REVIEW: Conversion of Death Signal into Survival Signal by Redox Signaling
D. K. Das* and N. Maulik
Cardiovascular Research Center, University of Connecticut School of Medicine, Farmington, CT 06030-1110, USA; fax: (860) 679-4606; E-mail: ddas@neuron.uchc.edu* To whom correspondence should be addressed.
Received April 30, 2003
Reperfusion of ischemic myocardium produces reactive oxygen species (ROS) and results in apoptotic cell death and DNA fragmentation. Several redox-sensitive anti- and pro- apoptotic transcription factors including nuclear factor kappaB (NF-kappaB) and heterodimeric transcription factor AP-1 progressively and steadily increase in the heart as a function of the duration of ischemia and reperfusion. When the heart is adapted to ischemic stress by repeated short-term ischemia and reperfusion, NF-kappaB remains high, while AP-1 is lowered to almost baseline value. The anti-apoptotic gene Bcl-2 is downregulated in the ischemic/reperfused heart, while it is upregulated in the adapted myocardium. Cardioprotective abilities of the adapted myocardium are abolished when heart is pre-perfused with N-acetyl cysteine to scavenge ROS, suggesting a role of redox signaling. Mammalian heart is protected by several defense systems, which include, among others, the redox-regulated protein thioredoxin. Reperfusion of ischemic myocardium results in the downregulation of thioredoxin 1 (Trx 1) expression, which was upregulated in the adapted myocardium. The increased expression of Trx 1 is completely blocked with an inhibitor of Trx 1, cis-diammine-dichloroplatinum, which also abolished cardioprotection afforded by ischemic adaptation. The cardioprotective role of Trx 1 is further confirmed with transgenic mouse hearts overexpressing Trx 1. The Trx 1 mouse hearts displayed significantly improved post-ischemic ventricular recovery and reduced myocardial infarct size and apoptosis compared to the corresponding wild-type mouse hearts. The results of this study implicate a crucial role of redox signaling in transmitting anti-death signal.
KEY WORDS: apoptosis, ischemia/reperfusion, adaptation, survival signal, redox signaling
Abbreviations: ROS) reactive oxygen species; Trx) thioredoxin; NF-kappaB) nuclear factor kappaB; CDDP) cis-diammine-dichloroplatinum; SOD) superoxide dismutase; GSHPx) glutathione peroxidase; TNF-alpha) tumor necrosis factor alpha; IL-1) interleukin 1; PLD) phospholipase D; PKC) protein kinase C; MAP kinase) mitogen-activated protein kinase; MAPKAP kinase 2) MAP kinase activated protein kinase 2; HSP) heat shock protein; JUN) Junas kinase; JNK) c-Jun NH2-terminal kinase; SAPK) stress-activated protein kinase; DMTU) dimethyl thiourea; iNOS) inducible nitric oxide synthase.
Pathophysiology of ischemic heart disease is multifactorial including
abnormal lipid metabolism and calcium homeostasis and production of
reactive oxygen species (ROS). These factors give rise to the
“death signal” resulting in apoptotic cell death leading to
an infracted heart. Mammalian heart is protected against ischemic
injury by several lines of defense. The first line of defense consists
of intracellular antioxidants such as superoxide dismutase (SOD),
catalase, glutathione peroxidase, and glutathione reductase. Oxidative
stress-inducible proteins produced by the heart in an attempt to
counteract the invading ROS can also function as a defense system.
Several redox-sensitive transcription factors including nuclear factor
kappaB (NF-kappaB) and AP-1 progressively and steadily
increase in the heart as a function of the duration of ischemia and
reperfusion. When the heart is adapted to ischemic stress by repeated
short-term ischemia and reperfusion, NF-kappaB remains high
while AP-1 binding is lowered [1]. The
anti-apoptotic gene Bcl-2 is downregulated in the
ischemic/reperfused heart, but upregulated in the adapted myocardium
[2]. Significant induction of p53 gene
occurs after ischemia/reperfusion [1]. Thus, during
ischemia/reperfusion, death signal becomes predominant resulting in
apoptotic cell death. Apoptotic cells are barely detected in the
adapted myocardium suggesting conversion of death signal into survival
signal. Cardioprotective abilities of the adapted myocardium are
completely abolished when heart is pre-perfused with N-acetyl cysteine
to scavenge ROS suggesting that redox signaling plays a crucial role in
generating survival signal during myocardial adaptation to ischemia [3].
ROS, REDOX SIGNALING, AND APOPTOSIS
Evidence is rapidly accumulating to support the role of ROS as intracellular signaling molecules. The long-held view of ROS being detrimental to the biological tissues was challenged after the recent discovery that the ROS can function as signaling molecules [4, 5]. The mitogenic signals mediated through the generation of ROS activate transcription factors including NF-kappaB, antioxidant enzymes, and Bcl-2 [6].
