
|
Pro/antioxidant Status in Murine Skin Following Topical Exposure to Cumene Hydroperoxide Throughout the Ontogeny of Skin CancerA. A. Shvedova1,2*, E. R. Kisin1, A. Murray2, C. Kommineni1, V. Vallyathan1, and V. Castranova1,21Pathology and Physiology Research Branch, National Institute for Occupational Safety and Health, 1095 Willowdale Road, m/s 2015, Morgantown, West Virginia, USA; fax: (304) 285-5938; E-mail: ats1@cdc.gov2Physiology and Pharmacology Department, West Virginia University, Morgantown, WV, USA * To whom correspondence should be addressed.
|
Received April 30, 2003
Organic peroxides used in the chemical and pharmaceutical industries have a reputation for being potent skin tumor promoters and inducers of epidermal hyperplasia. Their ability to trigger free radical generation is critical for their carcinogenic properties. Short-term in vivo exposure of mouse skin to cumene hydroperoxide (Cum-OOH) causes severe oxidative stress and formation of spin-trapped radical adducts. The present study was designed to determine the effectiveness of Cum-OOH compared to 12-O-tetradecanoylphorbol-13-acetate (TPA) in the induction of tumor promotion in the mouse skin, to identify the involvement of cyclooxygenase-2 (COX-2) in oxidative metabolism of Cum-OOH in keratinocytes, and to evaluate morphological changes and outcomes of oxidative stress in skin of SENCAR mice throughout a two-stage carcinogenesis protocol. Dimethyl-benz[a]anthracene (DMBA)-initiated mice were treated with Cum-OOH (32.8 µmol) or TPA (8.5 nmol) twice weekly for 20 weeks to promote papilloma formation. Skin carcinoma formed only in DMBA/Cum-OOH-exposed mice. Higher levels of oxidative stress and inflammation (as indicated by the accumulation of peroxidative products, antioxidant depletion, and edema formation) were evident in the DMBA/Cum-OOH group compared to DMBA/TPA treated mice. Exposure of keratinocytes (HaCaT) to Cum-OOH for 18 h resulted in expression of COX-2 and increased levels of PGE2. Inhibitors of COX-2 efficiently suppressed oxidative stress and enzyme expression in the cells treated with Cum-OOH. These results suggest that COX-2-dependent oxidative metabolism is at least partially involved in Cum-OOH-induced inflammatory responses and thus tumor promotion.
KEY WORDS: cumene hydroperoxide, 12-O-tetradecanoylphorbol-13-acetate, oxidative stress, tumor promotion, skin
INTRODUCTION
Skin cancer is by far the most common type of cancer, with a huge impact on morbidity, health, and health care economics [1]. Organic peroxides have become a subject of occupational safety research interest due to possible genotoxic and carcinogenic risks in the workplace. Peroxide compounds yield free radicals and are used extensively in industry as initiators of polymerization. Cumene hydroperoxide (Cum-OOH) is a catalyst of rapid polymerization and used in the production of styrene and acrylic monomers, in curing agents for unsaturated polyester resins, and as an intermediate for cross-linking agents [2, 3]. Cum-OOH is also widely used as a bulk material for the production of acetone and phenol. Humans are exposed to Cum-OOH during manufacturing processes as well as in polluted urban air environments since Cum-OOH can be produced from the photochemical reaction of nitrogen oxide with unsaturated hydrocarbons and peroxides present in the exhaust fumes of gasoline, diesel, and aviation engines. Dermal exposure to Cum-OOH can cause a number of toxic outcomes in skin, e.g., allergic and irritant dermatitis, rash, defattening of dermis and hair loss, burns, and epidermal hyperplasia [4, 5].
There is substantial evidence indicating the involvement of reactive oxygen species (ROS) in the initiation, promotion, and progression stages of chemical carcinogenesis [6-9]. A number of hydroperoxides and dialkylperoxides used in industry are effective tumor promoters in mouse skin [9-11]. Organic peroxide-induced lipid peroxidation was implicated as one of the essential mechanisms of toxicity in keratinocytes [12-15]. Free radicals are considered key factors contributing to skin tumor promotion by organic peroxides [11, 16-18]. Exposure of mouse keratinocytes and skin flaps in vitro to Cum-OOH was shown to form metal-catalyzed alkoxyl, alkyl, and aryl radicals [16, 17]. Since organic peroxides have a reputation for being potent skin tumor promoters and inducers of epidermal hyperplasia [19], their ability to trigger free radicals may be critical for their carcinogenic properties. Short-term topical exposure to Cum-OOH was shown to initiate formation in vivo of lipid-derived free radicals causing severe oxidative stress in skin [9]. The present study was designed to validate the effectiveness of cancer promotion properties of Cum-OOH in comparison to 12-O-tetradecanoylphorbol-13-acetate (TPA), a well-established cancer promoter, and to assess morphological changes and oxidative stress outcomes in the skin of SENCAR mice throughout a two-stage carcinogenesis protocol.
