local minimum of free energy due to significant value of the activation barrier separating this particular conformation from more ordered ones.
Data of spectroscopy and scanning calorimetry allow a detailed description of conformational changes in VL-barstar versus the individual proteins, the VL domain and barstar. These changes involve secondary structure that is significantly rearranged due to fusion of the two modules and is characterized by significant reduction in alpha-helical regions.Fig. 8. A scheme illustrating possible conformational states (folding “landscape”) of VL-barstar under the assumption that the partially disordered molecule is in a kinetic trap [36]. If the thermodynamic mechanism of stabilization (the thermodynamic trap) takes place, only one partially structured compact conformation (which was experimentally achieved) is attainable for the given protein. In this case, the more ordered hypothetical conformations (shown on the right of the dashed line) do not exist. Thus, in the case of the thermodynamic mechanism of the protein stabilization, folding “landscape” is represented by the left side of the scheme that depicts the region of the only two experimentally achieved conformations, the unfolded and partially folded ones.
Tertiary structure of VL-barstar is characterized by combination of compactness and significant destabilization. In this study, we have obtained several lines of evidence for the presence of a compact tertiary structure for the chimeric protein. First, the calorimetric curve has a small but reliably detected heat absorption peak. Second, the increase in heat capacity during the second heating in the calorimetric cell suggests that during the first heating in the pre-transition region at least part of the hydrophobic residues is solvent-inaccessible. Preferential location of hydrophobic amino acid residues inside the VL-barstar molecule is also confirmed by low fluorescence intensity of protein-bound ANS at neutral pH and increased ANS binding observed at a decrease in pH. The latter decrease indicates the progressive loss of tertiary interactions. However, we have not observed complete unfolding of the protein molecule, which would result in loss of hydrophobic probe binding at low pH. Finally, data of intrinsic VL-barstar fluorescence suggest that under physiological conditions at least some of the tryptophan residues of the chimeric protein are inaccessible to solvent. This provides an additional argument for compact tertiary structure of the VL-barstar molecule.
Destabilization of tertiary structure is further indicated by a low value of denaturation enthalpy that is less than 20% of the enthalpy obtained for each of the individual modules of VL-barstar (3.5 cal/g for VL domain [21] and 4.7 cal/g [18] or 7.2 cal/g [19] for wild type barstar). In agreement with these data, lack of asymmetry in aromatic residues (as evidenced by CD spectra in the 250-300 nm region) indicates the loss of a significant part of the tertiary interactions. The individual VL domain [21] and barstar [17, 33] exhibit pronounced optical activity within the 250-300 nm range due to asymmetry in the environment of aromatic residues. The loss of stability of tertiary structure due to fusion of the two components into a single polypeptide chain is also confirmed by the lack of cooperativity during urea-induced denaturation of the chimeric protein (Fig. 4b).
Significant changes in tertiary and secondary structures of the chimeric molecule result in the loss of functional activity of both antigenic and inhibitory structural modules. It is known that Asp35 and Asp39 of the barstar molecule are major determinants of its binding to barnase [10, 16]. These residues are involved in the alpha-helix [33]. Thus, the lack of functional activity of barstar in the chimeric protein may be attributed to the loss of well-defined alpha-helical regions. This is quite consistent with lower values of negative ellipticity in CD spectra of barstar in the far-UV region (Fig. 3).
Taken together, the calorimetric and spectroscopic data suggest that the conformation of the chimeric protein VL-barstar belongs to the group of so-called partially structured states, which are stable under physiological conditions. These intermediate states share one common feature, the loss of a part of tertiary interactions and destabilization of the protein molecule [37]. A large body of evidence has been recently accumulated indicating that the intermediate states may exist not only in partially denaturing but also under physiological conditions [38, 39]. It should be noted that partially structured protein conformation is not directly related to the loss of functional activity, because there are several examples demonstrating functionality in proteins with a variable extent of structural disorder [21, 40, 41]. According to a current concept, partially disordered conformations existing under physiological conditions involve two major classes. Members of the first class of “native molten globules” still have compact tertiary structure, together with a well-defined secondary structure, in spite of loss of part of tertiary interactions [38]. Proteins constituting the second class of “natively unfolded” proteins lack a compact spatial structure and are therefore conformationally close to random coil [42]. According to an extent of the loss of a secondary structure, these proteins may be further subdivided into pre-molten globules and intrinsic coils [43]. Precise conformational discrimination of the above classes and sub-classes is often complicated. Nevertheless, taking into consideration the presence of compact yet destabilized tertiary structure and well-defined secondary structure, the chimeric protein VL-barstar can be attributed to the class of “native molten globules”.