A large number of degenerative diseases, including coronary heart disease, have been linked with the overproduction of ROS. Numerous studies have demonstrated that excessive production of ROS in concert with drastic reduction in antioxidant reserve play a role in the pathophysiology of ischemic heart disease [7]. Myocardial ischemia/reperfusion causes cardiomyocytes to face conditions that shift their redox status to undergo a drastic change subjecting them to oxidative stress [8]. Interventions with ROS scavengers or antioxidants have been found to be cardioprotective against reperfusion injury [9].
Coronary heart diseases cause cardiomyocyte death not only by necrosis but also by apoptosis [10]. Reperfusion results in apoptosis and DNA fragmentation [3]. In concert, ischemia/reperfusion is associated with the induction of a number of both pro- and anti-apoptotic genes and transcription factors [11]. Ebselen, a glutathione peroxidase mimic, reduced cardiomyocyte apoptosis [12]. The hearts from the transgenic mice overexpressing GSHPx-1 gene were resistant to ischemia/reperfusion injury while those from the GSHPx-1 knockout animals, devoid of any copy of GSHPx-1, were extremely vulnerable to the cellular injury compared to wild-type controls [13]. Thus, it appears that cardiomyocyte death induced by ischemia/reperfusion is redox-regulated and that ROS not only trigger the cell death [14-17] but also function as intracellular signaling molecules.
INTRACELLULAR PATHWAYS FOR REDOX SIGNALING: RELATIONSHIP TO
SURVIVAL SIGNAL
Recent studies imply that ROS are not only destructive elements for cells, but also they are essential for the biological and physiological function of cells. Biological cells including cardiomyocytes contain enzymes that can simultaneously generate ROS and intracellular redox buffer in response to a specific stress. Depending on the amounts of antioxidant reserve and oxygen free radicals, the ROS are either destroyed or persist. Thus, ROS fulfill the definition of a second messenger, which are either upregulated or downregulated after physiologic stimuli like ischemia. A number of growth factors and cytokines induce oxidative stress and overproduce specific antioxidant enzymes [18, 19]. Environmental stresses including heat stress; oxidative stress and ischemia/reperfusion also produce oxidative stress, which is then translated into the induction of antioxidant enzymes [1].
Perhaps, the finding of the production of ROS during the agonist-induced activation of NF-kappaB provided the first concrete evidence for the role of ROS as second messenger. NF-kappaB regulates the inducible expression of a number of genes involved in cell survival and execution. For example, NF-kappaB has been found to control anti-apoptotic gene, Bcl-2, and pro-apoptotic factors, bax and p53, in the ischemic/reperfused myocardium [20]. Diverse extracellular signals from interleukin 1 (IL-1), tumor necrosis factor alpha (TNF-alpha), H2O2, etc. converge into development of oxidative stress, which leads to activation of NF-kappaB [21]. Such activation of NF-kappaB can be blocked by antioxidants such as vitamin E [22] or alpha-lipoic acid [23]. Antioxidants such as N-acetyl cysteine can prevent NF-kappaB activation by suppressing the generation of ROS [24].
Receptor tyrosine kinases play an important role in redox signaling by autophosphorylation of tyrosines along their own intracellular tails. Myocardial adaptation to ischemia occurs through the activation of several tyrosine kinases [25]. Phosphorylation of tyrosine kinases have been shown to be linked with the activation of both phospholipases C (PLC) and D (PLD) leading to the activation of multiple kinases including phosphokinase C (PKC) and mitogen-activated protein kinase (MAP kinase). PLD is critical in ischemic adaptation [26, 27]. There appears to be a link between adaptation and activation of PLD, eventually resulting in activation of PKC isoenzymes [28]. Pretreatment with tyrosine kinase inhibitors partially attenuates ischemic adaptation [29].
The MAP kinases play an essential role in mediating intracellular signal transduction. In response to extracellular stimulation, MAP kinases are rapidly activated and regulate cellular functions by inducing the phosphorylation of proteins, such as an oncogene product c-jun and S6 ribosomal protein kinase, and by activating MAPKAP kinase 2 (MAP kinase-activated protein kinase 2) [30, 31]. MAPKAP kinase 2 has been implicated in the stress activated signal transduction pathway [32]. A mitogen-activated protein kinase cascade has been identified in the heart [33]. The participation of MAP kinases in the adapted hearts involves a tyrosine kinase-phospholipase D-MAP kinase-MAPKAP kinase 2 signaling pathway [34].
