Role of Arginine 226 in the Mechanism of Tryptophan Indole-Lyase from Proteus vulgaris
V. V. Kulikova1, L. N. Zakomirdina1, N. P. Bazhulina1, I. S. Dementieva1, N. G. Faleev2, P. D. Gollnick3, and T. V. Demidkina1*
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, Moscow 119991, Russia; fax: (7-095) 135-1405; E-mail: tvd@eimb.ru2Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow 117813, Russia; fax: (7-095) 135-5085; E-mail: ngfal@ineos.ac.ru
3Department of Biological Sciences, State University of New York at Buffalo, Buffalo, NY 14260, USA; E-mail: gollnick@acsu.buffalo.edu
* To whom correspondence should be addressed.
Received May 27, 2003
In the spatial structure of tryptophanase from Proteus vulgaris the guanidinium group of arginine 226 forms a salt bridge with the 3´-oxygen atom of the coenzyme. The replacement of arginine 226 with alanine using site-directed mutagenesis reduced the affinity of the coenzyme for the protein by one order of magnitude compared to the wild-type enzyme. The catalytic activity of the mutant enzyme in the reaction with L-tryptophan was reduced 105-fold compared to the wild-type enzyme. The rates of the reactions with some other substrates decreased 103-104-fold. The mutant enzyme catalyzed exchange of the C-alpha-proton in complexes with some inhibitors with rates reduced 102-fold compared to the wild-type enzyme. Absorption and circular dichroism spectra of the mutant enzyme and the enzyme-inhibitor complexes demonstrate that the replacement of arginine 226 with alanine does not significantly affect the tautomeric equilibrium of the internal aldimine, but it leads to an alteration of the optimal conformation of the coenzyme-substrate intermediates.
KEY WORDS: tryptophan indole-lyase from Proteus vulgaris, pyridoxal 5´-phosphate, arginine 226, site-directed mutagenesis
Abbreviations: PLP) pyridoxal 5´-phosphate; DTT) dithiothreitol; LDH) lactate dehydrogenase; SOPC) S-o-nitrophenyl-L-cysteine.
Tryptophan indole-lyase (tryptophanase, EC 4.1.99.1) is a bacterial
pyridoxal 5´-phosphate (PLP)-dependent enzyme catalyzing
reactions of alpha,beta-elimination and
beta-substitution of L-tryptophan and a number of other
beta-substituted L-amino acids [1, 2].
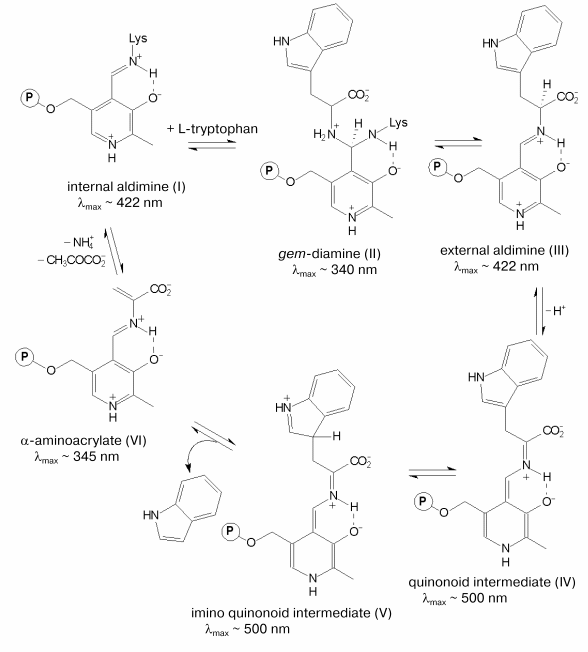
The chemical mechanism of the alpha,beta-elimination reaction [3, 4] is presented in the scheme. It includes the following main steps: formation of the external aldimine (III) with the substrate going through the formation of gem-diamine (II), C-alpha-proton abstraction yielding the quinonoid intermediate (IV), removal of the side group as a result of the concerted process of protonation of the indole fragment and cleavage of the C-C bond [4] resulting in the formation of the alpha-aminoacrylate intermediate (VI). The latter is attacked further by the epsilon-amino group of the lysine residue that binds the coenzyme resulting in regeneration of the holoenzyme. Due to the presence of the coenzyme, both the PLP-dependent enzymes and the intermediates of the reactions catalyzed by these enzymes possess characteristic