MATERIALS AND METHODS
Chemicals. Fatty acid-free human serum albumin (hSA), luminol, sodium dodecyl sulfate (SDS), cumene hydroperoxide (Cum-OOH), 12-O-tetradecanoylphorbol-13-acetate (TPA), 7,12-dimethylbenz[a]anthracene (DMBA), acetone, phenylmethylsulfonyl fluoride (PMSF), dithiothreitol (DTT), guaiacol, cetylmethylammonium bromide, 3-amino-1,2,4-triazole (3-AT), 2-thiobarbituric acid, Dulbecco's modified Eagle's medium (DMEM), aspirin and glutathione were obtained from Sigma Chemical Co (USA). ThioGlo-1TM was obtained from Covalent Inc. (USA). 2,2´-Azobis(2-amidinopropane)-dihydrochloride (AAPH) was purchased from Wako Chemicals (USA). The human keratinocyte (HaCaT) cell line was a kind gift from Dr. Travers (Indiana University School of Medicine, Indianapolis, USA). KGM basal medium was purchased from Clonetics Corporation (USA). Fetal bovine serum (FBS) was obtained from HyClone (USA). N-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide (NS-398), COX-2 Polyclonal Primary Antibody, and Prostaglandin E2 ELISA kit were purchased from Cayman Chemical Company (USA). 10% Tris-HCl Ready Gels and Opt-4CN Substrate Kit were obtained from Bio-Rad (USA). Goat Anti-Rabbit Immunoglobulin was purchased from Dako (USA).
Animals. SENCAR mice (n = 72) were purchased from the National Cancer Institute (Frederick, MD, USA). Each mouse was housed in an individual ventilated cage under controlled environmental conditions in an “Association for Assessment and Accreditation of Laboratory Animal Care” (AAALAC) accredited facility. The animals were weighed biweekly and the food consumption was recorded monthly.
Tumor promotion experiment. Six-week-old female SENCAR mice were randomly divided into six groups consisting of 12 mice in each group. The dorsal skin in the inter-scapular area was shaved with a surgical clipper 2 days before tumor initiation, and animals showing no hair re-growth were used in the experiment. DMBA (51.2 µg dissolved in 100 µl of acetone) was used as a tumor initiator and applied to the skins of mice in groups 4, 5, and 6 (table). In groups 1, 2, and 3, 100 µl of acetone was applied to the mouse skins. Fourteen days following tumor initiation, the mice were promoted twice a week for the next 20 weeks with 8.5 nmol TPA (groups 2 and 5) or 32.8 µmol Cum-OOH (groups 3 and 6) dissolved in 100 µl of acetone. Negative control mice were treated with acetone alone (groups 1 and 4). The body weight of each animal and papillomas/carcinomas appearing on the shaved area of the skin were recorded at weekly intervals. Tumor promoting activity was evaluated by both the ratio of tumor-bearing mice and the number of tumors, >1 mm in diameter, per mouse. Animals were sacrificed by inhalation of an excess of carbon dioxide after the termination of the treatments. Skin flaps from the inter-scapular area of the back of mice (1.5 × 2.0 cm) were excised and samples were taken for histopathology and biochemical analyses. Skin for biochemical studies was immediately frozen at -80°C until processing was performed.
Histopathology examination. Excised skin was fixed in 10% neutral buffered formalin, and then embedded in paraffin. Skin samples were cut at 3 µm, mounted on silanized slides, dewaxed in xylene, dehydrated though an ethanol series, and stained with hematoxylin and eosin. Hematoxylin and eosin stained slides were examined by light microscopy. Photomicrographs were prepared using an Olympus 300 double-headed microscope (Japan).
Homogenate preparation. The skin homogenates were prepared from frozen tissues (5-7 mg) with 1 ml of ice-cold phosphate-buffered saline (PBS, pH 7.4) using a tissue tearer (model 985-370, Biospec Products, Inc., USA). Homogenates were stored at -80°C until processed further.
Inflammatory biomarker determinations. The effect of Cum-OOH and TPA applications on inflammation was determined by two biomarkers, edema formation and myeloperoxidase (MPO) activity. To assess the extent of Cum-OOH- and TPA-induced edema in mouse skin 24 h after the last treatment, the skin bi-fold thickness was measured with a dial caliper (The Dyer Company, USA). Edema formation was expressed as the net increase in skin bi-fold thickness between experimental and control groups. For MPO activity, a spectrophotometric assay was used [20]. Briefly, skin homogenates were mixed with PBS (100 mM, pH 7.0) containing cetylmethylammonium bromide (0.02%), guaiacol (13 mM), and 3-AT (3.75 mM). The reaction was started by the addition of H2O2 (0.6 mM). Oxidation of guaiacol was monitored by changes of absorbance at 470 nm (epsilon = 26.6 mM-1*cm-1) using a Shimadzu UV 160U spectrometer (Japan). Activity of MPO was calculated in nmoles of tetraguaiacol per min per mg of protein.
Determination of peroxidative products (TBARS). Peroxidative products were determined using the procedure described by Buege and Aust [21]. The formation of thiobarbituric acid-reactive substances (TBARS) was measured in skin homogenates. Tissue homogenates containing 0.5 mg of protein were mixed with 1 ml of 0.67% thiobarbituric acid and 30% trichloroacetic acid (1 : 1). The samples were heated for 20 min at 100°C and centrifuged for 15 min at 5000g. The absorbency of the supernatant was determined at 535 nm using a UV 160 U Shimadzu spectrophotometer (Japan). A molar extinction coefficient of epsilon = 1.56*105 M-1*cm-1 was used for calculations.
