REVIEW: Short Forms of Membrane Receptors: Generation and Role in Hormonal Signal Transduction
O. V. Smirnova and R. L. Bogorad*
Faculty of Biology, Lomonosov Moscow State University, Moscow 119992, Russia; fax: (7-095) 939-4309; E-mail: rbogorad@yahoo.com* To whom correspondence should be addressed.
Received March 17, 2003; Revision received July 7, 2003
This review highlights the generation of various types of short forms of membrane hormonal receptors and the mode of regulation of their tissue-specific patterns. The short forms of membrane receptors are classified on the basis of localization of missing functional fragments. The review provides examples of tissues for which expression of short forms may serve as a marker of changes of ontogenetic stage, physiological state, or the development of pathological process. The short forms of receptors are shown to participate in determining tissue-specificity and efficacy of hormonal signal transduction, as well as in transport of hormones within cell, through physiological barriers, and in blood circulation. Peculiarities of signal transduction pathways for short receptor forms and potential physiological significance of these forms are analyzed. It is concluded that the ratio of long and short receptor forms may serve as a key marker of dynamic changes of differentiation stage and alterations of metabolic and proliferative activity of tissues under normal and pathologic conditions, and thus to be an important indicator of therapeutic effect for many pathological processes.
KEY WORDS: membrane hormone receptors, receptor short forms, alternative splicing, signal transduction, hormone transport
Abbreviations: CREM) cAMP responsive element modulator; CPSF) cleavage polyadenylation specific factor; hnRNP) heterogeneous nuclear ribonucleoproteins; IGFII) insulin like growth factor II; JAK) Janus kinases; MAP) mitogen activated protein kinases; NAPOR) neuroblastoma apoptosis-related RNA binding protein; NPR(A,B,C)) natriuretic peptide receptor (type A, B, C); PACAP) pituitary adenylate cyclase activating peptide; PBFII) polypurine binding factor II; PRAP) prolactin receptor associated protein; SF2/ASF) splicing factor 2/alternative splicing factor; STAT) signal transducer/activator of transcription.
According to the initial classification of membrane hormone receptors
based on the main principles of their structure-functional
organization, receptors were divided into several superfamilies, each
characterized by a “central” signal transduction pathway.
These include G-protein coupled receptors; receptors coupled with
tyrosine kinases, receptor tyrosine kinases, receptor serine/threonine
kinases, receptor guanylate cyclases, receptor phosphatases, receptor
ion channels, etc.
Later shorter homologs of known receptors lacking some functional domains have been recognized. In the present review, we define the short form of receptors as both protein products of shortened members of a homolog gene family, and also as protein products encoded by one gene, but generated due to various mechanisms. Gene polymorphism as the mechanism responsible for appearance of individual receptor forms is out of consideration in this review. Short forms have been found for the following receptor superfamilies: G-proteins coupled receptors (some receptor subtypes for dopamine, glutamate, vasoactive intestinal peptide, PACAP and other signaling compounds); receptors coupled with tyrosine kinases (receptors for growth hormone, prolactin, leptin, erythropoietin and other cytokines); receptor tyrosine kinases (receptors for insulin, epidermal growth factor, fibroblast growth factor, and other growth factors) [1-5]. Usually, their role and physiological importance have not been specially investigated because short forms cannot transduce a signal typical for full-length receptors.
However, recent data provide increasing evidence that short receptor forms may underline tissue specificity and efficacy of realization of hormonal effect; they also seem to be involved in intercellular (systemic and local) and intracellular hormone transport. In this review, we summarize current knowledge on the role of short forms of membrane receptors in additional specialization of hormone signal transduction and regulation of its magnitude depending on tissue type and physiological state of a tissue.
1. MECHANISMS OF GENERATION OF SHORT RECEPTOR FORMS
Short forms of receptors can be classified by localization of missing functional domains of a receptor molecule. Using this criterion, the following groups of short receptors can be recognized: forms with shortened extracellular domain, forms with shortened intracellular domain, and also special receptor forms lacking transmembrane and intracellular domain. These special forms are also known as soluble receptors or hormone transport proteins of receptor type.
During production of short receptor forms almost all ways of generation of related proteins with variations in amino acid sequence and domain composition described for eukaryotes are used [1-6] (see below).
1.1. Expression of various genes. Receptors for many hormones are often encoded by a family of genes (e.g., receptors for natriuretic peptide [3], somatostatin [7], etc.). In the case of neurotransmitter receptors, this is even a “rule of the game” (e.g., dopamine receptors [8], glutamate receptors [5], etc.). Natriuretic peptide receptors (NPR) represent an illustrative example of the use of various genes for generation of receptor forms that have homologous amino acid sequences but differ in the presence of some structural domains and in their functions. There are three genes encoding NPR-A, NPR-B, and NPR-C, respectively [3]. Two receptors (NPR-A and NPR-B) share high homology, and their C-terminus contains a catalytic guanylate cyclase domain. These two types of receptors contain all domains typical for this superfamily and transduce comprehensive signal of natriuretic peptides: atrial natriuretic peptide acting via NPR-A regulates natriuresis and water balance; C-type natriuretic peptide acting via NPR-B functions as a local regulator of endothelial functions. NPR-C (also known as clearance receptor) shares homology with NPR-A and NPR-B in its extracellular domain, but it practically lacks the cytoplasmic domain and guanylate cyclase activity [9]. In contrast to NPR-A and NPR-B, this type of receptor is considered as a negative regulator of natriuretic peptide signal transduction and a transporter of natriuretic peptides through physiological barriers (see Section 4) [10].
1.2. Alternative splicing of pre-mRNA. This is one of the most common modes for generation of various forms of receptors: e.g., for growth hormone [11], prolactin [12], leptin [13], insulin [14], luteinizing hormone [15, 16], dopamine (types 2 and 3) [17], PACAP [18, 19], etc.
Variants of alternative splicing of prolactin receptor pre-mRNA represent a good illustration. The mammalian gene encoding prolactin receptor (>100 kb) consists of 11 exons [20]. In various species, there are 3-4 alternative promoters and 3-4 corresponding non-coding first exons [21, 22]. There are many variants of alternative splicing of 3´-non-coding region, which significantly influence length of mature mRNA (from 1.5 to 9.5 kb) [23]. Different mammalian species are characterized by distinct arrangement of alternative splicing resulting in generation of various forms of prolactin receptor. In rodents long form of prolactin receptor is generated without the use of exon 11 (termination of translation occurs within exon 10 due to the presence of an internal stop-codon there). During generation of short form of prolactin receptor exon 10 is not used; a sequence from the end of exon 9 up to the beginning of exon 11 is excluded (the site of termination of translation localizes in exon 11) [24] (figure, (a)). Moreover, the existence of several variants of exon 11 in rodents serves as a basis for expression of several short forms of prolactin receptor in rodents (in mouse there are 1 long and 3 short forms, see figure, (c)) [25]. Besides short form, a pathological intermediate form of prolactin receptor expressed in Nb2 cell is found in rat. This intermediate form was generated due to deletion of 597 bp in exon 11 (figure, (b)) [26, 27]. Short form of prolactin receptor differs from long and intermediate forms by the structure of cytoplasmic domain: it lacks the so-called second box, required for activation of JAK/STAT cascade, which is ultimately important for the superfamily of receptors coupled with tyrosine kinases [28, 29]. In contrast to the long receptor form mediating almost all prolactin effects (expression of gene encoding beta-casein, cell proliferation, etc.), the short form of prolactin receptor is involved in negative regulation of receptor long form activity and in tissue specific signals (see Section 4) [30]. Long, intermediate, and two short forms of prolactin receptor are expressed in humans. The cytoplasmic domain of the first short form, S1a, contains amino acid residues encoded by a part of exon 10 and also a unique 39-membered amino acid sequence encoded by exon 11. Generation of intracellular domain of S1b form does not involve exon 10 at all; this domain contains only 3 amino acid residues formed due to reading frame shift in exon 11 resulting in formation of an early stop codon [31]. Generation of the intermediate form in humans involves the same mechanism as in rodents; the only exception is that in humans deletion causes reading frame shift, and the C-terminus of the receptor is non-homologous to the long form [27].