spectral properties (scheme). The electronic state of PLP plays an important role in the efficiency of the catalysis. It is mainly determined by the interaction of the coenzyme with the active site amino acid residues. Determination of the three-dimensional structure of tryptophanase from Proteus vulgaris [5] has opened the way to investigation of the mechanism of the enzyme by site-directed mutagenesis. In the active site of tryptophanase the 3´-hydroxy-group of PLP forms a salt bridge with the guanidinium group of arginine 226. This residue is conserved in the sequences of tryptophanase from different sources (Fig. 1). To elucidate the role of this amino acid residue in the mechanism of tryptophanase, arginine 226 was replaced with alanine using site-directed mutagenesis, and properties of the mutant form R226A were investigated.
Mechanism of the alpha,beta-elimination reaction
MATERIALS AND METHODSFig. 1. Alignment of partial amino acid sequences of tryptophanase from different sources: Proteus vulgaris (pvulgaris), Proteus inconstans (pinconst), Escherichia coli (ecoli), Enterobacter aerogenes (entaer), Haemophilus influenzae (haeminfl), Pasteurella multocida (pmult), Vibrio cholerae (vcholera), Vibrio vulnificus (vvulnif), Symbiobacterium thermophilum 1 (symbther1), Symbiobacterium thermophilum 2 (symbther2), Thermoanaerobacter tengcongensis (thermoanaer). The conservative arginine residue is shown in bold.

Materials. In this work we used the following chemicals: rabbit muscle lactate dehydrogenase (LDH), NADH, PLP, L-tryptophan, L-phenylalanine, L-methionine, L-serine, beta-chloro-L-alanine, dithiothreitol (DTT), and D2O from Sigma (USA); S-benzyl-L-cysteine, S-ethyl-L-cysteine, EDTA, beta-phenyl-DL-serine, and L-cysteine sulfinic acid from Serva (Germany); Lauria-Bertani (LB) medium from Amersham (USA); tryptone and yeast extract from Difco (USA); DEAE-cellulose from Whatman (USA); phenyl-Sepharose and Superdex S-200 from Pharmacia Biotech (Sweden). S-o-Nitrophenyl-L-cysteine (SOPC) was synthesized as described in [6]. Oxindolyl-L-alanine was obtained by oxidation of L-tryptophan according to the method described in [7]. We also used salts of domestic production of analytical grade.
Site-directed mutagenesis. Site-directed mutagenesis was performed using the pAVK2 plasmid containing the tryptophanase gene from Proteus vulgaris [8]. Single-stranded DNA of this plasmid was obtained using the M13K07 helper phage [9]. To replace arginine 226 with alanine, the synthetic oligonucleotide 5´-GATTCAGCTGCTTTTTGTG-3´ with substituted nucleotides in the corresponding codon was used. Site-directed mutagenesis was performed by the Eckshtein method [10] using a Sculptor in vitro mutagenesis kit from Amersham. Mutant clones obtained after mutagenesis were screened by sequencing the gene in the mutated region [11] using a Sequenase Version 2.0 DNA sequencing kit from Amersham and custom primers complementary to nucleotides 1047-1065 of the tryptophanase gene [8]. E. coli SVS370 cells deficient in the tryptophanase gene were used as the host cells.