Chemiluminescent measurements of total antioxidant reserve. A water-soluble azo-initiator, AAPH, was used to produce peroxyl radicals [22]. Oxidation of luminol by AAPH-derived peroxyl radicals was assayed by the chemiluminescent response. A delay in the chemiluminescent response caused by the interaction of endogenous antioxidants with AAPH-derived peroxyl radicals was observed upon addition of homogenates. Based on the known rate of peroxyl radical generation by AAPH, the amount of peroxyl radicals scavenged by endogenous antioxidants was evaluated. The incubation medium contained 0.1 M phosphate buffer (pH 7.4) at 37°C, AAPH (50 mM), and luminol (0.4 mM). The reaction was started by the addition of AAPH. A luminescent analyzer 633 (Coral Biomedical, Inc., USA) was employed for these determinations.
Determination of glutathione (GSH) and total protein thiols in cells and tissue. The total protein sulfhydryl concentration in homogenates of skin or cells was determined using ThioGloTM-1, a maleimide reagent which produces a highly fluorescent product upon reaction with SH-groups [23]. A standard curve was established by addition of GSH (0.02-1.0 mM) to 0.1 M phosphate buffer (pH 7.4) containing 10 µM ThioGloTM-1. GSH content was estimated from the immediate fluorescent response registered upon addition of ThioGloTM-1 to a tissue or cell homogenate. Total protein sulfhydryls were determined from the augmentation of the fluorescence response after addition of SDS (4 mM) to the same homogenate. A Shimadzu RF-5000 U spectrofluorophotometer was employed in the assay (excitation 388 nm and emission 500 nm).
HaCaT cell culture. Human keratinocyte (HaCaT) cells were plated into 96-well plates or 75 cm2 flasks and grown in DMEM with 10% FBS at 37°C in a tissue culture incubator (5% CO2) until 80% confluent monolayers were obtained. HaCaT cells were then incubated with various concentrations of Cum-OOH (50, 100, or 200 µM) in the presence or in the absence of COX-2 inhibitors (2 µM NS-398 or 50 µM aspirin) in phenol red free KGM basal medium for 18 h at 37°C. Cells were harvested by trypsinization and pellets were used for the protein extraction. HaCaT homogenates were prepared by freezing the cells in the plates at -80°C and then thawing the cells.
ELISA assay. The concentration of PGE2 in cultured supernatants was measured using a Prostaglandin E2 ELISA Kit. Each sample was assayed at two dilutions and each dilution was assayed in duplicate. A 50-µl standard or sample was incubated with PGE2-acetylcholinesterase tracer and PGE2 monoclonal antibody for 18 h at 4°C in a plate covered with plastic film. Plates were developed with Ellman's reagent for 60-90 min at room temperature using an orbital shaker. The absorbency was determined at 405 nm using a UV Spectra Max 250 spectrophotometer (Molecular Devices, USA).
Western blot analysis of COX-2 in keratinocyte cells. Protein was extracted from the pellet using buffer (62.5 mM Tris, pH 6.8, 0.5 mM DTT, 2% SDS, 1 mM PMSF). Samples were separated using 10% Tris-HCl Ready Gels. After transfer of proteins, the membrane was blocked with 5% nonfat milk in 0.1% Tween-20 for 1 h. The blocked membrane was incubated with COX-2 Polyclonal Primary Antibody (1 : 500) and 5% nonfat milk solution in 0.1% Tween-20 at 4°C for 18 h and then washed three times with PBS. Primary antibody sites were detected using a secondary antibody, Goat Anti-Rabbit Immunoglobulin (1 : 500) with 5% nonfat milk solution in 0.1% Tween-20 for 1 h. Bands were visualized with an Opt-4CN Substrate Kit (USA). Band intensities were quantified using a Fluor-S MultiImager (Bio-Rad Laboratories, USA).
Protein assays. Measurements of protein in homogenates of tissues and cells were conducted using a Bio-Rad protein assay kit, catalog No. 500-0006 (USA). Protein concentrations for Western blot analysis were measured using a Microdetermination of Total Protein Kit (Sigma).
Statistics. Data were expressed as the mean ± standard error of the mean for each group. One-way ANOVA with Tukey test was employed to compare the responses between treatments. Statistical significance was set at p < 0.05.
RESULTS
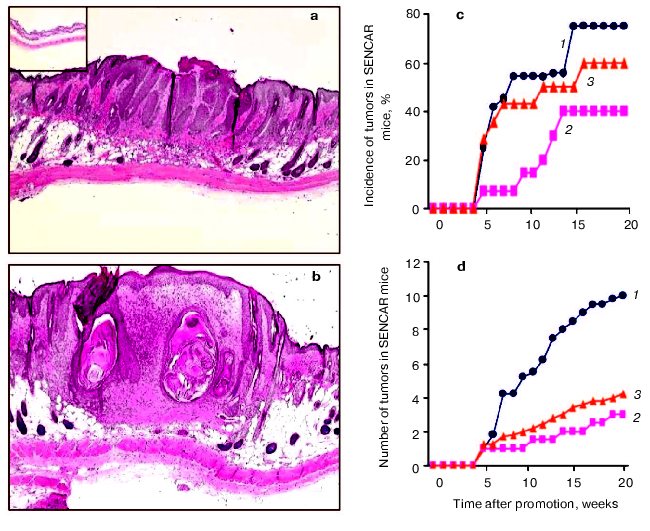
Effects of Cum-OOH or TPA on morphological alterations in the skin of SENCAR mice. Initially, we examined the histopathological changes in the skins of SENCAR mice 24 h after the last topical application of Cum-OOH and TPA. We observed epidermal hyperplasia in DMBA/Cum-OOH and DMBA/TPA groups (20 weeks of treatment) but not in control groups given DMBA/acetone (Figs. 1a and 1b, see color insert) or acetone/acetone (data not shown). In DMBA-initiated mice, topical treatment with Cum-OOH resulted in increased epidermal thickness compared with TPA-treated mice. Histological observation of mouse skin exposed to DMBA/Cum-OOH displayed muscle degeneration and a high degree of meiotic activity. DMBA/TPA-exposed mice showed less fibrosis in adipose tissues than the DMBA/Cum-OOH-treated group. Both DMBA/Cum-OOH and DMBA/TPA applications resulted in an increase in the number of blood vessels and inflammatory infiltrates compared to control (DMBA/acetone). However, accumulation of inflammatory cells in the skins of mice given DMBA/Cum-OOH was greater compared to that in the skins of mice treated with DMBA/TPA (Fig. 1b).