In humans, besides alternative variants of intracellular domain a form with shortened extracellular domain due to alternative splicing has been revealed. This form known as DeltaS1 form is generated by deletion of exons 4 and 5 resulting in the absence of N-terminal fibronectin-like domain (one of two fibronectin-like domains in the prolactin receptor molecule) in the mature receptor molecule; such deletion significantly influences receptor affinity [32].Scheme illustrating generation of various forms of prolactin receptor in rodents: a) generation of long and short forms of rat prolactin receptor by alternative splicing; b) generation of intermediate (Nb2) form of rat prolactin receptor by joining of exons 9 and 10 and deletion (597 bases) within exon 10; c) generation of 1 long and 3 short forms (due to use of three alternative 11th exons) of mouse prolactin receptor by alternative splicing. Rectangles show exons: white and black rectangles designate coding and non-coding regions. Dashed lines show introns. SC and DL designate stop codons located within reading frame and deletions, respectively
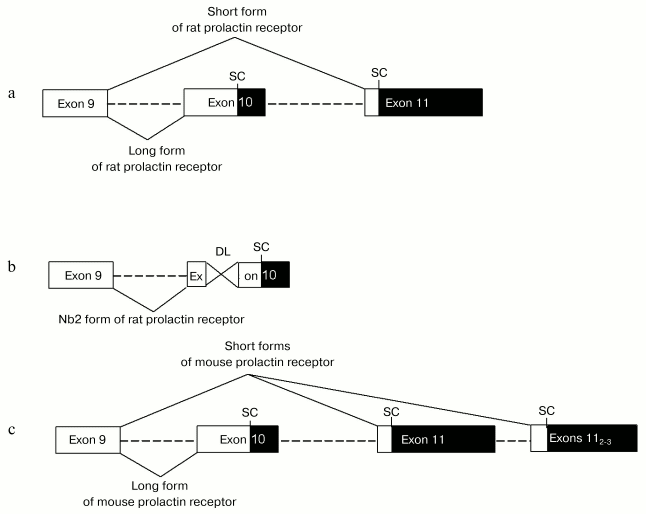
In rodents (and even-toed ungulates), along with anchored membrane bound forms soluble secretory form of receptor is also formed by alternative splicing. This form is produced by exclusion of exons 7 and 8; reading frame shift accompanying this exclusion results in formation of a short hydrophilic C-terminal domain of the receptor molecule, which differs from the corresponding domain of the long form of prolactin receptor [33]. In humans, generation of the secretory form of prolactin receptor involves proteolysis rather than alternative splicing (see Section 1.4)1. The soluble form of prolactin receptor is not involved in signal transduction; it is considered as a transport prolactin binding protein, which negatively influences activity of the long form.
1 Recently, the possibility of generation of soluble forms of human prolactin receptor via alternative splicing has been demonstrated in direct experiments (Trott, J. F., et al. (2003) J. Mol. Endocrinol., 30, 31-47). 1.3. Alternative polyadenylation. Polyadenylation of pre-mRNA is the other mechanism of alternative form generation at the stage of pre-mRNA processing. Conservative sequences representing polyadenylation signals are now well characterized. Some eukaryotic genes possess several alternative polyadenylation signals; their “use” may change length of the coding site [34]. For example, in mammals generation of secretory forms of epidermal growth factor receptors [35, 36] involves usage of a polyadenylation site localized in the intron between exons 12 and 13; the resultant mature mRNA lacks a site corresponding to exon 17, encoding a transmembrane domain. Generation of membrane-unbound form of mouse growth hormone receptor (which is a receptor-type transport protein for growth hormone) may be not only a result of alternative splicing, but also involve alternative polyadenylation site localized in exon 8a [37]. Alternative polyadenylation may serve as an effective mechanism responsible for regulation of expression of alternative receptor forms at the stage of translation. For example, use of proximal polyadenylation sites leads to removing of mRNA instability signals and thus increasing of expression level of such form [38].
1.4. Proteolysis. Generation of receptor forms by proteolytic cleavage is typical for soluble receptors (transport hormone-binding proteins). For example, in humans (in contrast to many other mammalian species) transport proteins for growth hormone [39], leptin [40], and prolactin [41] are formed mainly due to proteolysis of full-size receptor. Proteolysis of many receptors is performed by metalloproteases of metzicin family, known as TACE and ADAM17. These proteases cleave an extracellular domain of receptor expressed on the cell surface [42]. However, currently specific sequences of sites for cleavage remain unrevealed.
2. REGULATORY MECHANISMS UNDERLYING EXPRESSION OF RECEPTOR
FORMS
2.1. Regulation of transcription. In the case of generation of receptor forms as protein products of some different genes, the expression of each element of a receptor system can be regulated independently. For example, expression of active natriuretic peptide receptors (NPR-A and NRP-B) and clearance receptor (NPR-C) is determined by various transcription factors [43]. Such a mechanism seems to be required for effective regulation of receptor forms in neurotransmitter systems, which are characterized by the existence of a set of genes encoding various receptor forms.
2.2. Regulation of RNA processing. The main mechanism of control of alternative splicing involves the action of factors (responsible for selection of exons) interacting with small nuclear RNA spliceosomes finally leads to preferential use of proximal or distal exon. These factors are universal for all pre-mRNAs, including receptor pre-mRNAs. SF2/ASF belongs to a family of splicing trans-factors, known as SR proteins, which are enriched with serine and cysteine residues. In vivo and in vitro experiments have demonstrated that this factor is responsible for preferential use of proximal acceptor exon [44, 45]. Proteins of the hnRNP family exhibit an antagonistic effect resulting in preferential use of distal acceptor exons [46]. Thus, “choice” of exon during alternative splicing may be determined by ratio of expression and activity of proteins of SR and hnRNP families. Regulation of such ratio can be controlled at the transcriptional level or by change of phosphorylation degree and also by some other factors [47]. Participation of several types of serine/threonine kinases--calmodulin-dependent kinase, MAP-kinase, and some others--in phosphorylation of these proteins has been revealed at the present time.
Besides universal regulatory mechanisms, there are tissue-specific factors regulating alternative splicing of some pre-mRNA. For example, NAPOR protein (neuroblastoma apoptosis-related RNA binding protein) regulates splicing of NMDA 1 glutamate receptors in rat forebrain increasing probability of exclusion of exon 21 and inclusion of exon 5 [48].