Growth of cells and purification of the enzymes. Growth of E. coli SVS370 cells containing the pAVK2 plasmid with the gene of the wild-type enzyme and the plasmid with the gene of the mutant enzyme and isolation of the enzymes were performed as described previously [12]. The mutant enzyme was isolated using gel filtration as an additional step of purification. The enzyme solution (20 mg/ml, 2 ml) was applied to a Superdex S-200 column (1.5 × 70 cm) equilibrated with 0.15 M potassium phosphate buffer, pH 7.0, containing 5 mM DTT and 0.1 mM PLP. The enzyme was eluted with the same buffer. After the chromatography the enzyme was precipitated by the addition of ammonium sulfate to 75% saturation and stored at -20°C. The purity of the enzymes was checked using SDS-PAGE as described by Laemmli [13].
Protein determination. Protein concentration during the purification was determined by the Lowry method [14]. Concentration of the purified enzyme was determined spectrophotometrically at 278 nm, using A1%1 cm = 9.19 [15].
Enzyme assays. Activity of the enzymes during the purification was determined using SOPC as a substrate. The rate of the reaction was monitored by a decrease in the optical density at 370 nm (epsilon = 1860 M-1*cm-1) [16]. The measurements were performed at 30°C in 0.1 M potassium phosphate buffer, pH 7.8, in the presence of 0.1 mM PLP. One unit of the tryptophanase activity was defined as the amount of the enzyme catalyzing the decomposition of 1 µmol of SOPC per min.
Kinetic studies. Kinetic parameters of the alpha,beta-elimination reaction were determined at 30°C in the coupled system with LDH by measuring a decrease in the optical density of NADH at 340 nm (epsilon = 6220 M-1*cm-1) [17]. The reaction mixtures contained 0.1 M potassium phosphate buffer, pH 7.8, 0.1 mM PLP, 0.2 mM NADH, 8 units of LDH, and variable concentrations of the substrates. The steady-state kinetic parameters of the alpha,beta-elimination reaction were determined by the Michaelis-Menten equation using the Enzfitter program (Elsevier Biosoft) [18].
Inhibition of alpha,beta-elimination of SOPC by different amino acids was studied under conditions described above. Values of the inhibition constants were determined using the Enzfitter program [18].
Kinetics of isotope exchange of the C-alpha-proton of L-phenylalanine and L-methionine catalyzed by the wild-type tryptophanase and the mutant enzyme was investigated in 0.1 M potassium phosphate buffer in D2O in the presence of 0.1 mM PLP. The pH value of the buffer solution was 7.4 that corresponded to pD = 7.8 [19]. Tryptophanase (1-5 mg) was added to the buffer solution containing the amino acid, and then the mixture was incubated at 30°C. For the wild-type enzyme L-methionine concentration in samples was 38.5 mM. For the mutant enzyme concentrations of L-methionine and L-phenylalanine were 640 and 180 mM, respectively. After known time intervals aliquots of reaction mixture were withdrawn and heated at 90°C for 5 min to stop the reaction and analyzed by proton magnetic resonance (NMR). The NMR spectra were taken using a Brucker AMXIII-400 unit (Germany) at 400 MHz. The ratio of deuterated to non-deuterated products was calculated from the relative integral intensities of the C-alpha-proton and beta-CH2 group signals, the latter being used as an internal standard.
Taking the Km value as equal to the Ki value for the competitive inhibition of the enzyme in the reaction with SOPC and determining the ratio [S]0/([S]0 - [P]) from the NMR spectrum, the rate of deuterium exchange was calculated from the integrated dependence of the substrate transformation degree on time allowing for the inhibition by the product [20] using the equation:
![]()
where [S]0 is the concentration of the inhibitor, [P] is the concentration of the product, and t is time.
Spectral studies of the enzymes. Absorption spectra of the holoenzymes and their complexes with inhibitors were taken using a Cary-50 spectrophotometer (Varian, USA). CD spectra were taken using a Mark III dichrograph (Yobin-Yvon, France). Concentration of the enzymes was 1-2 mg/ml, and the spectra were taken in 0.1 M potassium phosphate buffer, pH 7.8.