Tumor promotion in mouse skin exposed to Cum-OOH or TPA. Tumor promotion activity was evaluated by both the ratio of tumor-bearing mice (Fig. 1c) and the number of tumors per mouse (Fig. 1d). As shown in Figs. 1c and 1d, tumors appeared at 5 weeks after exposure to Cum-OOH or TPA following initiation. In DMBA-initiated mice, treatment with Cum-OOH or TPA for 20 weeks produced a 40 or 75% incidence of papillomas, respectively (Fig. 1c). No carcinomas were observed in the skins of mice treated with TPA following DMBA initiation. In contrast, in mice treated with Cum-OOH, we observed a 60% incidence of carcinomas following DMBA initiation. In addition, Cum-OOH treatment following initiation with DMBA resulted in 4.0 ± 0.2 carcinomas per mouse and 3.0 ± 0.2 papillomas per mouse (Fig. 1d). After TPA treatment following DMBA initiation, we observed 10.0 ± 1.5 papillomas per mouse.Fig. 1. Morphological alterations and tumor promotion in skin of SENCAR mice treated with DMBA/TPA (a) or DMBA/Cum-OOH (b) (20 weeks); insert, DMBA/acetone (magnification ×4). c) Incidence of tumors in SENCAR mice treated with DMBA/Cum-OOH or DMBA/TPA: 1, 2) percentage of animals having one or more papillomas induced by TPA (1) and Cum-OOH (2); 3) percentage of animals having one or more Cum-OOH-induced carcinomas. d) Number of tumors in SENCAR mice treated with DMBA/TPA and DMBA/Cum-OOH: 1, 2) number of papillomas per mouse induced by TPA (1) and Cum-OOH (2); 3) number of Cum-OOH-induced carcinomas per mouse. Values are means of 12 mice per group.
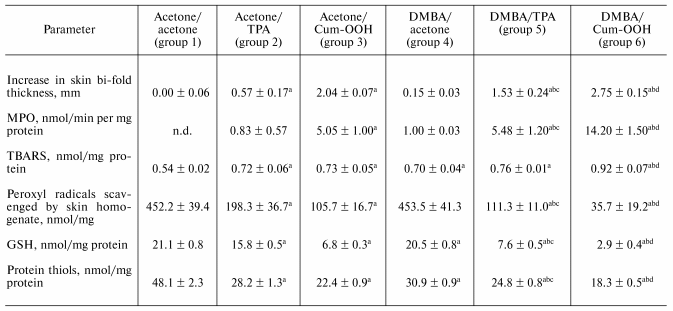
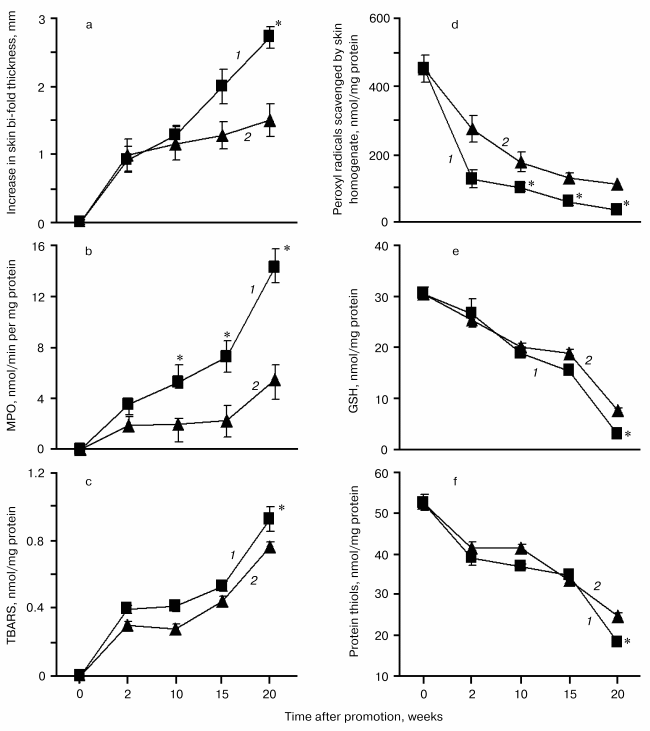
Inflammatory biomarkers in the skin of SENCAR mice exposed to Cum-OOH or TPA. Topical application of DMBA/Cum-OOH or DMBA/TPA to mouse skin resulted in an 18.3-fold or a 10.2-fold net increase, respectively, in the skin bi-fold thickness at the termination of the experiment (20 weeks) compared to control (DMBA/acetone) (table). After topical application with DMBA/Cum-OOH or DMBA/TPA, MPO activity increased 14.2- or 5.5-fold, respectively, above the levels seen in the control (table). This increase in skin bi-fold thickness and MPO activity was time-dependent after DMBA/Cum-OOH or DMBA/TPA exposure (Figs. 2a and 2b).