Along with transcription trans-factors, selection of exon ultimately requires cis-elements of mRNA sequence (splicing enhancers and silencers) as well as 5´- and 3´-untranslated regions [49]. Thus, it is relevant to suggest that the use of various promoters (even with non-coding alternative 5´-exons) can finally influence amino acid sequence of protein product via modulation of alternative splicing. Although the interrelationship between use of some promoter and generation of certain forms remains to be demonstrated, we can suggest that in the case of prolactin receptor such interrelationship actually exists. A correlation between tissue specific promoters and domination of certain forms of prolactin receptor in some tissues indirectly supports such hypothesis. In rat gonads, promoter I and long form of prolactin receptor dominate, whereas in liver, promoter II and short form of prolactin receptor dominate [50-53].
Hormonal regulation is one of the most important tools controlling alternative splicing of many mRNAs, particularly receptor mRNA (see Table 1) [54-62]. For example, two forms of insulin receptor are expressed in humans and rat; one form contains an amino acid sequence encoded by exon 11 (11+ form), whereas the other one lacks such amino acid sequence. Insulin increases the proportion of 11+ form of the receptor [56]. Estrogens increase the proportion of dopamine receptors type 2 with included exon [54]. Sex steroids also regulate alternative splicing of prolactin receptor mRNA. For example, in rats estradiol promotes preferential use of distal exon (this results in synthesis of short receptor form), whereas testosterone promotes inclusion of proximal exon, resulting in decreased generation of short form [61].
Table 1. Hormonal regulation of alternative
splicing of membrane receptors
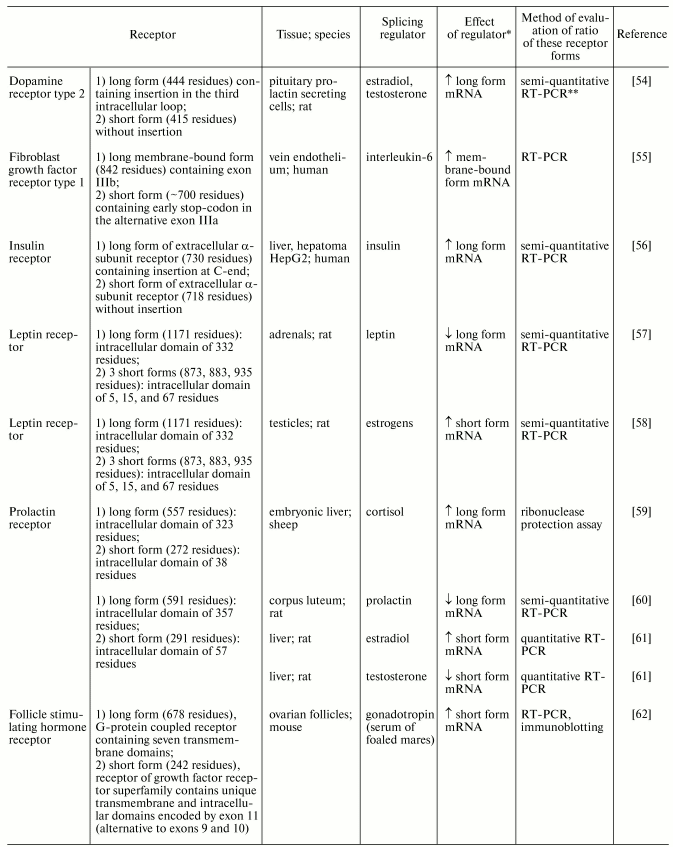
*Arrows ^ and v show increase or decrease,
respectively.
**RT-PCR, reverse transcription-polymerase chain
reaction.
All the abovementioned hormones act on SF2/ASF and hnRNP splicing regulators at the level of their transcription, phosphorylation, and change of cellular localization [63]. The role of hormones activating cascade of serine/threonine kinases in the regulation of alternative splicing has been studied in detail [64]. Taking into consideration the fact that many hormones (insulin, growth hormone, leptin, growth factors, etc.) activate receptor tyrosine kinases or receptors associated with tyrosine kinases, it is very important to study the role of tyrosine kinases in regulation of splicing.
It should be noted that transition of cell cycle stages is closely related to change in alternative splicing pattern. Many regulatory factors of the cell cycle share structural resemblance and functions with factors regulating alternative splicing; it is believed that some factors may share both functions. For several factors, cell cycle-dependent expression profile was demonstrated [65]. Thus, the ratio of expressed forms may change from one stage of the cell cycle to another.
Polyadenylation is controlled by CPSF (cleavage polyadenylation specific factor) and PBFII (poly(A) binding factor) [66]. These factors can influence the coding sequence of mRNA and also length of 3´-untranslated region, regulating mRNA stability. Both mechanisms are used for control of generation and expression of receptor forms by alternative polyadenylation (see Section 4.1). Several examples illustrating hormonal regulation of polyadenylation factors are known: follicle stimulating hormone increases stability of CREM-tau (cAMP responsive element modulator) mRNA. It influences activity of polyadenylation factors, and this results in preferential use of weaker proximal polyadenylation signal (and thus increases expression level of a given mRNA due to cleavage of mRNA instability signal) [38]. Similar mechanisms may also be employed for regulation of expression of soluble receptor forms, which are often formed by alternative polyadenylation.
2.3. Regulation of proteolysis. No convincing evidence exists on the role of proteolysis in regulation of generation of soluble receptor forms. However, it was demonstrated that activation of phospholipase C can increase the proportion of cleavage of extracellular domain of growth hormone receptor [67]. It is possible, that proteolysis may be controlled indirectly via regulation of expression of substrates for proteolysis. For example, short forms of leptin and growth hormone receptors formed by alternative splicing are more readily subjected to proteolysis than long forms [42, 67].
3. TISSUE SPECIFIC EXPRESSION OF RECEPTOR FORMS
Various regulatory factors are responsible for generation and maintenance of tissue-specific ratios of expression of different receptor forms. Some tissues are characterized by predomination of certain form(s) of receptors, whereas in other tissues such predomination has not been recognized. Table 2 shows examples of tissues where short receptor forms dominate [4, 13, 27, 53, 68-74]. For example, the expression of prolactin receptor forms was qualitatively evaluated in many human and rat tissues. In the rat, long form predominates in gonads and hypothalamus, whereas short form predominates in liver [53]. Characteristic distribution of short and long forms of some neurotransmitter receptors was found in pre- and postsynaptic membranes: short form of dopamine receptor type 2 is the predominant form in bodies and axons of dopaminergic neurons and long form is the main type of postsynaptic receptor [69]. The existence of hormone-sensitive tissues with predomination of short receptor forms suggests existence of tissue-specific effects realized by these short receptors.
Table 2. Tissues with predominant expression
of short forms of membrane receptors
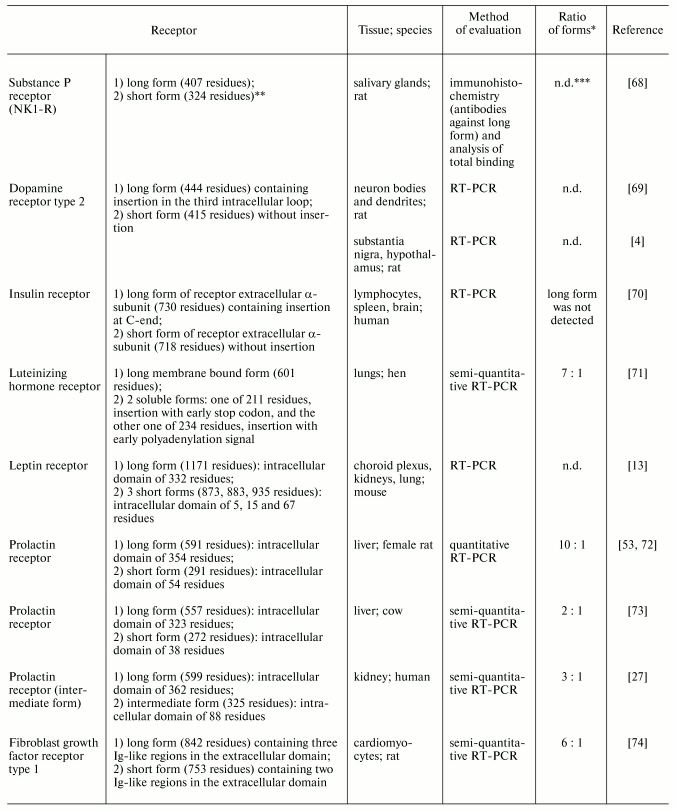
*Ratio of mRNA (protein) of short to long forms considered to
be predominant when it significantly exceeds 1.