To explain formation of intermediate structural state of the chimeric protein, the following data should be taken into consideration. During refolding from inclusion bodies in vitro, the isolated VL domain formed compact and stable tertiary structure typical for native proteins [21, 22]. Consequently, intermediate conformation of VL-barstar resulted from fusion of heterologous domains through a linker, with the addition of the C-terminal histidine hexapeptide. A linker peptide of 12-15 residues provides sufficient flexibility for each module (see for example [44, 45]). The linkers and the C-terminal fragment (tag) comprising a few histidine residues are widely used for construction of chimeric proteins. Their employment does not generally result in conformational alterations similar to these reported here for VL-barstar. Thus, the structural source of incomplete folding found for VL-barstar might be attributed to properties of barstar itself and its interaction with the VL domain in the chimeric protein.
In this regard, it should be noted that in previous studies of individual barstar it was isolated from soluble fraction of cell lysate rather than from inclusion bodies [10, 16-19]. In our case, refolding of individual barstar solubilized from inclusion bodies resulted in protein aggregation (unpublished observation). It is possible that barstar folding in vivo critically depends on chaperones that promote formation of a stable and functional conformation of the soluble form of this protein. In this context, VL-barstar is distinct from another VL-derived fusion, VL-barnase [23]. Barnase is a hydrophilic and highly stable protein; the amino acid sequence of wild type barnase lacks cysteine residues. In VL-barnase fusion, barnase plays a role of an intramolecular chaperone, which allows expression of the soluble chimeric construct into the cytoplasm [23] rather than into inclusion bodies (where individual VL domain is expressed to [21]). The fact that the VL domain can be correctly (re)folded in vitro as an individual protein but does not attain complete folding after fusion to C-terminus of barstar, strongly suggest that folding of VL-barstar is blocked at the stage preceding formation of a complete system of specific tertiary interactions. This implies the presence of early interactions of two premature structures of the heterologous modules at the stage of incomplete folding of the two constituents of the VL-barstar molecule.
Thus, the conformational study of VL-barstar provides evidence for interdomain interactions in chimeric constructions comprising proteins that belong to phylogenetically distant taxonomic groups (in our case, mammals and bacteria). VL-barstar represents the first example of the recombinant immunoconjugate derived from barstar and a single antibody domain. Interaction of the two heterologous modules of VL-barstar at the early stage of the spatial structure formation arrests subsequent folding and results in altered secondary and partially unfolded tertiary structures, which preclude functional activity of the two constituting modules. Since fusion proteins comprising antibody fragments and barstar/barnase are considered as complementary partners for a new class of bivalent/bispecific proteins of potential biomedical applications, our results demonstrate the necessity of detailed conformational study of new constructs based on barstar.
This work was supported by the Belarussian-Russian Foundation for Basic Research (grant B02R-044), Belarussian Foundation for Basic Research (grant B01-132), INTAS (grant 2000-0554), Russian-Belarussian Foundation for Basic Research (02-04-81016), Russian Foundation for Basic Research (04-04-49024), Foundation for Support of Russian Science, Programs for Support of Leading Scientific Schools (grant 2077.2003.4), and Program of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology”.
REFERENCES
1.Brinkmann, U. (1996) Mol. Med. Today,
2, 439-446.
2.Thrush, G. R., Lark, L. R., Clinchy, B. C., and
Vietta, E. S. (1996) Annu. Rev. Immunol., 14, 49-71.
3.Carter, P. (2001) Nature Rev., 1,
118-129.
4.Chaudhary, V. K., Queen, C., Junghans, R.,
Waldmann, T., FitzGerald, D., and Pastan, I. (1989) Nature,
339, 394-397.
5.Rybak, S. M., Hoogenboom, H. R., Meade, H. M.,
Raus, J. C. M., Schwartz, D., and Youle, R. J. (1992) Proc. Natl.
Acad. Sci. USA, 89, 3165-3169.
6.Deonarain, M. P., and Epenetos, A. A. (1995)
Tumor Targeting, 1, 177-182.
7.Newton, D. L., Futami, J., Ruby, D., and Rybak, S.
M. (2003) Meth. Mol. Biol., 207, 283-304.
8.Makarov, A. A., and Ilinskaya, O. N. (2003) FEBS
Lett., 540, 15-20.
9.Deyev, S. M., Waibel, R., Lebedenko, E. N.,
Schubiger, A. P., and Plückthun, A. (2003) Nature
Biotechnol., 21, 1486-1492.
10.Hartley, R. W. (2001) Meth. Enzymol.,
341, 599-611.
11.Lai, J., Quadri, S. M., Borchardt, P. E., Harris,
L., Wucher, R., Askew, E., Schweichler, L., and Vriesendorp, H. M.
(1999) Clin. Cancer Res., 5, 3315-3323.
12.Vriesendorp, H. M., Quadri, S. M., Wyllie, C. T.,
Lai, J., Borchardt, P. E., Harris, L., Wucher, R., Askew, E., and
Schweichler, L. (1999) Clin. Cancer Res., 5,
3324-3329.
13.Collinet, B., Herve, M., Pecorari, F., Minard,
P., Eder, O., and Desmadril, M. (2000) J. Biol. Chem.,
275, 17428-17433.
14.Blondel, A., Nageotte, R., and Bedouelle, H.
(1996) Protein Eng., 9, 231-238.