Before taking spectra, the protein preparations were incubated in the presence of 50-fold molar excess of PLP at 30°C for 30 min. The excess of PLP was then removed by dialysis.
Pyridoxal-5´-phosphate content. The PLP content in the enzyme preparations was determined in 0.1 M NaOH by the method of Peterson and Sober [21] using molar absorption coefficient for PLP at 390 nm epsilon = 6600 M-1*cm-1.
Preparation of apoenzyme. Apoenzyme was prepared by treatment of the enzyme preparations with DL-penicillamine [22].
Determination of dissociation constant of PLP for the mutant enzyme. The dissociation constant of PLP was determined by spectrophotometric titration. Aliquots of PLP solution were added to a solution of the apoenzyme in 0.1 M potassium phosphate, pH 7.8. After 30 min of incubation at 30°C, changes in the absorption spectra were detected at 425 nm. The amount of PLP bound to the enzyme was determined using the molar absorption coefficient for the internal aldimine (epsilon = 10,680 M-1*cm-1) that was calculated from the spectra of the holoenzyme with a known content of PLP. The data were fitted to the Scatchard type plot [23].
Alignment of sequences. Alignment of amino acid sequences of tryptophanase from different sources was performed using the CLUSTALX program [24].
RESULTS AND DISCUSSION
Enzyme purification. Expression of the wild-type enzyme in the pAVK2 plasmid yields about 11% of tryptophanase of total protein in a cell extract [12]. In the case of the mutant enzyme, its expression constituted about 4%. Thus, the method of purification developed for obtaining the homogeneous wild-type enzyme did not yield homogeneous preparation of the mutant enzyme. To purify the mutant enzyme, Superdex gel filtration was used as the additional step of purification. According to the SDS-PAGE the purity of the mutant enzyme after the gel filtration was ~95%.
Determination of dissociation constant of pyridoxal-5´-phosphate. The value of dissociation constant of PLP for tryptophanase from Proteus vulgaris is 2.1*10-6 M [12]. We determined that Kd value for the mutant enzyme is equal to 5.5*10-5 M. The replacement of arginine 226 with alanine in tryptophanase increases Kd value a little more than 10-fold, this corresponding to a decrease in the free energy of binding by 2.7 kJ/mol.
Kinetic parameters. The steady-state kinetic parameters of the alpha,beta-elimination of a number of substrates catalyzed by the wild-type enzyme and by the R226A mutant form are presented in Table 1. The mutant enzyme was virtually incapable of catalyzing the reaction with the natural substrate L-tryptophan. The rates of the reactions with other substrates decreased 3-4-fold compared to those catalyzed by the wild-type enzyme, but for S-methyl-L-cysteine we could not detect the formation of pyruvate even qualitatively. The kcat/Km value decreased 3-6-fold for all substrates except for L-serine. For the latter the kcat/Km value decreased by a factor of 102, the Km value being decreased more than 102-fold. Thus, the replacement of the arginine 226 with alanine significantly affected both the rate of alpha,beta-elimination of all investigated substrates and binding of some of them.
Table 1. Steady-state kinetic parameters of
the alpha,beta-elimination reaction catalyzed by the
wild-type tryptophanase and the R226A mutant enzyme*
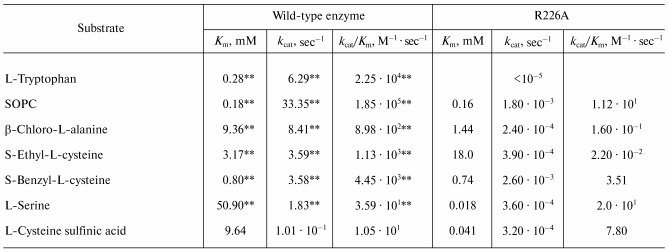
*Measurement errors constituted no more than 10%.