**No data on mechanism of formation.
***n.d., no data or authors did not provide quantitative
results (in Tables 2 and 3).
An intriguing illustration underlying the importance of short receptor is a change in predominant receptor form during development of the fetus. For example, it is shown that the long form of fibroblast growth factor receptor type 1 predominant in embryonic rat cardiomyocytes is replaced by the short form in adult heart [74]. Similar changes are also found in the case of prolactin receptors of rat and bovine liver: the long form dominates early embryogenesis, whereas predominance of the short form develops during late embryogenesis [73, 75]. Short form of insulin receptor lacking alpha-subunit encoded by exon 11 of the insulin receptor gene dominates in embryonic but not in adult hepatocytes [76]. The replacement of a predominant receptor form in the course of organogenesis is a perfect natural model for investigation of change of both regulatory factors pattern and direction of hormonal effects realized in these tissues at different stages of pre- and postnatal development. This replacement emphasizes the importance of short receptor forms in the development of tissues. Study of characteristic features of expression of receptor forms under pathological conditions is especially important from both theoretical and practical viewpoints. Under pathological conditions (e.g., tumor development) both receptor forms typical for normal tissues, as well as new, altered forms may be expressed. Rat lymphoma Nb2 cells express a specific intermediate form of the prolactin receptor, which is not typical for normal tissues [26]. Analysis of expression of receptor forms can be used to evaluate sensitivity of tissues to hormones under pathological conditions and to diagnose disease. For example, in the case of pancreatic adenocarcinoma predominance of short form of fibroblast growth factor receptor type 1 is an important marker of high rate of tumor growth [77]. Rat erythroleukemic cells (partially lost erythroid characteristics) differ from normal cells by dramatically reduced level of expression of short form of erythropoietin receptor [78]. Diabetes mellitus type 2 is characterized by reduced level of short form of insulin receptor [79]. Under development of thyroid gland cancer an increased content of short form of insulin receptor and local production of IGFII is one of the leading way for automitogenic stimulations [80]. We also found change in expression pattern of receptor forms under experimental pathological conditions: development of obstructive cholestasis in female rats caused reduction in expression of short (predominant) form and increase in expression of long form in liver [72].
The peculiarities of short receptor form distribution in tissues under normal and pathological conditions, increase of their expression at certain stages of ontogenesis, as well as their possible importance for diagnostics and therapy attract an interest in the role of short receptors in signal transduction. Results of such studies are summarized in the next section.
4. MECHANISMS UNDERLYING ROLES OF SHORT RECEPTOR FORMS IN SIGNAL
TRANSDUCTION AND THEIR PHYSIOLOGICAL IMPORTANCE
Short receptor forms (including soluble ones, see Table 3) lacking some functionally important domain cannot transduce a signal that is normally mediated by the full-length receptor. However, these short forms are involved (together with long form or independently) in modulation of magnitude of regulatory signal and/or modification of certain stages of signal transduction such as specific ligand binding to receptor, oligomerization, activation of cascade of downstream high molecular weight and/or low molecular weight messengers, and also internalization of hormone-receptor complexes.
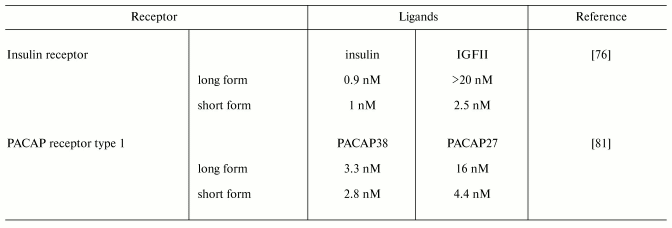
Since the extracellular domain of membrane receptors determines specificity and affinity of a ligand binding, short receptor variants significantly influence these parameters. PACAP receptor type 1 containing full-length sequence of extracellular domain binds preferentially PACAP38, whereas the shorter form (lacking 21 amino acid residues due to exclusion of exon 4) binds PACAP38 and PACAP27 with nearly equal effectiveness [81, 82]. The short form of insulin receptor binds insulin molecule with the same effectiveness as the long form and it is 10-fold more effective in IGFII binding ([70]) (Table 4). Specificity and affinity of ligand binding may also depend on the receptor form which is a partner for oligomerization. For example, formation of heterodimers between NPR-A and NPR-B (receptor guanylate cyclases) with short clearance receptor (NRP-C) results in preferential binding of a certain type of natriuretic peptide. Homodimers of receptor forms with shortened extracellular domain can modulate rate of response to hormonal signal due to differences in oligomerization rate. Affinity of human prolactin receptor DeltaS1 lacking one of the extracellular subdomains for this hormone is two times in magnitude less than that of full-length form (dissociation rate constants of hormone-receptor complex are 1.3*10-6 and 1.3*10-8 M, respectively). However, the short form is more active in signal transduction than the long one (maximal effect is observed after 10 and 15 min, respectively) due to effective homodimerization [32].
Table 3. Modes of generation of membrane
receptor soluble forms
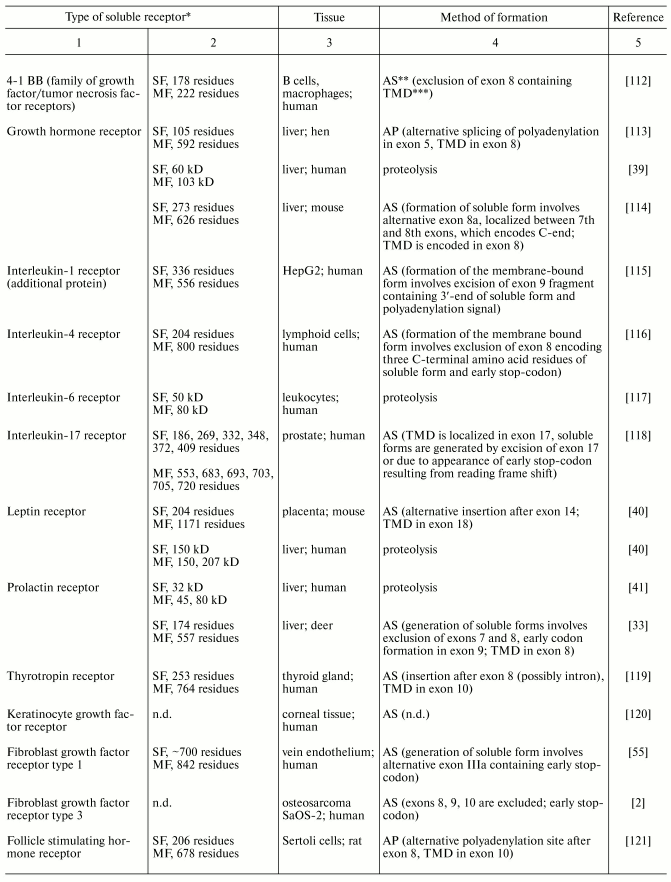
*SF and MF are soluble and membrane-bound forms,
respectively.