15.Chenal, A., Nizard, P., Forge, V., Pugniere, M.,
Roy, M.-O., Mani, J.-C., Guillain, F., and Gillet, D. (2002) Protein
Eng., 15, 383-391.
16.Schreiber, G. (2001) Meth. Mol. Biol.,
160, 213-226.
17.Khurana, R., and Udgaonkar, J. B. (1994)
Biochemistry, 33, 106-115.
18.Wintrode, P., Griko, Y. V., and Privalov, P. L.
(1995) Protein Sci., 4, 1528-1534.
19.Schoppe, A., Hinz, H.-J., Agashe, V. R.,
Ramachandran, S., and Udgaonkar, J. B. (1997) Protein Sci.,
6, 2196-2202.
20.Dubnovitsky, A. P., Kravchuk, Z. I., Chumanevich,
A. A., Cozzi, A., Arosio, P., and Martsev, S. P. (2000) Biochemistry
(Moscow), 65, 1011-1018.
21.Martsev, S. P., Chumanevich, A. A., Vlasov, A.
P., Dubnovitsky, A. P., Tsybovsky, Ya. I., Deyev, S. M., Cozzi, A.,
Arosio, P., and Kravchuk, Z. I. (2000) Biochemistry, 39,
8047-8057.
22.Martsev, S. P., Dubnovitsky, A. P., Vlasov, A.
P., Hoshino, M., Hasegawa, K., Naiki, H., and Goto, Yu. (2002)
Biochemistry, 41, 3389-3395.
23.Martsev, S. P., Tsybovsky, Y. I., Stremovskiy, O.
A., Odintsov, S. G., Balandin, T. G., Arosio, P., Kravchuk, Z. I., and
Deyev, S. M. (2004) Protein Eng., Design Selection,
17, 85-93.
24.Thannhauser, T. W., Konishi, Y., and Scheraga, H.
A. (1984) Analyt. Biochem., 138, 181-188.
25.Ellman, G. L. (1959) Arch. Biochem.
Biophys., 82, 70-77.
26.Kunitz, M. (1946) J. Biol. Chem.,
164, 563-568.
27.Gill, S. C., and von Hippel, P. H. (1989)
Analyt. Biochem., 182, 319-326.
28.Tsumoto, K., Ejima, D., Kumagai, I., and Arakawa,
T. (2003) Prot. Express. Purif., 28, 1-8.
29.Hartley, R. W. (1993) Biochemistry,
32, 5978-5984.
30.Lubienski, M., Bycroft, M., Freund, S. M. V., and
Fersht, A. R. (1994) Biochemistry, 33, 8866-8877.
31.Wong, K.-B., Fersht, A. R., and Freund, S. M. V.
(1997) J. Mol. Biol., 268, 494-511.
32.Protasevich, I. I., Schulga, A. A., Vasilieva, L.
I., Polyakov, K. M., Lobachov, V. M., Hartley, R. W., Kirpichnikov, M.
P., and Makarov, A. A. (1999) FEBS Lett., 445,
384-388.
33.Notling, B., Golbik, R., Soler-Gonzalez, A. S.,
and Fersht, A. (1997) Biochemistry, 36, 9899-9905.
34.Martsev, S. P., Dubnovitsky, A. P., Stremovsky,
O. L., Chumanevich, A. A., Tsybovsky, Y. I., Kravchuk, Z. I., and
Deyev, S. M. (2002) FEBS Lett., 518, 177-182.
35.Nymalm, Y., Kravchuk, Z. I., Salminen, T.,
Chumanevich, A. A., Dubnovitsky, A. P., Kankare, J., Pentikoinen, O.,
Lehtonen, J., Arosio, P., Martsev, S. P., and Johnson, M. S. (2002)
J. Struct. Biol., 138, 171-186.
36.Dill, K. A., Bromberg, S., Yue, K., Fiebig, K.
M., Yee, D. P., Thomas, P. D., and Chan, H. S. (1995) Protein
Sci., 4, 561-602.
37.Creighton, T. F. (1990) Biochem. J.,
270, 1-16.
38.Ptitsyn, O. B. (1995) Adv. Prot. Chem.,
47, 83-229.
39.Privalov, P. L. (1996) J. Mol. Biol.,
258, 707-725.
40.Reddy, G. B., Srinivas, V. R., Ahmad, N., and
Surolia, A. (1999) J. Biol. Chem., 274, 4500-4503.
41.Bose, H. S., Whittal, R. M., Baldwin, M. A., and
Miller, W. L. (1999) Proc. Natl. Acad. Sci. USA, 96,
7250-7255.
42.Uversky, V. N., Gillespie, J. R., and Fink, A. L.
(2000) Proteins, 41, 415-427.
43.Uversky, V. N. (2002) Eur. J. Biochem.,
269, 2-12.
44.Atwell, J. L., Breheny, K. A., Lawrence, L. J.,
McCoy, A. J., Kortt, A. A., and Hudson, P. J. (1999) Protein
Eng., 12, 597-604.
45.George, R. A., and Heringa, J. (2003) Protein
Eng., 15, 871-879.