Expression and Refolding of Tobacco Anionic Peroxidase from E. coli Inclusion Bodies
D. M. Hushpulian1, P. A. Savitski2, A. M. Rojkova1, T. A. Chubar1, V. A. Fechina2, I. Yu. Sakharov1, L. M. Lagrimini3, V. I. Tishkov1*, and I. G. Gazaryan1
1Department of Chemical Enzymology, Faculty of Chemistry, Lomonosov Moscow State University, Moscow 119992, Russia; fax: (7-095) 939-2742; E-mail: vit@enz.chem.msu.ru; igazaryan@hotmail.com2Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, Moscow 119071, Russia; fax: (7-095) 954-2804; E-mail: zherdev@inbi.ras.ru
3Syngenta Biotechnology, 3054 Cornwallis Rd., Research Triangle Park, NC 27709-2257, USA; fax: (919) 541-8585; E-mail: mark.lagrimini@syngenta.com
* To whom correspondence should be addressed.
Received June 24, 2003; Revision received July 28, 2003
Coding DNA of the tobacco anionic peroxidase gene was cloned in pET40b vector. The problem of 11 arginine codons, rare in procaryotes, in the tobacco peroxidase gene was solved using E. coli BL21(DE3) Codon Plus strain. The expression level of the tobacco apo-peroxidase in the above strain was ~40% of the total E. coli protein. The tobacco peroxidase refolding was optimized based on the earlier developed protocol for horseradish peroxidase. The reactivation yield of recombinant tobacco enzyme was about 7% with the specific activity of 1100-1200 U/mg towards 2,2´-azino-bis(3-ethylbenzothiazoline-6-sulfonate) (ABTS). It was shown that the reaction of ABTS oxidation by hydrogen peroxide catalyzed by recombinant tobacco peroxidase proceeds via the ping-pong kinetic mechanism as for the native enzyme. In the presence of calcium ions, the recombinant peroxidase exhibits a 2.5-fold decrease in the second order rate constant for hydrogen peroxide and 1.5-fold decrease for ABTS. Thus, calcium ions have an inhibitory effect on the recombinant enzyme like that observed earlier for the native tobacco peroxidase. The data demonstrate that the oligosaccharide part of the enzyme has no effect on the kinetic properties and calcium inhibition of tobacco peroxidase.
KEY WORDS: recombinant tobacco peroxidase, expression, refolding, purification, calcium effect
Abbreviations: PCR) polymerase chain reaction; IPTG) isopropyl-beta-D-thiogalactopyranoside; ABTS) ammonium 2,2´-azino-bis(3-ethylbenzothiazoline-6-sulfonate); TOP) tobacco peroxidase; HRP) horseradish peroxidase; DTT) dithiothreitol; RZ) ratio of peroxidase solution absorbance at 403 and 280 nm.
The study of native tobacco peroxidase (TOP) isolated and characterized
in this laboratory [1] revealed a number of unusual
properties of the enzyme. First is its high stability both during
storage and in the reaction course, in particular at extreme pH values
in the presence of calcium ions. High stability of TOP over a wide
range of pH allows its activity towards veratryl alcohol to be detected
at pH < 2 [2], which is impossible in the case
of horseradish peroxidase. The enzyme is also stable in the course of
its chemical modification in alkaline medium resulting in more active
conjugates of antibodies and tobacco peroxidase compared to those of
horseradish peroxidase [3, 4].
Second, this enzyme has a different profile of substrate specificity
compared to peroxidases from other sources. For instance, contrary to
horseradish peroxidase, TOP is almost inactive towards iodide and
phenol, and extremely active towards luminol even in the absence of
chemiluminescence enhancers, which makes the enzyme promising for the
purposes of chemiluminescent assay [5]. Third, the
properties of intermediate compounds of tobacco peroxidase, i.e., high
stability of Compound I and low stability of Compound II in acidic
medium, are very close to the properties of intermediate compounds of
fungal peroxidase Coprinus cinereus (Arthromyces ramosus)
that has been patented as a chemiluminescent reagent [6]. Fourth, contrary to other peroxidases, spectral
and catalytic properties of TOP are regulated by calcium ions [2, 7]. Computer modeling of the
tobacco peroxidase tertiary structure based on peanut and horseradish
structures [7] revealed the presence of Glu141 at
the active center entrance, while other peroxidases have Phe in this
position. The presence of negatively charged glutamic acid residue 141
at the TOP active center entrance may be the cause of its unusual
properties.