Biomarkers of inflammation and oxidative stress in the skin of SENCAR
mice exposed to Cum-OOH or TPA (20 weeks)
Note: ap < 0.05 vs acetone/acetone mouse group;
bp < 0.05 vs DMBA/acetone mouse group;
cp < 0.05 vs acetone/TPA mouse group;
dp < 0.05 vs acetone/Cum-OOH mouse group; n.d.,
not determined.
Biomarkers of oxidative stress in the skin of mice exposed to Cum-OOH or TPA. Topical application of DMBA/Cum-OOH or DMBA/TPA induced oxidative stress in mouse skin, as indicated by the accumulation of peroxidative products, depletion of total antioxidant reserve, and a decrease in the levels of glutathione (GSH), and protein SH-groups (table).
After 20 weeks of treatment with DMBA/Cum-OOH or DMBA/TPA, levels of lipid peroxidation products (TBARS) in the skin of SENCAR mice were increased by 31.4 or 8.6%, respectively, above the levels seen in the control (DMBA/acetone) (table). Chemiluminescent assay revealed a 92.1 or 75.5% decrease in the total antioxidant reserve in skin of mice treated with DMBA/Cum-OOH or DMBA/TPA compared to control (table). Levels of GSH were decreased by 85.9 or 62.9% after DMBA/Cum-OOH or DMBA/TPA topical application compared to DMBA/acetone (control). Along with GSH depletion, the levels of protein thiols decreased by 40.8 or 19.7% after DMBA/Cum-OOH or DMBA/TPA topical application compared to DMBA/acetone treated mice (table). The increase in lipid peroxidation products (TBARS) and decrease in total antioxidant reserve, GSH, and protein SH-groups was time-dependent after DMBA/Cum-OOH or DMBA/TPA exposure (Figs. 2c, 2d, 2e, 2f).
Levels of GSH and protein thiols in HaCaT cells exposed to Cum-OOH. Effect of COX-2 inhibitors. As shown in Fig. 3a, the exposure of HaCaT cells to 100 and 200 µM Cum-OOH produced a significant dose-dependent decrease in GSH levels by 88.9 and 95.7%, respectively. Similarly, the cellular content of protein SH-groups decreased by 55.1 and 62.3% after treatment with Cum-OOH (100 and 200 µM), respectively (Fig. 3a, inset).Fig. 2. Time-course of inflammation and oxidative stress parameters in skin of SENCAR mice during tumor promotion (DMBA/Cum-OOH (1) or DMBA/TPA (2)). Values are means ± SEM of 12 mice/experiment. * p < 0.05 versus DMBA/TPA treated mice.
To assess the involvement of cyclooxygenase-2 (COX-2) in oxidation of Cum-OOH, cells were co-exposed to inhibitors of COX-2. In the presence of NS-398 or aspirin, the decrease of the level of GSH was 20 or 37% less than that in the group treated with 100 µM Cum-OOH alone. When cells were exposed to 200 µM of Cum-OOH and NS-398 or aspirin, the decrease in GSH level was 3.9 or 4.0% less than that after incubation with Cum-OOH (Fig. 3a). Similarly, cell pretreatment with Cum-OOH and NS-398 or aspirin decreased protein thiol levels by 18 or 21% less than after incubation with 100 µM of Cum-OOH alone. When cells were exposed to 200 µM of Cum-OOH in the presence of NS-398 or aspirin, the decrease in protein thiol levels were 11 or 10% less than after incubation with Cum-OOH (Fig. 3a, inset).Fig. 3. Effect of COX-2 inhibitors (NS-398 or aspirin) on the levels of GSH, protein thiols, and TBARs in HaCaT cells exposed to Cum-OOH for 18 h: a) GSH (inset, protein thiols); b) TBARs in the absence (1) and presence of aspirin (2) and NS-398 (3). Values are means ± SEM of three experiments. * p < 0.05 versus cells treated with Cum-OOH alone.
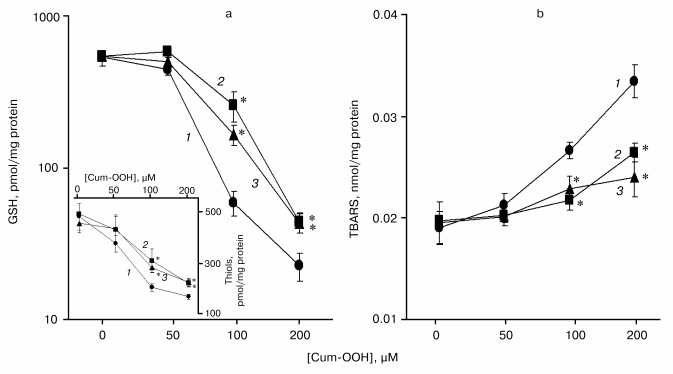
Level of lipid peroxidation products in HaCaT cells exposed to Cum-OOH. Effect of COX-2 inhibitors. Exposure of HaCaT cells to Cum-OOH (100 and 200 µM) increased the level of TBARs by 40 and 76%, respectively (Fig. 3b).