The intracellular signaling mechanisms that lead to adaptation require one or more members of MAP kinase cascades. Among the three distinct MAP kinase families, stress-activated protein kinase (SAPK), also known as c-Jun NH2-terminal kinases (JNK), and p38 MAP kinase are known to be regulated by extracellular stresses [35]. JNKs and p38 MAP kinase appear to be involved in distinct cellular functions, because they possess different cellular targets and are located on different signaling pathways. Adaptation triggered a tyrosine kinase-regulated signaling pathway leading to the translocation and activation of p38 MAP kinase and MAPKAP kinase 2 [36]. Ischemia/reperfusion increased the induction of JNK1, c-JUN, and p38 MAP kinase proteins [37]. Ischemic adaptation also enhanced these kinases; but subsequent ischemia/reperfusion-mediated increase in JNK1, p38, and c-Jun was blocked by adaptation. It appears that activation of stress-activated protein kinases are obligatory for myocardial adaptation to ischemia, but the activation is only transient, and the activities rapidly come down after subsequent ischemia and reperfusion [37].
Evidence suggests that MAPKAP kinase 2 is a crucial step leading to gene expression and myocyte adaptation resulting in adaptive cardioprotection. This unique protein kinase is highly expressed in heart muscle and has been shown to increase when subjected to oxidative stress or heat shock [33, 36]. In cultured myocytes, the activity of MAPKAP kinase 2 was also found to increase when the myocytes were subjected to oxidative stress or heat shock [33]. Heat shock proteins HSP-27, HSP-32, and HSP-70 can be induced by oxidative stress and adaptation [38].
Cellular PKC activation is an important step in the mechanism of adaptive protection of heart [39]. For example, phenylephrine (an alpha1 agonist), angiotensin AT1, and bradykinin B2 receptors can activate PKC [40], and they can also precondition the hearts when infused prior to ischemia [39, 41]. A variety of stress signals can also activate PKC. For example, mechanical stress induced by stretching can activate PKC in cultured myocytes [42]. Both alpha?-receptor stimulation and Ca2+ can translocate and activate PKC [43].
REGULATION OF EXECUTION AND REPAIR BY REDOX SIGNALING
Although ischemia/reperfusion has been found to cause apoptosis, the precise physiological role of redox signaling is not clear. Ischemia/reperfusion, especially intermittent ischemia commonly known as ischemic preconditioning, leads to the activation of both G-proteins and receptor tyrosine kinases potentiating a signaling cascade resulting in the activation of multiple kinases that leads to the induction of the activation of several redox-sensing transcription factors and genes. Such intracellular events ultimately dictate the cells to survive or die. The changes in gene expression are likely to influence the physiologic function of the cardiomyocytes during post-ischemic survival.
Redox signaling appears to play a physiological role in myocardial survival during post-ischemic period. As mentioned earlier, ROS are generated during ischemia/reperfusion. When heart is adapted to ischemia, the generation of ROS is rapidly increased, but does not increase at the same rate during subsequent ischemia/reperfusion. The same pattern of the development of oxidative stress is observed when hearts are treated with endotoxin, IL-1, or TNF-alpha. Interestingly, these interventions lead to the development of oxidative stress within a very short period, but reduce subsequent oxidative stress when hearts are subjected to lethal ischemia/reperfusion. Such adaptive response is associated with the induction of several stress proteins and antioxidant enzymes as well as increased binding of NF-kappaB [36]. Increased activity of NF-kappaB and induction of the protective proteins can be blocked with a ROS scavenger such as dimethyl thiourea (DMTU) [44].
Thus, it seems likely that one of the major functions of redox signaling in the ischemic myocardium is to synthesize stress-inducible proteins through the activation of transcription factors such as NF-kappaB or by potentiating induction of other inducible proteins such as iNOS (inducible nitric oxide synthase) (Fig. 1). However, more study is necessary to completely unveil the physiologic function of redox cycling.
Fig. 1. Redox signaling by ischemia/reperfusion.
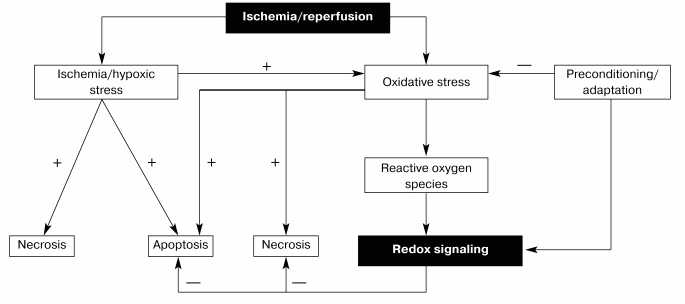
REDOX-SENSING TRANSCRIPTION FACTORS AND GENES: TRANSMISSION OF SURVIVAL SIGNAL
NF-kappaB and AP-1 are two well-known redox-sensitive transcription factors. NF-kappaB is a critical regulator for gene expression induced by diverse stress signals including mutagenic, and also oxidative and ischemic stresses. NF-kappaB is an oxidative stress responsive transcription factor, and ROS play a crucial role in the activation of the factor. Adaptation can increase NF-kappaB in the heart [45]. DMTU inhibits NF-kappaB translocation from cytosol to nucleus [44]. AP-1 is another redox-sensitive signaling molecule, which also plays an important regulatory role in cellular responses to stress induced by UV radiation, phorbol esters, and TNF-alpha [46]. Stress induced by ischemia/reperfusion causes simultaneous activation of c-Jun and AP-1, which is inhibited by DMTU [47, 48].