Systematic study of structure-function relationships for tobacco peroxidase by site-directed mutagenesis is ongoing in this laboratory. The current expression systems of TOP in transgenic tobacco and tomatoes [8], being laborious, time consuming, and expensive, are not suitable for these purposes. The most efficient system for production of peroxidase mutants is an E. coli expression system, which, in addition, results in the expression of a non-glycosylated enzyme. This allows us to obtain information on the role of the oligosaccharide part in catalysis and stability. It is well known that expression of peroxidase genes in E. coli yields a non-soluble product in the form of inclusion bodies, which can be reactivated into an active and soluble enzyme via a special refolding procedure. The refolding conditions for each peroxidase are very different. This article presents the results on the expression of anionic tobacco peroxidase protein in E. coli cells and optimization of its refolding procedure.
MATERIALS AND METHODS
The gene of anionic tobacco peroxidase was cloned in the laboratory of Prof. L. M. Lagrimini [9] and expressed in transgenic plants [8]. To obtain a construct expressing tobacco peroxidase in E. coli cells, cDNA of the TOP gene was amplified by polymerase chain reaction (PCR) with the following primers:
5´-CATATGCAATTAAGTGCAACATTTTATGATACC-3´
and
5´-GTCGACGAA-TTCTTAATTAACCCTCTT-3´
containing NdeI and XhoI restriction sites, respectively (shown in bold italic). The PCR product was cloned via “blunt” ends into an intermediate vector pCRII-TOPO. The NdeI-XhoI restrict of 0.9 kb containing the gene of tobacco peroxidase was then cloned into the expression vector pET40b via NdeI and SalI restriction sites (XhoI and SalI give the same “sticky” ends). Plasmids were isolated from two independent clones and sequenced to confirm the absence of any additional mutation. The plasmids were used to transform E. coli BL21(DE3) and BL21(DE3)CdPlus.
Transformed E. coli BL21(DE3)CdPlus cells were grown in 2-liter flasks in 400 ml LB-medium prepared with 10 mM Tris-HCl buffer, pH 8.0, containing 0.04 mg/ml kanamycin at 37°C. Expression was initiated by 2*10-4 M isopropyl-beta-D-thiogalactopyranoside (IPTG) in the mid-log phase of cell growth (0.5-0.7 absorbance unit at 600 nm), and the cultivation was continued for 4-6 h. The biomass was collected by centrifugation at 5000g and resuspended in 10 mM Tris-HCl buffer, pH 8.0, and disrupted by sonication (22 kHz, 10 min) in the presence of 2 M NaCl and 10 mM dithiothreitol (DTT). The mixture was incubated for 1.5 h at room temperature and then sonication was repeated. The supernatant was removed, and the precipitate was washed with 0.05 M Tris-HCl buffer, pH 8.5, with subsequent solubilization in 60 ml of 6 M urea containing 1 mM DTT. The solubilized TOP apoprotein (95% purity, 1 mg/ml) was added drop by drop to 600 ml of the refolding medium and incubated at 4°C. The optimization of refolding medium was performed by varying the concentration of urea (1.8-2.3 M), oxidized glutathione (0.1-0.7 mM), DTT (0.1-1 mM) with fixed 5 mM CaCl2, 5 µM hemin, 5% glycerol in 50 mM Tris-HCl buffer, pH 8.4-9.5. The enzyme activity was measured towards ammonium 2,2´-azino-bis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) during the incubation course. When the activity reached the plateau, the mixture was saturated with ammonium sulfate (60%). The precipitate was isolated by centrifugation and dissolved in 30 ml water. Alternatively, the protein was concentrated by ultrafiltration of the refolding medium through an Amicon YM-10 membrane (Amicon, USA). The resulting solution was applied in 15-ml portions to a Sephacryl S-200 (5.2 × 80 cm) column equilibrated with 0.005 M Tris-HCl buffer, pH 8.5. Active fractions were pooled and concentrated.
The protein content was determined spectrophotometrically using the formula: c (µg/ml) = 183*A230 - 75.8*A260 [10] which, as preliminarily shown, gives the same results as the Lowry method [11]. The purity of recombinant TOP preparations was monitored by SDS-PAGE.