**As described in [12].
L-Phenylalanine and L-methionine, competitive inhibitors of the wild-type enzyme [12], were also found to be competitive inhibitors for the mutant enzyme. The values of the inhibition constants for L-phenylalanine and L-methionine were determined using SOPC as a substrate, since the mutant enzyme did not catalyze decomposition of L-tryptophan. The values of Ki for L-phenylalanine and L-methionine increased 4- and 16-fold, respectively, compared to those for the wild-type enzyme (Table 2).
Table 2. Inhibition of the reaction of
alpha,beta-elimination of SOPC and the rates of the
C-alpha-proton exchange (kex) in the
enzyme-inhibitor complexes

*As described in [12].
Investigation of the rate of the C-alpha-proton exchange in complexes of the mutant enzyme with L-phenylalanine and L-methionine (Table 2) showed that the mutant enzyme catalyzed the exchange of the C-alpha-proton of L-phenylalanine and L-methionine in D2O with rates 102-103-fold lower than wild-type tryptophanase.
Spectral studies. Figures 2 (a and b) and 3 (a and b) present the absorption and CD spectra of the wild-type and mutant holoenzymes and their complexes with L-phenylalanine and L-methionine.
Fig. 2. Absorption (a) and CD (b) spectra of the wild-type tryptophanase (3*10-5 M) in 0.1 M potassium phosphate buffer, pH 7.8, and of the enzyme-inhibitor complexes: 1) holoenzyme; 2) complex with L-phenylalanine (0.1 M); 3) complex with L-methionine (0.09 M).
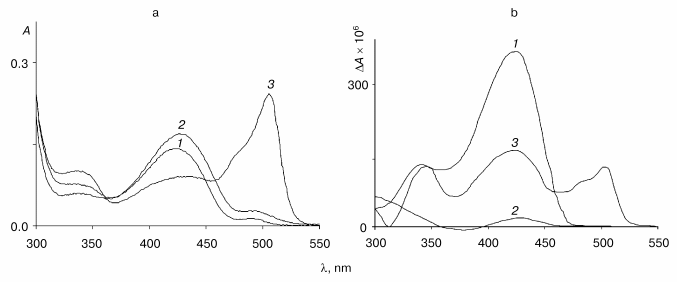
The absorption spectrum of the wild-type enzyme has characteristic absorption bands with maxima at 340 and 422 nm exhibiting positive dichroism (Fig. 2, a and b) and corresponding to two tautomeric forms of the internal aldimine - enolimine (340 nm) and ketoenamine (422 nm) [12]:Fig. 3. Absorption (a) and CD (b) spectra of the mutant tryptophanase (3*10-5 M) in 0.1 M potassium phosphate buffer, pH 7.8, and of the enzyme-inhibitor complexes: 1) holoenzyme; 2) complex with L-phenylalanine (0.15 M); 3) complex with L-methionine (0.2 M).
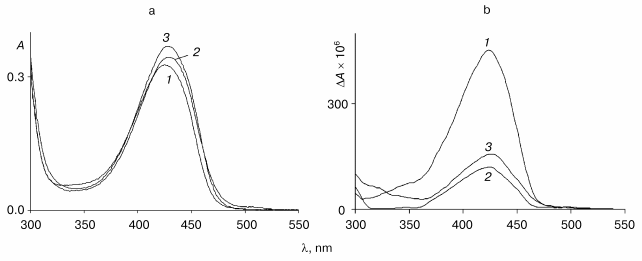
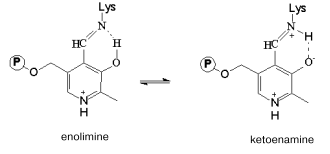
The spectrum of the wild-type holoenzyme also exhibits a low intensity band with a maximum at 490 nm. The replacement of arginine 226 with alanine did not result in significant changes in the absorption spectrum, the mutant enzyme retaining a predominant ketoenamine band as in the case of the wild-type enzyme (Fig. 3a). This band exhibits a positive CD, as shown for the wild-type tryptophanase (Fig. 3b). Consequently, replacement of arginine 226 with alanine did not affect the tautomeric equilibrium of the internal aldimine in the mutant enzyme. However, the difference in the values of the anisotropy factors of the ketoenamine bands for the wild-type holoenzyme (DeltaA/A = 2.5*10-3) and for the mutant holoenzyme (DeltaA/A = 1.39*10-3) indicates that the microenvironment of the coenzyme in the mutant enzyme differs from that in the wild-type enzyme.