Augmentation of peroxidative products was significantly reduced in the presence of COX-2 inhibitors. As shown in Fig. 3b, in the presence of NS-398 or aspirin, the increase in the level of TBARs was 20 or 25% less than that in the group treated with 100 µM Cum-OOH alone. When cells, exposed to 200 µM of Cum-OOH, were treated with NS-398 or aspirin, the increase in the level of TBARs was 50 or 37% less than that after incubation with Cum-OOH.
Levels of PGE2 in HaCaT cells exposed to Cum-OOH. Effect of COX-2 inhibitors. After treatment with Cum-OOH (50, 100, and 200 µM), the levels of PGE2 in HaCaT cells increased by 2.9-, 6.5-, and 9.7-fold compared to control (Fig. 4a).
Inhibitors of COX-2 decreased the level of PGE2 in cells treated with Cum-OOH. In the presence of NS-398 or aspirin, the increase in the level of PGE2 was 1.7- or 1.5-times less than that in the group treated with 50 µM Cum-OOH. When cells, exposed to 100 µM Cum-OOH, were treated with NS-398 or aspirin, the increase in the level of PGE2 was 2.6- or 1.9-times less than after incubation with Cum-OOH alone. The exposure of cells to 200 µM Cum-OOH in the presence of NS-398 or aspirin was associated with increases in the levels of PGE2 which were 2.7- or 2.1-times less than that in group treated with Cum-OOH (Fig. 4a).Fig. 4. Levels of prostaglandin E2 (a) and expression of COX-2 (b) in HaCaT cells exposed to Cum-OOH for 18 h. a) Effects of COX-2 inhibitors (NS-398 (2) or aspirin (3); 1) control). Values are means ± SEM of three experiments. * p < 0.05 versus cells treated with Cum-OOH only. b) COX-2 (1), NS-398/Cum-OOH (100 µM) (2), Cum-OOH (100 µM) (3), NS-398/Cum-OOH (200 µM) (4), Cum-OOH (200 µM) (5), control (6).
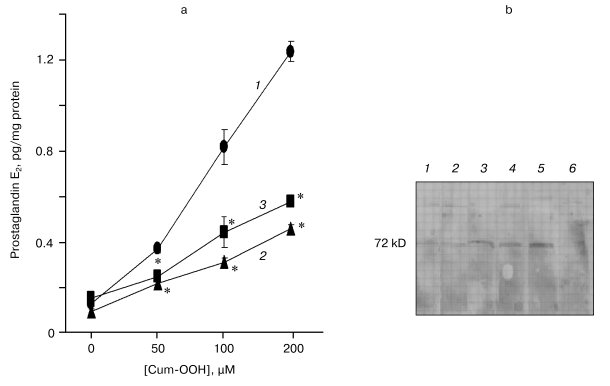
Expression of COX-2 in HaCaT cells exposed to Cum-OOH. Effect of COX-2 inhibitors. As shown in Fig. 4b, treatment of HaCaT cells with 100 and 200 µM Cum-OOH increased expression of constitutive COX-2 in a dose-dependant fashion. Expression of COX-2 in Cum-OOH treated cells was reduced by approximately 50% after co-incubation with NS-398 (Fig. 4b).
DISCUSSION
Chemically-induced skin cancer in mice has three chronological stages: initiation, promotion, and progression [24, 25]. Tumor initiation is a rapid and irreversible process, whereas promotion is a long-term process requiring chronic exposure to a tumor promoter. The application of tumor promoters to skin causes a significant reduction in antioxidant defense [26-29]. Oxidative DNA damage has gained increasing attention as a mechanism triggering genetic instability and mutation [30]. This mutagenic sequence is thought to occur via de-regulation of metabolic events giving rise to the formation of reactive oxygen species, e.g., hydroxyl and superoxide anion radicals, singlet oxygen, hydrogen peroxide, organic free radicals, and peroxides.
Since organic peroxides have a reputation for being potent skin tumor promoters and inducers of epidermal hyperplasia [19], their ability to trigger free radicals may be critical for their carcinogenic properties. Organic peroxide-induced lipid peroxidation has been implicated as one of the essential mechanisms of toxicity in keratinocytes [12-14]. Free radicals are considered key factors contributing to skin tumor promotion by organic peroxides [11, 16-18]. Exposure of mouse keratinocytes and skin flaps in vitro to Cum-OOH has been shown to form metal-catalyzed alkoxyl, alkyl, and aryl radicals [16, 17]. We observed that topical exposure of Cum-OOH induced lipid-derived peroxidation associated with free radical formation detected by ESR in skin of vitamin E-deficient mice but not in vitamin E-sufficient Cum-OOH treated controls [9]. Analysis of the ESR spectrum of lipid extracts obtained from mouse skin revealed formation of two spin-trapped radical species assigned to methyl and methoxyl radicals formed from Cum-OOH metabolic oxidation [9].