In various cell lines, wild-type p53 can cause DNA damage and induce apoptosis. p53 functions as an active transcription factor in lesioned brain [49]. How activation of p53 promotes apoptosis is unclear, but it might involve bax, a series of p53-inducible genes, or signaling through Fas-related pathway [50]. There are other p53 effectors including caspases that execute apoptotic cell death [51]. Several studies on the master controller gene of apoptosis, p53, reveal conflicting results [52]. Our recent study showed an increase in p53 activity in the ischemic/reperfused myocardium [47]. Adapting the heart to ischemia prevented such an increase in p53 activity, and thus, reduced p53-mediated death signal.
bcl-2 is an anti-death gene, which functions as an intracellular antioxidant. Recent analysis of the bcl-2 gene family reveals a complex network regulating apoptosis. Within this bcl-2 gene family, some of the candidates can suppress apoptosis, while the others can induce apoptosis [53]. Among the proteins coded by the genes of this family, Bcl-2 and Bcl-xL act as cell death repressors [54], whereas Bax and an alternatively sliced Bcl-x product, Bcl-xs, promote cell death [7]. When in excess over Bcl-2, Bax counteracts the repressive action of Bcl-2 on apoptosis. Similarly, excess Bcl-xs antagonizes the function of Bcl-xL. Thus, a critical balance between the Bcl-2, Bax, and Bcl-xL/S molecules may determine the fate of cells in response to cytotoxic agents or environmental stress. It has been reported that p53 activates the transcription of the bax gene via p53-response elements while downregulating bcl-2 expression at the same time [50].
Overexpressing bcl-2 can block apoptosis initiated by various different stimuli. For example, the activation of bcl-2 was associated with the inhibition of apoptosis in the adapted myocardium [47, 48]. A downregulation of this anti-death gene occurred in concert with a significant amount of apoptosis in the ischemic/reperfused myocardium. Thus, by inducing bcl-2 expression and reducing bax expression, preconditioning converts the death signal triggered by ischemia/reperfusion (which decreases bcl-2 and increases bax) into survival signal.
DEATH SIGNAL BY ROS
Apoptosis is recognized as a physiological counterpart of cell replication and is the contributing cause of cardiomyocyte cell death during ischemia/reperfusion and heart failure [55]. It is an energy requiring process, needs de novo pro apoptotic gene expression (p53, bax, etc.), and is directed by an in born genetic program. The common inducers of apoptosis include ROS and Ca2+, which are also implicated in the pathogenesis of myocardial ischemic/reperfusion injury. Cardiomyocytes exposed to hypoxia revealed apoptotic cell death in conjunction with the expression of Fas-mRNA [56]. Apoptotic and necrotic myocyte cell deaths associated with ischemia/reperfusion were shown to be independent contributing variables of infarct size [31]. Apoptosis constitutes a feature of human vascular pathology suggesting that it may modulate the cellularity of lesions that produce human vascular obstruction [57].
Apoptosis is a function of the duration of reperfusion. Up to 1 h of ischemia does not induce apoptosis [48]. Apoptotic cells become apparent after 90-min reperfusion. These results were corroborated with DNA fragmentation, which showed increased DNA ladders in 120-min reperfused hearts. The presence of apoptotic cells was abolished with Ebselen, a glutathione peroxidase mimic that also reduced the ischemic reperfusion injury [8]. In another study, SOD plus catalase were found to ameliorate the apoptotic cell death [58].
Although apoptosis occurs only after prolonged reperfusion, the signal for apoptosis is initiated during ischemia. Translocation of phosphatidylserine (PS) and phosphatidylethanolamine (PE), a hallmark for apoptosis, was found to occur during ischemia, but apoptosis did not become apparent until hearts were reperfused [59]. Assay of phosphatidylethanolamine and phosphatidylserine topography in cardiomyocytes demonstrated that about 30% of phosphatidylethanolamine and 5% of phosphatidylserine were available in the control hearts suggesting that ischemia results in a significant loss of normal asymmetric sarcolemmal phospholipid distribution due to an outward migration of PE and PS.