Previously we demonstrated that wild-type tryptophanase binds with L-alanine and oxindolyl-L-alanine and the absorption and CD spectra of the complexes of tryptophanase with these inhibitors exhibited maximum in the region of 495-505 nm, which is characteristic for the quinonoid intermediate [12]. In the present work we investigated the spectral characteristics of complexes of the wild-type enzyme with L-methionine and L-phenylalanine and the spectral properties of complexes of the mutant enzyme with L-alanine, oxindolyl-L-alanine, L-methionine, and L-phenylalanine.
In the absorption spectrum of the complex of the wild-type tryptophanase with L-methionine the characteristic band of the quinonoid structure predominates (Fig. 2a). The CD spectrum of this complex is better resolved and contains three positive bands with maxima at 340, 430, and 505 nm (Fig. 2b). The presence of the band corresponding to the quinonoid intermediate suggests that the bands with the maxima at 340 and 430 nm correspond to the gem-diamine and external aldimine. Thus, interaction of tryptophanase with L-methionine yields an equilibrium mixture of the three consecutive intermediates of the enzymatic reaction.
In the absorption and CD spectra of the complex of the wild-type tryptophanase with L-phenylalanine (Fig. 2, a and b), the characteristic band of the quinonoid structure is absent, and an increase in the intensity of the ketoenamine band with a maximum at 430 nm is observed with a simultaneous sharp decrease in the intensity of the corresponding band in the CD spectrum. Consequently, the wild-type enzyme interacts with L-phenylalanine to yield the external aldimine, while the quinonoid complex is formed with L-alanine, oxindolyl-L-alanine, and L-methionine.
In contrast to the wild-type enzyme, the characteristic band of the quinonid intermediate is absent in the absorption and CD spectra of the complexes of mutant enzyme with L-methionine, L-phenylalanine (Fig. 3, a and b), L-alanine, and oxindolyl-L-alanine (spectra not shown). Interaction of the mutant enzyme with these amino acids results in significant changes in the anisotropy factors of the bands with the maximum at 420-430 nm (DeltaA/A = 1.39*10-3 for the holoenzyme, DeltaA/A = 4.5*10-4 for the complex with L-methionine, and DeltaA/A = 3.3*10-4 for the complex with L-phenylalanine), suggesting that these bands are due to the external aldimines. The anisotropy factors of the ketoenamine band of the complexes of the mutant enzyme with L-methionine and L-phenylalanine differ from the corresponding values for the wild-type tryptophanase (DeltaA/A = 1.7*10-3 for the complex with L-methionine and DeltaA/A = 1.1*10-4 for the complex with L-phenylalanine). Thus, in the complexes of the mutant enzyme with the indicated amino acids deceleration of the reaction is observed on the stage of the C-alpha-proton abstraction from the external aldimines. This is in agreement with the lower value of the rate of the C-alpha-proton exchange in the complexes of the mutant enzyme with these amino acids compared to that obtained for the wild-type enzyme.