**AS, alternative splicing; AP, alternative
polyadenylation.
***TMD, transmembrane domain.
Table 3. (Contd.)
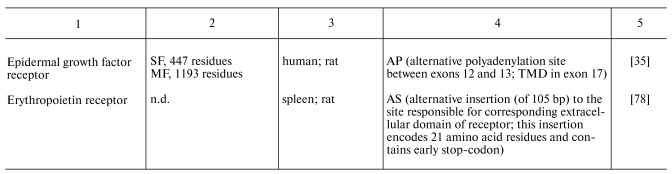
Table 4. Effective ligand concentrations
required for 50% inhibition of PACAP receptor type 1 and insulin
receptor binding
The existence of several receptor forms influences such stage of signal transduction as oligomerization of hormone-receptor complexes. For many receptor superfamilies (receptor tyrosine kinases, receptor serine/threonine kinases, receptors associated with tyrosine kinases) oligomerization is the ultimate precondition for signal transduction pathways because it promotes reciprocal phosphorylation of effector domains of these receptors (or proteins associated with these receptors) due to their bringing together. Mutations inducing ligand-independent dimerization of receptors of these superfamilies cause activation of corresponding cascades. For example, substitution of Gly380 in fibroblast growth factor receptor type 3 with Arg results in hormone-independent dimer stabilization and finally to some variant of dwarfism. Mutations in glial neurotrophic factor receptor (RET) leading to substitution of one of six cysteines localized in peri-membrane extracellular domain of receptor (609, 611, 618, 620, 630, 634) for other amino acids result in disulfide bonding between two receptor molecules; this causes constant activation of RET receptors and development of syndrome of multiple endocrine neoplasia type 2. Similar mutations are also known for other type of receptors [83].
It is believed that heterodimer formation between short and long forms (and also soluble and long forms) of receptors of these superfamilies cause inhibition of signal transduction realized by a long receptor form. For example, dimers of long form of prolactin receptor may effectively activate promoter of beta-casein gene. In in vitro experiments, co-transformation with short and long forms of prolactin receptor taken in various proportions has resulted in proportional decrease in activation of beta-casein gene promoter with increasing proportion of short form [30]. Decrease in signal efficiency may be achieved by decreasing the concentration of long homodimers and parallel increasing of expression of short isoform. However, correct evidence of the existence of such inhibition of hormonal signal in vivo requires demonstration of co-expression of these receptor forms within the same cell. At present, there are a limited number of studies on co-expression of several forms of various receptors in one cell. Nevertheless, co-localization of short and long forms of dopamine receptor type 2 was demonstrated in neurons [54]. Co-localization of PACAP receptor type 1 differing in structure of the third intracellular loop was found in rat gonads; however, single-cell PCR analysis revealed that only 10% of individual cells simultaneously contain mRNA of both short and long forms of this receptor [84].
It should be noted that short forms partially lacking active subdomains of a cytoplasmic domain of a receptor realize some but not all variants of classic signal transduction pathways typical for full-length receptor. For example, a short form of PACAP receptor (with shortened third intracellular loop responsible for binding to G-proteins) may activate adenylate cyclase but not phospholipase C [85]. A short form of the prolactin receptor lacking a part of the intracellular domain essential for JAK kinase activation does not phosphorylate STAT proteins. Nevertheless, the short isoform is involved in signal transduction pathway, which leads to proliferation of epithelial cells [86]; this signaling is mediated via cascade activation of MAP-kinases and some other proteins (Vav, phospholipase Cgamma, etc.). Thus, signal transduction pathways coupled with short receptor forms influences the ratio of activated second messengers and, consequently, the spectrum of modified effector proteins. These events determine changes in cell response to hormonal signal.
Recently, it was demonstrated that short receptor forms also possess their own downstream pathways of signal transduction that are not involved in long form signal transduction. For example, in corpus luteum short form of prolactin receptor is associated with PRAP protein (prolactin receptor associated protein), which is 17-ketosteroid dehydrogenase [87, 88]. Short form of dopamine receptor type 2 can inhibit both adenylate cyclase (as in the case of the long form) and phospholipase C [89]. Moreover, a short form of follicle stimulating hormone receptor (generated due to exclusion of exons 9 and 10 and use of additional exon 11) belongs to the superfamily of growth factor receptors rather than to the superfamily of receptors associated with G-proteins (as is for its long form). Activation of this form of receptor requires dimerization and activated receptor stimulates cascades of serine/threonine kinases and finally L-calcium channels [62].
At the stage of internalization the role of short and long receptor forms may differ. The rate of internalization of short forms of some receptors is higher than that of long ones (e.g., prolactin [90], leptin [91, 92], and insulin [93] receptors). Differences in the rate of internalization of short and long forms of prolactin receptors may be attributed to the fact that the former has two motifs increasing the internalization rate, whereas the latter has only one such motif [90]. Short receptor forms play an important role in intracellular transport of hormones between functionally distinct membranes of certain cells. In some cases internalization of hormone-receptor complexes into cells is accompanied not only by degradation of both hormone and its receptor but also by transportation of hormone and/or hormone-receptor complex to the nuclear membrane; such intracellular route of hormone (or hormone-receptor complex) may induce activation of “non-classic” nuclear pathways of peptide/protein hormone signal transduction pathways [94]. Good evidence for the presence of such a pathway is the demonstration that nuclei contain receptors for insulin [95], prolactin [96], growth hormone [97], and numerous growth factors [98, 99]. Results of in vitro experiments on prolactin effects on purified cell nuclei [100, 101] and binding of insulin [95, 102] and prolactin [103-105] receptors in cell nuclei also support this viewpoint.
A physiological role of short forms may also consist of transcytosis, specific intracellular transport of hormones from one functional site of a cell membrane to another; this leads to active transfer of high molecular weight hormones through physiological barriers (e.g., the blood-brain barrier). For example, it was shown that short form of leptin receptor can transfer this hormone through membrane-cultivated monolayers of kidney cells of Madin-Darby dogs in vitro [106]. High level of expression of receptor short form for leptin and prolactin in vivo in choroid plexus, a region of intensive liquor generation and putative site of hormone transfer through the blood-brain barrier [53, 107], and a decreased efficacy of leptin transfer in brain of rats lacking short form of leptin receptor [108] additionally support this viewpoint. Clearance natriuretic peptide receptor (NPR-C) is widely distributed in endothelial membranes, where it is involved into natriuretic peptide internalization; this mechanism may effectively reduce blood concentration of natriuretic peptides [109-111].