In the present study, long-term topical exposure to Cum-OOH induced time-dependent oxidative stress in the skin of SENCAR mice. We observed that biomarkers of oxidative stress and inflammation in the skin were higher in the group treated with Cum-OOH compared to the TPA-exposed group (table and Fig. 2). Antioxidant reserves and TBARS were shown to be the most sensitive biomarkers with oxidative changes starting as early as two weeks and progressing until 20 weeks of exposure to DMBA/Cum-OOH or DMBA/TPA (Figs. 2d and 2c). Changes in GSH and protein thiols were only significant after 15-20 weeks of exposure to DMBA/Cum-OOH or DMBA/TPA (Figs. 2e and 2f). Bhasin and coauthors [31] reported a 30% increase of MDA/g tissue and no change in antioxidant levels after a single exposure to TPA or Cum-OOH using a two-stage cancer promotion protocol. Significant changes were found only in catalase activity in the skin of Swiss albino mice after 24 h of exposure to Cum-OOH [31]. To the best of our knowledge, our data show for the first time the time course of accelerated oxidative stress in skin during 20 weeks of exposure to DMBA/TPA or DMDA/Cum-OOH.
Inflammatory leukocyte-induced oxidative stress is associated with the biological processes of certain cancers [32-34]. We observed that the inflammatory index determined by skin thickness and MPO activity were 1.8- and 2.6-fold higher in the skin of mice treated with DMBA/Cum-OOH compared to DMBA/TPA-treated skin. Despite a slightly higher incidence of papillomas and number of papillomas/mouse found after exposure to DMBA/TPA, we did not observed any carcinoma formation within this group. To the contrary, exposure of mice to DMBA/Cum-OOH resulted in the formation of carcinomas (4 per mouse, Fig. 1d). Morphological changes in the skin of mice treated with DMBA/Cum-OOH were distinct from the DMBA/TPA-exposed group. In particular, in DMBA-initiated mice, topical treatment with Cum-OOH resulted in increased epidermal thickness, muscle degeneration and a high degree of meiotic activity, higher fibrosis in adipose tissues, and greater accumulation of inflammatory cells compared with TPA-treated mice (Fig. 1b).
COXs and lipoxygenases (LOXs) are two important enzyme classes that metabolize polyunsaturated fatty acids and affect carcinogenesis [35]. The LOXs convert arachidonic, linoleic, and other polyunsaturated fatty acids into biologically active metabolites that influence cell signaling, structure, and metabolism [36]. In the classic pathway involving only arachidonic acid (the eicosanoid-generation pathway), arachidonic acid is a substrate for both LOX and COX enzymes to form various metabolites, such as HETEs (5-, 8-, 12-, and 15-S-HETE) and prostaglandins [2]. There is an increasing body of evidence indicating that COX-2 plays a critical role in multistage carcinogenesis and tumor promotion [34]. Organic peroxide and Cum-OOH, in particular, are substrates for intracellular peroxidases--COX-2 and lipoxygenase. COX-2 is barely detectable in basal cells of the interfollicular epidermis and of hair follicles. Inflammatory stimuli cause an increase in the number of basal cells expressing COX-2 [37]. Fischer [38] showed induction of COX-2 by benzoyl peroxide in keratinocytes. We found that overnight treatment of HaCaT cells with 200 µM Cum-OOH resulted in a 2-fold increase in the expression of constitutive COX-2 (Fig. 4b) and 9.7-fold increase in the level of PGE2 in cell supernatants (Fig. 4a). COX-2 specific inhibitors have been recently found to effectively suppress both TPA-induced prostaglandin synthesis and tumor promotion [39]. We observed that expression of COX-2 in Cum-OOH-treated cells was reduced when the cells were co-incubated with inhibitors of COX-2, e.g., aspirin and NS-398, showing a probable involvement of COX-2 in the oxidative metabolism of Cum-OOH. Similarly, accumulation of peroxidative products and PGE2 were significantly reduced in the presence of aspirin or NS-398 (Figs. 3b and 4a). Our present results suggest that COX-2 is involved in Cum-OOH-induced carcinogenesis. However, we cannot exclude involvement of LOX in oxidative metabolism of Cum-OOH and cancer promotion.
In conclusion, topical exposure to Cum-OOH induced time-dependent oxidative stress as well as papilloma and carcinoma formation in the skin of SENCAR mice during 20 weeks of a two-stage cancer promotion study. The suppressive effects of aspirin and NS-398 (COX-2 inhibitors) observed in dermal cell cultures exposed to Cum-OOH suggest involvement of COX-2 in oxidative metabolism and possibly cancer promotion.
REFERENCES
1.Stratton, S. P., Dorr, R. T., and Alberts, D. S.
(2000) Eur. J. Cancer, 36, 1292-1297.
2.Lewis, R. J. (1993) Hawley's Condensed Chemical
Dictionary, 12th Ed., Van Nostrand Reinhold, New York, p. 133.
3.Ward, D. J. (1993) in Encyclopedia of Chemical
Technology (Kirk-Othmer, J., ed.) Vol. 7, Academic Press, N.
Y., pp. 730-736.
4.Hathaway, G. J., Proctor, N. H., and Hughes, J. P.
(1996) in Proctor and Hughes' Chemical Hazards of the
Workplace, 4th Ed., Van Nostrand Reinhold, New York, pp.
169-170.
5.Adams, R. M. (1999) in Occupational Skin
Disease (Adams, R. M., ed.) W. B. Saunders, Philadelphia, PA, pp.
491-500.
6.Kensler, T. W., and Taffe, B. G. (1986) Adv.
Free Rad. Biol. Med., 2, 347-387.