Caspases are a family of cysteine proteases that play a central role in apoptosis. Recent studies have established that activation of proximal caspases leads to the activation of the distal caspases in the process of apoptosis [24]. Caspase 3 (ced-3) activation begins when caspase-9 binds to Apaf-1 (ced-4 is homologous to the recently identified human protein, Apaf-1), this reaction is initiated by the release of cytochrome c into the cytosol. Caspase-9 is directly activated by Apaf-1 and cytochrome c [60]. Significant accumulation of cytochrome c in the cytosol, over myofibrils, and near intercalated discs of cardiomyocytes in failing hearts is reported in a heart transplantation study. The cytochrome c was also associated with the activation of caspase-3 and cleavage of its substrate--protein kinase cdelta [61, 62]. A related study demonstrated selective cleavage of poly(ADP-ribose)polymerase (PARP) into apoptotic fragments in ischemic/reperfused myocardium [63]. Administration of a caspase inhibitor YVAD-cmk blocked caspase activation and reduced apoptotic cells. This study indicated that caspases are critical mediators of ischemia/reperfusion injury [62]. Deprivation of serum and glucose, components of ischemia in vivo, induced apoptosis, which was blocked with zVAD-fmk, a caspase inhibitor [63]. During ischemia, cytochrome c is released from mitochondria and this is one of the earliest events in ischemic myocardium. The possible scenario of ischemia-induced myocardial apoptosis suggests an elevation of cytosolic Ca2+, release of cytochrome c associated with Apaf-1 factor and pro-caspase-9, triggering the activation of caspase-3 and apoptosis. It is tempting to speculate that ischemia triggers an apoptotic signal, but execution does not occur until the ischemic myocardium is reperfused.
CONVERSION OF DEATH SIGNAL INTO SURVIVAL SIGNAL BY REDOX
SIGNALING
A variety of antioxidants play a major role in myocardial protection against acute stress such as ischemia or hypoxia. To defend itself against ROS, the heart is equipped with its own defense system consisting of antioxidants and antioxidant enzymes [64]. A SOD mimic, selenoperoxide derivative, Ebselen, reduced apoptosis and ameliorated ischemic/reperfusion injury [8]. The apoptotic process is increased by the addition of H2O2 and aminotriazole, a potent catalase inhibitor [65]. Addition of SOD and catalase in cultured neutrophils was effective in delaying the event of apoptosis [66]. Overexpression of GHSPx-1 gene was associated with reduced number of apoptotic cardiomyocytes, and GSHPx-1 gene knockout mice mouse hearts had significantly higher number of apoptotic cells.
The biochemical basis of the redox signaling is that oxidizing conditions are maintained by the stabilizing disulfides in the extracellular surface while the intracellular environment is maintained in the reduced state with the help of free sulfhydryl groups. The principle disulfide reductase responsible for maintaining the inside of the cell in the reduced state is a low molecular weight redox-active protein with two cysteine residues in its active sites, thioredoxin. Thioredoxin is ubiquitously present in mammalian cells including heart. Several reports exist indicating that thioredoxin is induced by oxidative stress. Thioredoxin appears to play a crucial role in redox regulation of the ROS signaling during ischemia/reperfusion. Thioredoxin may be an important component of the cellular defense against cardiac injury. Oxidized thioredoxin was found to be released into plasma of the patients undergoing cardiopulmonary bypass surgery. Endurance training by swimming accompanied by a reduction of ischemia/reperfusion-induced oxidative stress with a concomitant increase in thioredoxin reductase resulted in a protection against myocardial ischemia/reperfusion injury. Thioredoxin attenuated hypoxia-reoxygenation injury of murine endothelial cells in a thiol-free condition suggesting thioredoxin protection of myocardial injury through a novel redox signaling pathway.
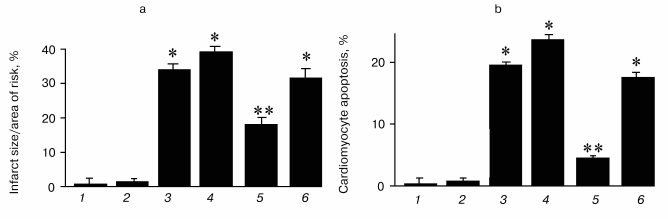
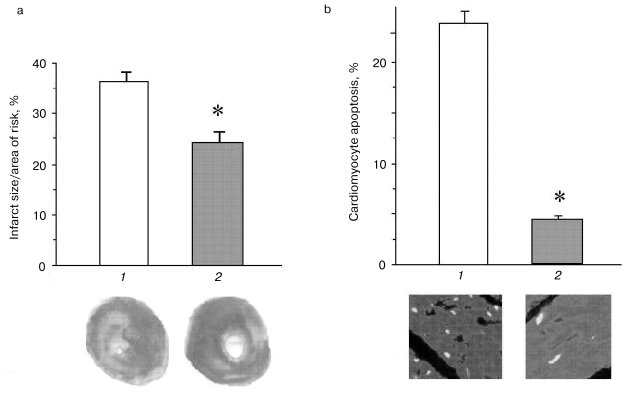
Thioredoxins are critical for redox regulation of protein function and signaling via thiol redox control. Thioredoxins are reduced by electrons from NADPH via thioredoxin reductase. A recent study has demonstrated that thioredoxin (Trx-1) plays an important role in redox signaling and in transmitting survival signal in ischemic myocardium. Trx-1 is downregulated after ischemia/reperfusion but upregulated in the adapted myocardium. Inhibition of Trx-1 with cisplatin (CDDP) abrogated cardioprotective effects of ischemic adaptation as evidenced from impaired post-ischemic ventricular recovery, increased myocardial infarct size, and cardiomyocyte apoptosis (Fig. 2). The role of Trx-1 in transmitting survival signal was further supported from the results that transgenic mouse hearts with extra copies of Trx-1 were resistant to apoptotic cardiomyocyte death (Fig. 3).