4.1. Mechanisms of action of soluble receptors. Many soluble forms of membrane receptors lacking transmembrane and cytoplasmic domains are known to date (Table 3) [2, 33, 35, 39-41, 55, 78, 112-121]. They may bind hormone ligand as effectively as full-length forms. Soluble receptors can be subdivided into two groups: receptors acting locally in tissues and acting systemically via the circulation. The first group includes soluble cytokine receptors, which are involved in hormone pre-uptake for subsequent binding to transmembrane receptor in the form of oligomeric complex with receptor for homologous and heterologous cytokine. Such mechanism is widely used for control of specificity of interaction of receptor complexes with hormone. Receptors of the second group can be referred to transport proteins (growth hormone binding protein [122], leptin binding protein [40], prolactin binding protein [123]). The possible role of these soluble receptors may consist of hormone storage and inhibition of ligand action in tissues [124]. However, there are some evidences that in vivo soluble receptors may potentiate hormone effect (e.g., growth hormone [125] and leptin [126]) due to increase in circulation time in blood. Besides the transport function, soluble receptors exhibit additional functions. For example, in in vitro experiments treatment of mouse osteoblasts with complex of interleukin-6 with its soluble receptor resulted in osteoclast formation, whereas treatment of these cells with interleukin-6 did not produce such effect. Stimulation of differentiation and protection of neurons against apoptosis are also attributed to a complex of soluble receptor with its ligand [127].
Thus, the existence of short forms of various receptors in combination with “usual” long forms allows modification of: 1) type of hormone signal; 2) rate of signal transduction; 3) efficacy of realization and proportion of various pathways involved in signal transduction. These parameters selectively “fit” to certain cell type, its physiological or pathological state, stage of differentiation, and/or stage of cell cycle. Short receptor forms may also serve as highly selective hormone transporters to various cell compartments and also to various organs and tissues that are protected by physiological barriers; they may be involved into systemic specific hormone transport and also in special signaling pathways realizing various hormone effects.
REFERENCES
1.Min, W., Lillehoj, H. S., and Fetterer, R. H.
(2002) Biochem. Biophys. Res. Commun., 299, 321-327.
2.Jang, J. H. (2002) Biochem. Biophys. Res.
Commun., 292, 378-382.
3.Lowe, D. G., Klisak, I., Sparkes, R. S., Mohandas,
T., and Goenddel, D. V. (1990) Genomics, 8, 304-312.
4.Dal Toso, R., Sommer, B., Ewert, M., Herb, A.,
Pritchett, D. B., Bach, A., Shivers, B. D., and Seeburg, P. H. (1989)
EMBO J., 8, 4025-4034.
5.Dingledine, R., Borges, K., Bowie, D., and
Traynelis, S. F. (1999) Pharmacol. Rev., 51, 7-61.
6.Burnashev, V., and Rozov, A. (2000) Cell. Mol.
Life Sci., 57, 1499-1507.
7.Cole, S. L., and Schindler, M. (2000) J.
Physiol. Paris, 94, 217-237.
8.Van Tol, H. H., Bunzow, J. R., Guan, H. C.,
Sunahara, R. K., Seeman, P., Niznik, H. B., and Civelli, O. (1991)
Nature, 350, 610-614.
9.Medvedev, A., Sandler, M., and Glover, V. (1999)
Eur. J. Pharmacol., 384, 239-241.
10.Bogorad, R. L., Sokolova, N. A., Smirnova, O. V.,
and Ashmarin, I. P. (2001) Neirokhimiya, 18, 182-186.
11.Ross, R. J., Esposito, N., Shen, X. Y., von Laue,
S., Chew, S. L., Dobson, P. R., Postel-Vinay, M. C., and Finidori, J.
(1997) Mol. Endocrinol., 11, 265-273.
12.Goffin, V., Bouchard, B., Ormandy, C. J.,
Weimann, E., Ferrag, F., Touraine, P., Bole-Feysot, C., Maaskant, R.
A., Clement-Lacroix, P., Edery, M., Binart, N., and Kelly, P. A. (1998)
Ann. N. Y. Acad. Sci., 840, 498-509.
13.Ghilardy, N., Ziegler, S., Wiestner, A., Stoffel,
R., Heim, M. R., and Skoda, R. C. (1996) Proc. Natl. Acad. Sci.
USA, 93, 6231-6235.
14.Kosaki, A., Nelson, J., and Webster, N. J. G.
(1998) J. Biol. Chem., 273, 10331-10337.
15.Abdennebi, L., Lesport, A. S., Remy, J. J.,
Grebert, D., Pisselet, C., Monniaux, D., and Salesse, R. (2002)
Reproduction, 123, 819-826.
16.Jiang, X., Russo, I. H., and Russo, J. (2002)
Int. J. Oncol., 20, 735-738.
17.Giros, B., Solkoloff, O., Martres, M. P., Riou,
J. F., Emorine, L. J., and Schwartz, J. C. (1989) Nature,
342, 923-926.
18.Spengler, D., Waeber, C., Pantaloni, C.,
Holsboer, F., Bockaert, J., Seeburg, P. H., and Journot, L. (1993)
Nature, 365, 170-175.
19.Pisegna, J. R., and Wank, S. A. (1993) Proc.
Natl. Acad. Sci. USA, 90, 6345-6349.
20.Bole-Feysot, C., Goffln, V., Edery, M., Binart,
N., and Kelly, P. A. (1998) Endocrine. Rev., 19,
225-268.
21.Hu, Z. Z., Zhuang, L., and Dafau, M. L. (1996)
J. Biol. Chem., 271, 10242-10246.
22.Hu, Z. Z., Zhuang, L., Meng, J., Leondires, M.,
and Dafau, M. L. (1999) J. Clin. Endocrin. Metab.,
84, 1153-1156.
23.Hu, Z. Z., Zhuang, L., and Dafau, M. L. (1998)
Trends Endocrinol. Metab., 9, 94-102.
24.Bignon, C., Binart, N., Ormandy, C., Schuler, L.
A., Kelly, P. A., and Djiane, J. (1997) J. Mol. Endocrinol.,
19, 109-120.
25.Davis, J. A., and Linzer, D. I. H. (1989) Mol.
Endocrinol., 3, 674-680.
26.Ali, S., Edery, M., Pellegrini, I., Paly, J.,
Djiane, J., and Kelly, P. A. (1992) Mol. Endocrinol., 6,
1242-1248.
27.Kline, J. B., Roehrs, H., and Clevenger, C. V.
(1999) J. Biol. Chem., 274, 35461-35468.
28.Kelly, P. A., Djiane, J., Postel-Vinay, M. C.,
and Edery, M. (1991) Endocr. Rev., 3, 235-251.
29.Lebrun, J. J., Ali, S., Ullrich, A., and Kelly,
P. A. (1995) J. Biol. Chem., 270, 10664-10670.
30.Perrot-Applanat, M., Gualillo, O., Pezet, A.,
Vincent, V., Edery, M., and Kelly, P. A. (1997) Mol.
Endocrinol., 11, 1020-1032.
31.Hu, Z. Z., Meng, J., and Dufau, M. L. (2001)
J. Biol. Chem., 276, 41086-41094.
32.Clevenger, C. V., and Kline, J. B. (2001)
Lupus, 10, 706-718.
33.Jabbour, H. N., Clarke, L. A., Bramley, T.,
Postel-Vinay, M. C., Kelly, P. A., and Edery, M. (1998) J. Mol.
Endocrinol., 21, 51-59.
34.Edwalds-Gilbert, G., Verladi, K. L., and
Milcarek, C. (1997) Nucleic Aids Res., 25, 2547-2561.
35.Callaghan, T., Antczak, M., Flickinger, T.,
Raines, M., Myers, M., and Kung, H. J. (1993) Oncogene,
8, 2939-2948.
36.Reiter, J. L., and Maihle, N. J. (1996)
Nucleic Acids Res., 24, 4050-4056.
37.Edens, A., and Talamantes, F. (1998)
Endocrine. Rev., 19, 559-582.