The activity was assayed with a Shimadzu UV 120-02 spectrophotometer (Japan) at 25°C in 0.1 M Na-acetate buffer (pH 5.0, if not indicated otherwise) with 0.4 mM ABTS and 1 mM H2O2. Rate constants in the reaction of ABTS oxidation were determined from steady-state data by varying the concentration of substrates in the range 0.015-0.15 mM for ABTS and 0.01-0.5 mM for H2O2 in the presence and in the absence of 50 mM CaCl2 in 0.05 M Na-acetate buffer, pH 4.5. The extinction coefficient for ABTS oxidation product at 405 nm was taken equal to 36.8 mM-1*cm-1 [12]. Hydrogen peroxide concentration was determined spectrophotometrically (epsilon240 = 43.6 M-1*cm-1) [13].
RESULTS AND DISCUSSION
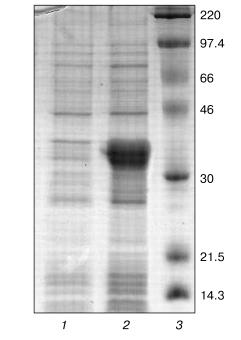
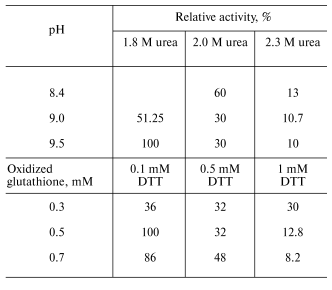
Optimization of refolding. The tobacco peroxidase gene contains 11 arginine codons AGA and AGG, which are rare for prokaryotes. As a rule, genes containing these codons cannot be expressed in regular E. coli strains, and we observed that this rule holds for E. coli BL21(DE3) in the case of tobacco peroxidase gene. Its expression was observed only in E. coli BL21(DE3)CdPlus, which solves the problem of rare codons due to the introduced genes of the corresponding tRNA. The electrophoresis data of E. coli whole cells before and after IPTG induction are shown in Fig. 1 (lanes 1 and 2, respectively). The expression level of recombinant TOP is higher than 40% of the total E. coli protein. Like other peroxidases, tobacco peroxidase is expressed in the form of inclusion bodies. The primary purification step, i.e., centrifugation of disrupted cells, removes soluble E. coli proteins. The further treatment of the precipitate with 2 M NaCl provides further purification of the inclusion bodies. The purity of TOP apoprotein solubilized from inclusion bodies in 6 M urea is ~85-90% and is sufficient for the subsequent refolding procedure. The optimal conditions for tobacco peroxidase refolding were slightly different from those for horseradish peroxidase. The maximum activity yield for the recombinant TOP was observed at pH 9.5 and 1.8 M urea (pH 9.3 and 2.0 M urea, respectively, for horseradish peroxidase [14]), and with changed ratio for DTT and oxidized glutathione from 1 : 7 to 1 : 5 (0.1 and 0.5 mM, respectively) (Table 1). The decrease in the concentration of oxidized glutathione slows down the rate of refolding compared to horseradish peroxidase, i.e., the refolding procedure takes 6-7 days instead of 36-48 h for horseradish peroxidase. The specific feature of TOP refolding is in hemin addition just after the addition of the solubilized (in 6 M urea) enzyme apoprotein. In the case of horseradish peroxidase, hemin is added at the end of refolding to the apoenzyme with the restored tertiary structure. The entrapment of hemin by the apoenzyme active center resulting in the formation of the horseradish peroxidase holoenzyme takes only 2 h. One of the possible reasons for the poor entrapment of the negatively charged hemin by the active center of TOP can be the presence of a Glu residue in position 141 while other peroxidases have a Phe residue in this position. We showed earlier that Phe143Glu mutation in the molecule of horseradish peroxidase in an attempt to mimic the active center of TOP resulted in the impossibility of hemin entrapment by the mutant apoenzyme at the end of refolding. In that case, like in the one with the wild-type TOP, we had to add hemin in the refolding medium in the very beginning of the process [14]. However, even under this condition, the yield of the Phe143Glu mutant of horseradish peroxidase was much lower (5-10%) than that of the wild-type enzyme (up to 50%) [14]. The correctness of the proposed effect of Glu141 residue on hemin entrapment by recombinant TOP in the process of its refolding can be tested by Glu141Phe mutation. Currently, the mutant has been constructed and expressed and the study of its refolding is under way.