Fig. 2. Effects of ischemia/reperfusion (I/R) and ischemic adaptation on myocardial infarct size (a) and cardiomyocyte apoptosis (b) (the results are expressed as mean ± SEM of six animals per group): 1) control; 2) cisplatin (CDDP) (an inhibitor of Trx-1); 3) I/R; 4) CDDP + I/R; 5) adapted (preconditioned by four cyclic episodes of 5-min ischemia, each followed by another 10 min of reperfusion and then subjected to 30-min ischemia and 2-h reperfusion); 6) CDDP + adapted; * p < 0.05 vs control; ** p < 0.05 vs I/R.
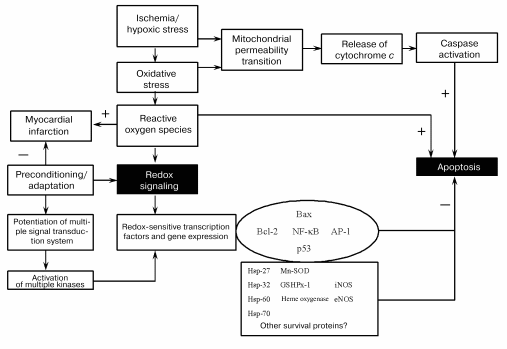
It appears from the above discussion, survival and death of the cardiomyocytes depend critically on their redox state (Fig. 4). While sufficient antioxidant reserve warrants a reducing environment for the survival of the myocytes, the reserve is exhausted during ischemic heart disease leading to a change in redox state. Such changes in the redox state determine whether the cardiomyocytes should die, and if they die, whether they die by necrosis or apoptosis. Ultimate survival or death depends on the redox signaling, transcription regulation of several redox-sensing genes, and DNA repair processes.Fig. 3. Effects of ischemia/reperfusion on the myocardial infarct size (a) and cardiomyocyte apoptosis (b). The hearts were made ischemic for 30 min followed by 2 h of reperfusion. At the end of the reperfusion, myocardial infarct size was measured as indicated in “Materials and Methods”. The results are shown as means ± SEM of six hearts per group; 1) normal amount of thioredoxin 1; 2) increased number of copies of thioredoxin 1; * p < 0.05 versus wild-type.
This study was supported by the National Institutes of Health grants NIH HL 56803, HL 22559, HL 33889, HL 5642, HL 63317, and P30 ES06639.Fig. 4. Redox regulation of anti-death signal (Mn-SOD is Mn2+-dependent superoxide dismutase).
REFERENCES
1.Maulik, N., Watanabe, M., Engelman, D.,
Engelman, R. M., and Das, D. K. (1995) Mol. Cell. Biochem.,
144, 67-74.
2.Maulik, N., Engelman, R. M., Rousou, J. A., Flack,
J. E., Deaton, D., and Das, D. K. (1999) Circulation, 100
(Suppl. II), 369-375.
3.Maulik, N., Yoshida, T., and Das, D. K. (1998)
Free Rad. Biol. Med., 24, 869-875.
4.Herrlich, P., and Bohmer, F. D. (2000) Biochem.
Pharmacol., 59, 35041.
5.Rosette, C., and Karin, M. (1996) Science,
274, 1194-1197.
6.Dalton, T. P., Shertzer, H. G., and Puga, A. (1999)
Annu. Rev. Pharmacol. Toxicol., 39, 67-101.
7.Das, D. K., and Maulik, N. (1994) Meth.
Enzymol., 233, 601-610.
8.Meerson, F. Z., Kagan, V. E., Kozlov, P., Beekina,
L. M., and Khipenko, Y. V. (1982) Basic Res. Cardiol.,
77, 465-472.
9.Tosaki, A., Droy-Lefaix, M. T., Pali, T., and Das,
D. K. (1993) Free Rad. Biol. Med., 14, 361-370.
10.Olivetti, G., Abbi, R., Quaini, F., Kajstura, J.,
Cheng, W., Nitahara, J. A., Quaini, E., Loreto, C. Di, Beltrami, C. A.,
Krajewski, S., Reed, J. C., and Anversa, P. (1997) New Eng. J.
Med., 336, 1131-1141.