38.Foulkes, N. S., Schlotter, F., Pevet, P., and
Sassone-Corsi, P. (1993) Nature, 363, 264-267.
39.Martini, J. F., Pezet, A., Guezennec, C. Y.,
Edery, M., Postel-Vinay, M. C., and Kelly, P. A. (1997) J. Biol.
Chem., 272, 18951-18958.
40.Maamra, M., Bidlingmaier, M., Postel-Vinay, M.
C., Wu, Z., Strasburger, C. J., and Ross, R. J. (2001)
Endocrinology, 142, 4389-4393.
41.Kline, J. B., and Clevenger, C. V. (2001) J.
Biol. Chem., 276, 24760-24767.
42.Baumann, G. (2001) J. Pediatr. Endocr.
Metab., 14, 355-375.
43.Matsukawa, N., Grzesik, W. J., Takahashi, N.,
Pandey, K. N., Pang, S., Yamauchi, M., and Smithies, O. (1999) Proc.
Natl. Acad. Sci. USA, 96, 7403-7408.
44.Jumaa, H., and Nielsen, P. J. (1997) EMBO
J., 16, 5077-5085.
45.Jumaa, H., Guenet, J. L., and Nielsen, P. J.
(1997) Mol. Cell. Biol., 17, 3116-3124.
46.Matter, N., Marx, M., Weg-Remers, S., Ponta, H.,
Herrlich, P., and Konig, H. (2000) J. Biol. Chem., 275,
35353-35360.
47.Murray, M. V. (1999) in Post-transcriptional
Processing and the Endocrine System (Chew, S. L., ed.) Karger,
Basel, pp. 83-100.
48.Zhang, W., Liu, H., Han, K., and Grabowski, P. J.
(2002) RNA, 8, 671-685.
49.Akker, S. A., Smith, P. J., and Chew, S. L.
(2001) J. Mol. Endocrinol., 27, 123-131.
50.Hu, Z. Z., Zhuang, L., Meng, J., Tsai-Moris, C.
H., and Dafau, M. L. (2002) Endocrinology, 143,
2139-2142.
51.Shirota, M., Banville, D., Ali, S., Jolicoeur,
C., Boutin, J. M., Edery, M., Djiane, J., and Kelly, P. A. (1990)
Mol. Endocrinol., 4, 1136-1143.
52.Moldrup, A., Ormandy, C., Nagano, M., Murthy, K.,
Banville, D., Tronche, F., and Kelly, P. A. (1996) Mol.
Endocrinol., 10, 661-671.
53.Nagano, M., and Kelly, P. A. (1994) J. Biol.
Chem., 269, 13337-13345.
54.Guivarch, D., Vincent, J. D., and Vernier, P.
(1998) Endocrinology, 139, 4213-4221.
55.Zhao, X. M., Frist, W. H., Yeoh, T. K., and
Miller, G. G. (1994) J. Clin. Invest., 94, 992-1003.
56.Sell, S. M., Reese, D., and Ossowski, V. M.
(1994) J. Biol. Chem., 269, 30769-30772.
57.Tena-Sempere, M., Pinilla, L., Gonzalez, L. C.,
Casanueva, F. F., Dieguez, C., and Aguliar, E. (2000) J.
Endocrinol., 167, 479-486.
58.Tena-Sempere, M., Pinilla, L., Zhang, F.-P.,
Gonzalez, L. C., Huhtaniemi, I., Casanueva, F. F., Dieguez, C., and
Aguliar, E. (2001) Biol. Reprod., 64, 634-643.
59.Philips, I. D., Anthony, R. V., Buter, T. G.,
Ross, J. T., and McMillen, I. C. (1997) Endocrinology,
138, 1351-1354.
60.Bowen, J. M., Telleria, C. M., Towns, R., and
Keyes, P. L. (2000) Eur. J. Endocrinol., 143,
285-292.
61.Sakaguchi, K., Ohkubo, T., Sugiyama, T., Tanaka,
M., Ushiro, H., and Nakashima, K. (1994) J. Endocrinol.,
143, 383-392.
62.Babu, P. S., Krishnamurthy, H., Chedrese, P. J.,
and Sairam, M. R. (2000) J. Biol. Chem., 275,
27615-27626.
63.Diamond, R. H., Du, K., Lee, V. M., Mohn, K. L.,
Haber, B. A., Tewar, D. S., and Taub, R. (1993) J. Biol. Chem.,
268, 15185-15192.
64.Colwill, K., Feng, L. L., Yeakley, J. M.,
Songyang, Z., Cantley, L. C., and Fu, X. D. (1996) J. Biol.
Chem., 271, 24569-24575.
65.Burns, C. G., and Gould, K. L. (1999) in
Post-transcriptional Processing and the Endocrine System (Chew,
S. L., ed.) Karger, Basel, pp. 83-100.
66.Santra, B., and Carter, D. A. (1999) in
Post-transcriptional Processing and the Endocrine System (Chew,
S. L., ed.) Karger, Basel, pp. 122-140.
67.Mantyh, P. W., Rogers, S. D., Ghilardi, J. R.,
Maggio, J. E., Mantyh, C. R., and Vigna, S. R. (1996) Brain.
Res., 719, 8-13.
68.Alele, J., Jiang, J., Goldsmith, J. F., Yang, X.,
Maheshwari, H. G., Black, R. A., Baumann, G., and Frank, S. J. (1998)
Endocrinology, 139, 1927-1935.
69.Khan, Z., Mrzljak, L., Gutierrez, A., and de la
Calle, A. (1998) Proc. Natl. Acad. Sci. USA, 95,
7731-7736.
70.Seino, S., and Bell, G. I. (1989) Biochem.
Biophys. Res. Commun., 159, 312-316.
71.You, S., Kim, H., Hsu, C. C., El Halawani, M. E.,
and Foster, D. N. (2000) Biol. Reprod., 62, 108-116.
72.Bogorad, R. L., Smyslova, V. S., Rubtsov, P. M.,
and Smirnova, O. V. (2002) Mol. Biol. (Moscow), 36,
91-93.
73.Schuler, L. A., Nagel, R. J., Gao, J., Horseman,
N. D., and Kessler, M. A. (2001) Endocrinology, 138,
3187-3194.
74.Sheikh, F., Fandrich, R. R., Kardami, E., and
Cattini, P. A. (1999) Cardiovasc. Res., 42, 696-705.
75.Freemark, M., Nagano, M., Edery, M., and Kelly,
P. A. (1995) J. Endocrinol., 144, 285-292.
76.Frasca, F., Pandini, G., Scalia, P., Sciacca, L.,
Mineo, R., Costantino, A., Goldfine, I. D., Belfiore, A., and Vigneri,
R. (1999) Mol. Cell. Biol., 19, 3278-3288.
77.Vickers, S. M., Huang, Z. Q., McMillan-Crow, L.,
Greendorfer, J. S., and Thomson, J. A. (2002) J. Gastrointest.
Surg., 6, 546-553.
78.Fujita, M., Takahashi, R., Kitada, K., Watanabe,
R., Kitazawa, S., Ashoori, F., Liang, P., Saya, H., Serikawa, T., and
Maeda, S. (1997) Cancer Lett., 112, 47-55.
79.Sesti, G., Federici, M., Lauro, D., Sbraccia, P.,
and Lauro, R. (2001) Diabetes Metab. Res. Rev., 17,
363-373.
80.Vella, V., Pandini, G., Sciacca, L., Mineo, R.,
Vigneri, R., Pezzino, V., and Belfiore, A. (2002) J. Clin.