Table 1. Effect of urea concentration, pH, and the ratio between DTT and oxidized glutathione concentrations on the yield of active enzyme in the course of refolding of recombinant tobacco peroxidaseFig. 1. SDS-PAGE of E. coli BL21(DE3)CdPlus cells before (1) and after IPTG induction (2); 3) marker proteins (on the right, their molecular masses in kD).
Purification of recombinant TOP. Isolation of the active recombinant enzyme from the refolding medium and its subsequent purification by ultrafiltration and gel filtration demonstrated the difference between the recombinant forms of tobacco and horseradish peroxidases (TOP and HRP, respectively) even more.
In the case of HRP, the first purification step is performed by precipitation with ammonium sulfate, which concentrates the enzyme and provides the removal of refolding medium components and improperly folded or aggregated protein. For the recombinant TOP, precipitation with ammonium sulfate results in a significant loss in activity (70-80%), which can be avoided by using ultrafiltration. In this case, there is no detected loss in activity at the concentrating step (Table 2).
Table 2. Production and purification of
recombinant wild-type tobacco peroxidase (from 1.8 liters of culture
medium)
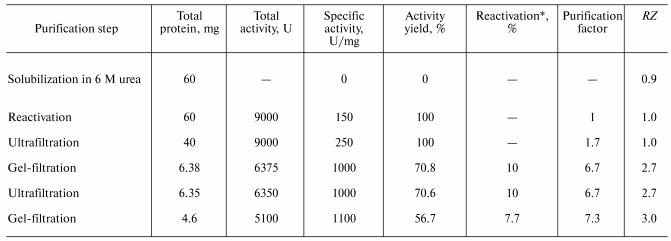
*Calculated as the ratio of the active enzyme to the total
peroxidase apoprotein solubilized from inclusion bodies.
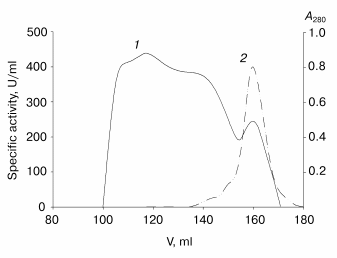
Further purification is achieved by gel filtration. The elution profile of the recombinant TOP (Fig. 2) shows that more than 80% of the protein is present in the high molecular weight form with low activity towards ABTS. It is likely that this is apoenzyme aggregates with partial entrapment of the holoenzyme. Unfortunately, the overlapping peaks of aggregated and non-aggregated enzyme forms (Fig. 2) does not allow their separation to be achieved in one step. High-purity enzyme preparations (>95% purity) have been obtained only after an additional gel filtration step. The results of recombinant TOP purification are presented in Table 2.
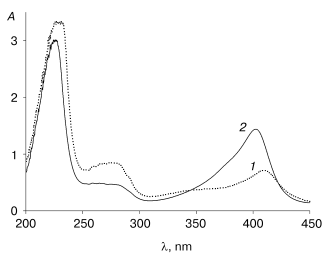
An interesting feature of the aggregated enzyme is the shift of its Soret band to 409 nm (Fig. 3). As shown earlier [2], native tobacco peroxidase undergoes a shift in the Soret band in the presence of calcium at extremely low pH values; however, this shift is accompanied with the growth of the enzyme extinction up to 1.5-fold, to 150 mM-1*cm-1, and the corresponding increase in RZ, from 3 to 4.5 [2], which is not observed in this case.Fig. 2. Elution profile of the recombinant wild-type tobacco peroxidase at the first gel filtration step: 1) absorbance at 280 nm (A280); 2) enzyme activity (U/ml).
The additional gel filtration step yields a homogeneous recombinant TOP preparation with specific activity of 1100 U/mg protein and RZ 3.0. The yield of the active enzyme with respect to the total recombinant TOP protein solubilized from inclusion bodies is about 8%, which is 3-4-fold lower compared to the recombinant HRP (Table 2). The cause of the lower reactivation yield of the recombinant TOP is still unknown. To clarify the problem and provide higher yields of the active enzyme, more experiments are needed.Fig. 3. Absorbance (A) spectra for recombinant wild-type tobacco peroxidase: 1) high-molecular-weight fraction; 2) highly purified preparation.