11.Nishio, Y., Kashiwagi, A., Taki, H., Shinozaki,
K., Maeno, Y., Kojima, H., Maegawa, H., Haneda, M., Hidaka, H.,
Yasuda, H., Horiike, K., and Kikkawa, R. (1998) Diabetes,
47, 1318-1325.
12.Das, D. K., and Maulik, N. (1998) in
Biological Oxidants and Antioxidants (Packer, L., ed.) ASH Ong
AOCS Press, IL, pp. 165-177.
13.Maulik, N., Yoshida, T., and Das, D. K. (1999)
Mol. Cell. Biochem., 196, 13-21.
14.Das, D. K., and Maulik, N. (1995) in Exercise
and Oxygen Toxicity (Sen, C. K., Packer, L., and Hanninen, O.,
eds.) Elsevier Science, Amsterdam.
15.Kukreja, R. C., and Hess, M. L. (1999) in
Pathophysiology of Reperfusion Injury (Das, D. K., ed.) CRC
Press, Boca Raton, FL, pp. 221-242.
16.Das, D. K., George, A., Liu, X., and Rao, P. S.
(1989) Biochem. Biophys. Res. Commun., 165,
1004-1009.
17.Halestrap, A. P., Kerr, P. M., Javadov, S., and
Woodfield, K. Y. (1998) Biochim. Biophys. Acta, 1366,
79-94.
18.Maulik, N., Watanabe, M., Engelman, D.,
Engelman, R. M., Kagan, V. E., Kisin, E., Tyurin, V., Cordis, G.
A., and Das, D. K. (1995) Am. J. Physiol. (Cell), 269,
C907-C916.
19.Sharma, H. S., and Das, D. K. (1997) Mediators
Inflamm., 6, 175-184.
20.Bromme, H. J., and Holz, J. (1996) Mol. Cell.
Biochem., 163/164, 261-275.
21.Beg, A. A., and Baltimore, D. (1996)
Science, 274, 787-789.
22.Suzuki, Y. J., and Packer, L. (1993) Biochem.
Mol. Biol. Int., 31, 693-700.
23.Suzuki, Y. J., Aggarwal, B. B., and Packer, L.
(1992) Biochem. Biophys. Res. Commun., 189,
1709-1715.
24.Screck, R., Rieber, P., and Baeuerle, P. A.
(1991) EMBO J., 10, 2247-2258.
25.Das, D. K. (1998) in Adv. Org. Biol. (Das,
D. K., ed.) Jay Press, CT.
26.Cohen, M. V., Liu, Y., Liu, G. S., Wang, P.,
Cordis, G. A., Das, D. K., and Downey, J. M. (1996) Circulation,
94, 1713-1718.
27.Moraru, I. I., Popescu, L., Maulik, N., Liu, X.,
and Das, D. K. (1992) Biochim. Biophys. Acta, 1139,
148-154.
28.Eskildensen, K., Helmond, Y. E. G., Gho, B. C.
G., Bezstarosti, K., Dekkers, D. H. W., Loe, K. S., and van Heusten, H.
(1998) Ann. N.Y. Acad. Sci., 793, 210-225.
29.Fryer, R. M., Schultz Hsu, J. E., and Gross, G.
J. (1998) Am. J. Physiol., 275, H2009-H2015.
30.Anderson, N. G., Maller, J. I., Tonks, N. K., and
Sturgill, T. W. (1990) Nature (Lond.), 343, 651-653.
31.Seger, R., and Krebs, E. G. (1995) FASEB
J., 9, 726-735.
32.Cobb, M. H., and Goldsmith, E. J. (1995) J.
Biol. Chem., 270, 14843-14846.
33.Zu, Y. L., Ai, Y., Gilchrist, A., Maulik, N.,
Watras, J., Sha'afi, R. I., Das, D. K., and Huang, C. K. (1997) J.
Mol. Cell. Cardiol., 29, 2150-2168.
34.Maulik, N., Watanabe, M., Zu, Y. L., Huang, C.
K., Cordis, G. A., Schley, J. A., and Das, D. K. (1996) FEBS
Lett., 396, 233-237.
35.Kyriakis, J. M., Banerjee, P., Nikolakaki,
E., Dai, T., Rubie, E. A., Ahmad, M. F., Avruch, J., and Woodgett, J.
D. (1994) Nature, 369, 156-160.
36.Maulik, N.,Yoshida, T., Zu, Y. L., Sato, M.,
Banerjee, A., and Das, D. K. (1998) Am. J. Physiol., 275,
H1857-H1864.
37.Sato, M., Cordis, G. A., Maulik, N., and
Das, D. K. (2003) Am. J. Physiol., in press.
38.Benjamin, I. J., McMillan, R., Scholich, K.,
Mullenix, J. B., Wittpoth, C., Poppleton, H. M., Pierre, S. C.,
Lindorfer, M. A., and Garrison, J. C. (1999) Science,
283, 1328-1331.