Endocrinol. Metab., 87, 245-254.
81.Harmar, A. J., Arimura, A., Gozes, I., Journot,
L., Laburthe, M., Pisegna, J. R., Rawlings, S. R., Robberecht, P.,
Said, S. I., Sreedharan, S. P., Wank, S. A., and Waschek, J. A. (1998)
Pharmacol. Rev., 50, 265-270.
82.Pantaloni, C., Brabet, P., Bilanges, B., Dumuis,
A., Houssami, S., Spengler, D., Bockaert, J., and Journot, L. (1996)
J. Biol. Chem., 271, 22146-22151.
83.Robertson, S. C., and Tynan, I. A. (2000)
Trends Genet., 16, 265-271.
84.Bresson-Bepoldin, L., Jacquot, M. C., Schlegel,
W., and Rawlings, S. R. (1998) J. Mol. Endocrinol., 21,
109-120.
85.Lu, N., Zhou, R., and DiCicco-Bloom, E. (1998)
J. Neurosci. Res., 53, 651-662.
86.Das, R., and Vonderhaar, B. K. (1995) Mol.
Endocrinol., 9, 1750-1759.
87.Duan, W. R., Farmer, T. G., Albarracin, C. T.,
Zhong, L., and Gibori, G. (1997) Endocrinology, 138,
3216-3221.
88.Nikelainen, P., Peltoketo, H., Vihko, R., and
Vihko, P. (1998) Mol. Endocrinol., 12, 1048-1059.
89.Ben-Jonathan, N., and Hnasko, R. (2001)
Endocr. Rev., 22, 724-763.
90.Vincent, V., Goffin, V., Rozakis-Adcock, M.,
Mornon, J. P., and Kelly, P. A. (1997) J. Biol. Chem.,
272, 7062-7068.
91.Barr, V. A., Lane, K., and Taylor, S. I. (1999)
J. Biol. Chem., 274, 21416-21424.
92.Uotani, S., Bjorbaek, C., Tornoe, J., and Flier,
J. S. (1999) Diabetes, 48, 279-286.
93.Choice, C. V., Howard, M. J., Poy, M. N., Hankin,
M. H., and Najjar, S. M. (1998) J. Biol. Chem., 273,
22194-22200.
94.Smirnova, O. V. (1999) Biol. Membr.
(Moscow), 16, 199-211.
95.Radulescu, R. T. (1995) J. Endocrinol.,
146, 365-368.
96.Orlova, A. N., Smirnova, O. V., Turovetsky, V.
B., and Smirnov, A. N. (1999) Byul. Eksp. Biol. Med.,
127, 579-582.
97.Lobie, P., Wood, T., Chen, C., Waters, M., and
Norstedt, G. (1994) J. Biol. Chem., 269, 31735-31746.
98.Marti, U., Burwen, S., Wells, A., Barker, M.,
Huling, S., Feren, A., and Jones, A. (1991) Hepatology,
13, 15-20.
99.Keresztes, M., and Boonstra, J. (1999) J.
Cell. Biol., 145, 421-424.
100.Buckley, A., Crowe, P. D., and Russell, D. H.
(1988) Proc. Natl. Acad. Sci. USA, 85, 8649-8653.
101.Buckley, A., Montgomery, D., Hendrix, M.,
Zukoski, C., and Putman, C. (1992) Arch. Biophys., 296,
198-206.
102.Radulescu, R. (1995) Med. Hypoth.,
45, 107-111.
103.Knopp, J., Zaliberova, Y., Jurcovicova, J.,
Torda, T., and Brtko, J. (1993) Endocr. Regul., 27,
26-28.
104.Ouhtit, A., Ronsin, B., Kelly, P., and Morel,
G. (1994) Biol. Cell, 82, 169-176.
105.Rao, Y., Buckley, D., and Buckley, A. (1995)
Arch. Biochem. Biophys., 322, 506-515.
106.Hileman, S. M., Tornoe, J., Flier, J. S., and
Bjorbaek, C. (2000) Endocrinology, 141, 1955-1961.
107.Bjorbaek, C., Elmquist, J. K., Michl, P.,
Ahima, R. S., van Bueren, A., MsCall, A. L., and Flier, J. S. (1998)
Endocrinology, 139, 3485-3491.
108.Kastin, A. J., Pan, W., Maness, L. M.,
Koletsky, R. J., and Ernsberger, P. (1999) Peptides, 20,
1449-1453.
109.Maack, T., Suzuki, M., Almeida, F. A.,
Nussenzveig, D., Scarborough, R. M., McEnroe, G. A., and Lewicki, J. A.
(1987) Science, 238, 675-678.
110.Fuller, F., Porter, J. G., Arfsten, A. E.,
Miller, J., Schilling, J. W., Scarborough, R. M., Lewicki, J. A., and
Schenk, D. B. (1988) J. Biol. Chem., 263, 9395-9401.
111.Almeida, F. M., Suzuki, M., Scarborough, R. M.,
Lewicki, J. A., and Maack, T. (1989) Am. J. Physiol.,
256, R469-475.
112.Setareh, M., Schwarz, H., and Lotz, M. (1995)
Gene, 164, 311-315.
113.Oldham, E. R., Bingham, B., and Baumbach, W. R.
(1993) Mol. Endocrinol., 7, 1379-1390.
114.Smith, W. C., Kuniyoshi, J., and Talamantes, F.
(1989) Mol. Endocrinol., 3, 984-990.
115.Jensen, L. E., Muzio, M., Mantovani, A., and
Whitehead, A. S. (2000) J. Immunol., 164, 5277-5286.
116.Kruse, S., Forster, J., Kuehr, J., and
Deichmann, K. A. (1999) Int. Immunol., 11, 1965-1970.
117.Montero-Julian, F. A. (2001) Cell. Mol.
Biol., 47, 583-597.
118.Haudenschid, D., Moseley, T., Rose, L., and
Reddi, A. H. (2002) J. Biol. Chem., 277, 4309-4316.
119.Takeshita, A. (1994) Nippon. Rinsho,
52, 974-978.
120.Wordinger, R. J., Clark, A. F., Agarwal, R.,
Lambert, W., and Wilson, S. E. (1999) Invest. Ophthalmol. Vis.
Sci., 40, 242-247.
121.Walker, W. H., Delfino, F. J., and Habener, J.
F. (1999) in Post-transcriptional Processing and the Endocrine
System (Chew, S. L., ed.) Karger, Basel, pp. 34-58.
122.Baumann, G., Stolar, M. W., Amburn, K.,
Barsano, C. P., and de Vries, B. C. (1986) J. Clin. Endocrinol.
Metab., 62, 134-141.
123.Amit, T., Dibner, C., and Barkey, R. J. (1997)
Mol. Cell. Endocrinol., 130, 167-180.
124.Lim, L., Spenser, S. A., McKay, P., and Waters,
M. J. (1990) Endocrinology, 127, 1287-1291.
125.Clark, R. G., Mortensen, D. L., Carlsson, L.
M., Spencer, S. A., McKay, P., Mulkerrin, M., Moore, J., and
Cunningham, B. C. (1996) Endocrinology, 137,
4308-4315.
126.Ge, H., Huang, L., Pourbahrami, T., and Li, C.
(2002) J. Biol. Chem., 277, 45898-45903.
127.Jones, S. A., Horiuchi, S., Topley, N.,
Yamamoto, N., and Fuller, G. M. (2001) FASEB J., 15,
43-58.