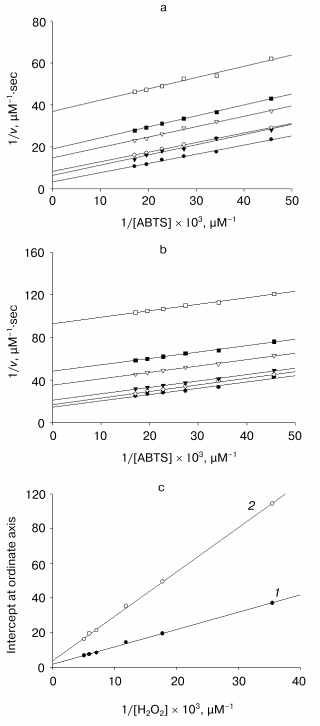
Effect of calcium cations on kinetic parameters of ABTS oxidation. The native TOP, as shown earlier, is sensitive to calcium ions: the second order rate constant for the enzyme interaction with hydrogen peroxide decreases 3 times upon addition of [Ca2+] > 40 mM at pH 5 [15]. As can be seen from the data in Fig. 4, the activity of the recombinant TOP is also dependent on the presence of calcium ions in the reaction medium. The slope of the dependence of the reaction rate on hydrogen peroxide concentration in double reciprocal plots increases by 2.5-fold upon addition of 50 mM calcium ions to the solution.
Enzymatic reactions catalyzed by heme-containing peroxidases, including tobacco peroxidase [16], proceed via a ping-pong mechanism [17]. In accordance with it, the dependence of the reaction rate on the concentration of one substrate at any fixed concentration of the second one (“primary” plots) presents a series of parallel lines with a slope linearly proportional to the inversed second-order rate constant for the enzyme interaction with varied substrate and fits the equation:Fig. 4. Kinetics of ABTS oxidation with hydrogen peroxide catalyzed by recombinant wild-type tobacco peroxidase. a, b) Dependences of the initial reaction rate on ABTS concentration at different fixed concentrations of hydrogen peroxide (28, 56, 84, 141, 169, and 197 µM) in double-reciprocal plots in the presence (a) and in the absence (b) of 50 mM Ca2+; c) secondary plots of the dependence of intercepts at the ordinate axis of the primary plots (a and b) on the inverse concentration of hydrogen peroxide in the absence (1) and in the presence calcium ions (2).
1/v = 1/[E]0{(1/kH2O2[H2O2]) + (1/kS[S])},
where [E]0 is the initial enzyme concentration, kH2O2 and kS are the second-order rate constants for H2O2 and the donor substrate (in this case, ABTS), respectively. In accordance with the above equation, the intercepts at the ordinate axis in the “primary” plots are linearly proportional to the inversed concentration of the second substrate, and the second-order rate constant for the varied “fixed” substrate can be calculated from the slope of the “secondary” plot.
As it is obvious from the data in Fig. 4, the double reciprocal plots of the reaction rate dependence on the concentration of ABTS in the presence of any fixed concentration of hydrogen peroxide present a series of parallel lines both in the presence and in the absence of calcium ions. This confirms the ping-pong mechanism of the reaction of ABTS oxidation with hydrogen peroxide catalyzed by the recombinant non-glycosylated TOP and remains unchanged in the presence of calcium ions. However, the addition of calcium ions decreases the rate constant for hydrogen peroxide by 2.5-fold (from 3.6*106 to 1.4*106 M-1*sec-1) and for ABTS by 1.5-fold (from 8.2*106 to 6.0*106 M-1*sec-1) (Fig. 4). It is interesting that the rate-limiting step of ABTS oxidation catalyzed by TOP is hydrogen peroxide cleavage rather than the substrate oxidation step, as occurs for all other peroxidases including the horseradish enzyme.