39.Mitchell, M. B., Meng, X., Brown, J., Harken, A.
H., and Banerjee, A. (1995) Circ. Res., 76, 73-81.
40.Dixon, B. S., Sharma, R. V., Dickerson, T., and
Fortune, J. (1994) Am. J. Physiol., 266, C1406-C1420.
41.Sato, M., Engelman, R. M., Rousou, J. A.,
Flack, J. E., Deaton, D., Cordis, G. A., Maulik, N., and Das, D. K.
(1998) Surg. Forum, 48, 212-215.
42.Yazaki, Y., Komuro, I., Yamazaki, T., Tobe, K.,
Maemura, K., Kadowaki, T., and Nagai, R. (1993) Mol. Cell.
Biochem., 119, 11-16.
43.Henrich, C. J., and Simpson, P. C. (1988) J.
Mol. Cell. Cardiol., 20, 1081-1085.
44.Das, D. K., Maulik, N., Sato, M., and Ray,
P. (1999) Mol. Cell. Biol., 196, 59-67.
45.Maulik, N., Goswami, S., Galang, N., and Das, D.
K. (1999) FEBS Lett., 443, 331-336.
46.McMahon, S. B., and Monroe, J. G. (1992) FASEB
J., 62, 2707-2715.
47.Artuc, M., Karman, D., Jurgovsky, K., and
Schadendorf, D. (1997) Anticancer, 17, 4359-4370.
48.Maulik, N., and Das, D. K. (1999) Heart
Failure Rev., 4, 165-173.
49.Hughes, P. E., Alexi, T., Yoshida, T., Schreiber,
S. S., and Knusel, B. (1996) Neuroscience, 74,
1143-1160.
50.Miyashita, T., Krajewski, S., Krajewska, Wang, M.
H., Lin, K., Liebermann, D., Hoffmann, B., and Reed, J. C. (1994)
Oncogene, 9, 1799-1805.
51.Thornberry, N. A., and Lazebnik, Y. (1998)
Science, 281, 1312-1316.
52.Clark, A. R., Purdie, C. A., Harrision, D. J.,
Morris, R. G., Bird, C. C., Hooper, M. L., and Wyllie, A. H. (1993)
Nature, 362, 849-852.
53.Williams, G. T., and Smith, C. A. (1993)
Cell, 74, 777-779.
54.Nunez, G., London, L., Hockenbery, D.,
Alexander, M., Mckearn, J. P., and Korsmeyer, S. J. (1999) J.
Immunol., 144, 3602-3610.
55.Maclellan, W. R., and Schneider, M. D. (1997)
Circ. Res., 81, 137-144.
56.Tanaka, M., Inada, T., Fujiwara, H., Ohtani,
S., Yamasaki, K., Fujiwara, T., Yokota, R., Sasayama, S., and Doyama,
K. (1998) Jap. Circ. J., 62, 512-516.
57.Kajstura, J., Cheng, W., Rreiss, K., Clark, W.
A., Sonnenblick, E. H., Krajewski, S., Reed, J. C., Olivetti, G., and
Anversa, P. (1996) Lab. Invest., 74, 86-107.
58.Galang, N., and Maulik, N. (2003) Nutr.
Environ. Interact. (Toxicol.), in press.
59.Maulik, N., Kagan, V. E., Tyrin, V. A., and Das,
D. K. (1998) Am. J. Physiol., 274, H242-H248.
60.Pang, G., O'Rourke, K., and Dixit, V. M. (1997)
J. Biol. Chem., 273, 584-590.
61.Narula, J., Pandey, P., Arbustini, E., Haider,
N., Narula, N., Kolodgie, F. D., Dal Bello, B., Semigran, M. J.,
Bielsa-Masdeu, A., Dec, G. W., Israels, S., Ballester, M.,
Virmani, R., Saxena, S., and Kharbanda, S. (1999) Proc. Natl. Acad.
Sci. USA, 96, 8144-8149.
62.Holly, T. A., Drincic, A., Byun, Y., Nakamura,
S., Harris, K., Klocke, F. J., and Cryns, V. L. (1999) J. Mol. Cell.
Cardiol., 31, 1709-1715.
63.Bialik, S., Cryns, V. L., Drincic, A., Miytata,
S., Wollowick, A. L., Srinivasan, A., and Kitsis, R. N. (1999) Circ.
Res., 85, 403-414.
64.Das, D. K., Maulik, N., and Moraru, I. I. (1995)
J. Mol. Cell. Cardiol., 27, 181-193.
65.Hanada, H., Kashiwagi, A., Takehara, Y., Kanno,
T., Yabuki, M., Sasaki, J., Inoue, M., and Utsumi, K. (1997)
Free Rad. Biol. Med., 23, 294-301.
66.Oishi, K., and Machida, K. (1997) Scand. J.
Immunol., 45, 21-27.