7.Nishi, J., Ogura, R., Sugiyama, M., Hidaka, T., and
Kohno, M. (1991) J. Invest. Dermatol., 97, 115-119.
8.Yagi, K. (1988) in The Biological Role of
Reactive Oxygen Species in Skin, University of Tokyo Press, Tokyo,
pp. 109-116.
9.Shvedova, A. A., Kisin, E. R., Murray, A. R.,
Kommineni, C., Castranova, V., Mason, R. P., Kadiiska, M. B., and
Gunther, M. R. (2002) Chem. Res. Toxicol., 15,
1451-1459.
10.Slaga, T. J., Klein-Szanto, A. J., Triplett, L.
L., Yotti, L. P., and Trosko, K. E. (1981) Science, 213,
1023-1025.
11.Gimenez-Conti, I. B., Binder, R. L., Johnston,
D., and Slaga, T. J. (1998) Toxicol. Appl. Pharmacol.,
149, 73-79.
12.Vessey, D. A., Lee, K. H., and Blacker, K. L.
(1992) J. Invest. Dermatol., 99, 859-863.
13.Vessey, D. A., Lee, K. H., and Boyer, T. D.
(1995) J. Invest. Dermatol., 104, 355-358.
14.Babich, H., Zuckerbraun, H. L., Wurzburger, B.
J., Rubin, Y. L., Borenfreund, E., and Blau, L. (1996)
Toxicology, 106, 187-196.
15.Shvedova, A. A., Tyurina, Y. Y., Kawai, K.,
Tyurin, V. A., Kommineni, C., Castranova, V., Fabisiak, J. P., and
Kagan, V. E. (2002) J. Invest. Dermatol., 118,
1008-1018.
16.Taffe, B. G., Zweier, J. L., Pannell, L. K., and
Kensler, T. W. (1989) Carcinogenesis, 10, 1261-1268.
17.Timmins, G. S., and Davis, M. J. (1993) J.
Photochem. Photobiol. B, 21, 167-173.
18.Kensler, T., Guyton, K., Egner, P., McCarthy, T.,
Lesko, S., and Akman, S. (1995) Progr. Clin. Biol. Res.,
391, 103-116.
19.Klein-Szanto, A. J., and Slaga, T. J. (1982)
J. Invest. Dermatol., 79, 30-34.
20.Nonaka, T., Mio, M., Doi, M., and Tasaka, K.
(1992) Biochem. Pharmacol., 44, 1115-1121.
21.Buege, J. A., and Aust, S. D. (1978) Meth.
Enzymol., 52, 302-310.
22.Niki, E. (1990) Meth. Enzymol.,
186, 100-108.
23.Shvedova, A. A., Kommineni, C., Jeffries, B. A.,
Castranova, V., Tyurina, Y. Y., Tyurin, V. A., Serbinova, E. A.,
Fabisiak, J. P., and Kagan, V. E. (2000) J. Invest. Dermatol.,
114, 354-364.
24.Bowden, G. T., Finch, J., Domann, F., and Krieg,
P. (1995) in Skin Cancer: Mechanisms and Human Relevance
(Mukhtar, H., ed.) CRC Press, Inc., Boca Raton, FL, pp. 99-111.
25.Hennings, H., Glick, A. B., Lowry, D. T.,
Krsmanovic, L. S., Sly, L. M., and Yuspa, S. H. (1993)
Carcinogenesis, 14, 2353-2358.
26.Perchellet, J. P., Abney, N. L., Thomas, R. M.,
Perchellet, E. M., and Maatta, E. A. (1987) Cancer Res.,
47, 6302-6309.
27.Solanki, V., Rana, R. S., and Slaga, T. J. (1981)
Carcinogenesis, 2, 1141-1146.
28.Amstad, P. A., Liu, H., Ichimiya, M., Berezesky,
I. K., and Trump, B. F. (1997) Carcinogenesis, 18,
479-484.
29.Isbir, T., Yaylim, I., Aydin, M., Ozturk, O.,
Koyuncu, H., Zeybek, U., Agachan, B., and Yilmaz, H. (2000)
Anticancer Res., 20, 219-224.
30.Ames, B. N., Gold, L. S., and Willett, W. C.
(1995) Proc. Natl. Acad. Sci. USA, 92, 5258-5265.
31.Bhasin, G., Kausar, H., Sarwar, A. M., and Athar,
M. (2003) Arch. Biochem. Biophys., 409, 262-273.
32.Ohshima, H., and Bartsch, H. (1994) Mutat.
Res., 305, 253-264.
33.Berhane, T., Halliday, G. M., Cooke, B., and
Barnetson, R. S. (2002) Br. J. Dermatol., 146,
810-815.
34.Marks, F., and Furstenberger, G. (2000) Eur.
J. Cancer, 36, 314-329.
35.Shureiqi, I., and Lippman, S. M. (2001) Cancer
Res., 61, 6307-6312.
36.Brash, A. R. (1999) J. Biol. Chem.,
274, 23679-23682.
37.Muller-Decker, K., Scholz, K., Neufang, G.,
Marks, F., and Furstenberger, G. (1998) Exp. Cell. Res.,
242, 84-91.
38.Fischer, S. M. (2002) J. Environ. Pathol.
Toxicol. Oncol., 21, 183-191.
39.Muller-Decker, K., Kopp-Schneider, A., Marks, F.,
Seibert, K., and Furstenberger, G. (1998) Mol. Carcinog.,
23, 36-44.