Thus, the interaction of TOP with calcium ions is likely to be determined by the peculiarities of the protein polypeptide chain folding because the recombinant enzyme synthesized in E. coli is non-glycosylated. The effect of calcium can reflect the instability of its distal binding site because the refolding proceeds with an excess of calcium ions in the medium, but the purification is performed in their absence. The lack of calcium in the distal binding site usually results in a decrease in the rate constant for hydrogen peroxide because of the shift of the distal histidine towards the heme plane and the formation of a hexa-coordinated iron [18]. The second order rate constant for hydrogen peroxide is of the same order of magnitude for both TOP and HRP, and thus, it is unlikely that the heme iron is hexa-coordinated in TOP in the absence of calcium. The lesser accessibility to the active site of TOP or non-optimal coordination of its catalytic histidine in the presence of calcium can result from structural changes caused by calcium cation adsorption on the protein surface, which has an excess of negatively charged residues (pI < 3.5). As noted above, one of the negatively charged residues, Glu141, contrary to Phe143 in horseradish peroxidase [15], controls the access to the enzyme active center. The interaction of this residue with calcium ions in solution may result in the active center screening and therefore, in its lower accessibility in the presence of calcium. Further work on mutagenesis of the active center residues will give a definite answer on the role of the Glu141 residue in the unusual behavior of anionic tobacco peroxidase induced by calcium.
Thus, the experiments resulted in the production of active non-glycosylated recombinant TOP from E. coli inclusion bodies. The comparison of the properties of native and recombinant tobacco peroxidase shows that the oligosaccharide part of the enzyme has no principal effect on the catalytic mechanism and calcium regulation of the enzymatic activity.
The work was funded by INTAS 991-1768 and INCO-Copernicus ICA2-CT-2000-10050 grants.
REFERENCES
1.Gazaryan, I. G., and Lagrimini, L. M. (1996)
Phytochemistry, 41, 1029-1034.
2.Gazarian, I. G., Lagrimini, L. M., George, S. J.,
and Thorneley, R. N. (1996) Biochem J., 320, 369-372.
3.Surugiu, I., Danielsson, B., Ye, L., Mosbach, K.,
and Haupt, K. (2001) Analyt. Chem., 73, 487-491.
4.Dzgoev, A. B., Gazaryan, I. G., Lagrimini, L. M.,
Ramanathan, K., and Danielsson, B. (1999) Analyt. Chem.,
71, 5258-5261.
5.Gazaryan, I. G., Rubtsova, M. Yu., Kapeliuch, Yu.
L., Rodriguez-Lopez, J. N., Lagrimini, L. M., and Thorneley, R. N. F.
(1998) Photochem. Photobiol., 67, 106-110.
6.Farhangrazi, Z. S., Copeland, B. R., Nakayama, T.,
Amachi, T., Yamazaki, I., and Powers, L. S. (1994) Biochemistry,
33, 5647-5652.
7.Mareeva, E. A., Orlova, M. A., Doseeva, V. V.,
Loginov, D. B., Galkin, A. G., Gazaryan, I. G., and Tishkov, V. I.
(1996) Appl. Biochem. Biotechnol., 61, 13-24.
8.Lagrimini, L. M., Joly, R. J., Dunlap, J. R., and
Liu, T. T. (1997) Plant Mol. Biol., 33, 887-895.
9.Diaz-De-Leon, F., Klotz, K. L., and Lagrimini, L.
M. (1993) Plant Physiol., 101, 1117-1118.
10.Kalb, V. F., Jr., and Bernlohr, R. W. (1977)
Analyt. Biochem., 82, 362-371.
11.Lowry, O. H., Rosebrough, N. J., Farr, A. L., and
Randall, R. J. (1951) J. Biol. Chem., 193, 265-275.
12.Childs, R. E., and Bardsley, W. G. (1975)
Biochem. J., 145, 93-103.
13.Arnao, M. B., Casas, J. L., del Rio, J. A.,
Acosta, M., and Garcia-Canovas, F. (1990) Analyt. Biochem.,
185, 335-338.
14.Gazaryan, I. G., Doseeva, V. V., Galkin, A. G.,
and Tishkov, V. I. (1994) FEBS Lett., 354, 248-250.
15.Gazaryan, I. G., Ouporov, I. V., Chubar, T. A.,
Fechina, V. A., Mareeva, E. A., and Lagrimini, L. M. (1998)
Biochemistry (Moscow), 63, 600-606.
16.Dunford, H. B., and Stillman, J. S. (1976)
Coord. Chem. Rev., 19, 187-251.
17.Dunford, H. B. (1999) Heme Peroxidases,
Wiley-VCH, N.Y.-Berlin.
18.Tanaka, M., Ishimori, K., and Morishima, I.
(1998) Biochemistry, 37, 2629-2638.