The role of arginine 226 in the mechanism of tryptophanase. Determination of the dissociation constant of PLP for the mutant enzyme demonstrated that the absence of the salt bridge formed by the guanidinium group of arginine 226 and the 3´-hydroxy-group of the coenzyme resulted in a decrease in the free energy of PLP binding by 2.7 kJ/mol. Such an insignificant decrease in the energy of binding suggests that in the mutant enzyme, the 3´-oxygen atom of PLP is capable of forming the hydrogen bond with some other residue of the active site or with a water molecule. As a result, the conformation of the internal aldimine in the mutant enzyme can differ from that in the wild-type enzyme, as demonstrated by the spectral data. Actually, the changes in the values of the anisotropy factors of the ketoenamine band in the complexes of the mutant enzyme with amino acids compared to the corresponding values for complexes of the wild-type tryptophanase indicate that the conformation (microenvironment) of the external aldimines is different in the wild-type and mutant enzymes. The conformation of the external aldimine in the mutant enzyme is likely to disturb the optimal for catalysis orientation of the C-alpha-proton of the substrate and the base abstracting this proton, this leading to deceleration of the enzymatic reaction at the stage of the formation of the quinonid intermediate.
The virtually complete absence of the catalytic activity of the mutant enzyme in the reaction with the natural substrate and the decrease in the rate of the reactions with other substrates that exceeds the decrease in the rate of the C-alpha-proton exchange in the enzyme-inhibitor complexes indicates that the absence of the interaction between the side group of arginine 226 and the 3´-oxygen atom of the coenzyme results in a decrease in the steady-state concentration of the quinoid intermediate and deceleration of the subsequent steps of beta-elimination. This is likely due to the disturbance of the optimal conformation of the coenzyme-substrate intermediates during all stages of the reaction.
The work was supported by the Russian Foundation for Basic Research (grants 01-04-48636 and 01-04-48719), the Program for the Support of Scientific Schools of Russia (grant 1800.2003.4), and the Howard Hughes Medical Institute (grant 751 95 54001).
REFERENCES
1.Snell, E. E. (1975) Adv. Enzymol.,
42, 287-333.
2.Newton, W. A., Morino, Y., and Snell, E. E. (1965)
J. Biol. Chem., 240, 1211-1218.
3.Davis, L., and Metzler, D. (1972) Enzymes,
7, 334-338.
4.Phillips, R. S., Sundararaju, B., and Faleev, N. G.
(2000) J. Am. Chem. Soc., 122, 1008-1114.
5.Isupov, M. N., Antson, A. A., Dodson, G. G.,
Dodson, E. J., Dementieva, I. S., Zakomirdina, L. N., Wilson, K. S.,
Dauter, Z., and Harutyunyan, E. H. (1998) J. Mol. Biol.,
276, 603-623.
6.Boyland, E., Manson, D., and Nery, R. (1962) J.
Chem. Soc., 2, 606-612.
7.Savige, W. E., and Fontana, A. (1980) Int. J.
Peptide Protein Res., 15, 285-297.
8.Kamath, A. V., and Yanofsky, C. (1992) J. Biol.
Chem., 267, 19978-19985.
9.Vieira, J., and Messing, J. (1987) Meth.
Enzymol., 153, 1-34.
10.Taylor, J. W., Ott, J., and Eckstein, F. (1985)
Nucleic Acids Res., 13, 8764-8785.
11.Sanger, F., Nicklen, S., and Coulsen, A. R.
(1977) Proc. Natl. Acad. Sci. USA, 74, 5463-5467.
12.Zakomirdina, L. N., Kulikova, V. V., Gogoleva, O.
I., Dementieva, I. S., Faleev, N. G., and Demidkina, T. V. (2002)
Biochemistry (Moscow), 67, 1189-1196.
13.Weber, K., and Osborn, M. (1969) J. Biol.
Chem., 244, 4406-4412.
14.Lowry, O. H., Rosenbrough, N. J., Farr, A. L.,
and Randall, R. J. (1951) J. Biol. Chem., 193,
265-275.
15.Phillips, R. S., and Gollnick, P. D. (1989) J.
Biol. Chem., 264, 10627-10632.
16.Suelter, C. H., Wang, J., and Snell, E. E. (1976)
FEBS Lett., 66, 230-232.
17.Morino, Y., and Snell, E. E. (1970) Meth.
Enzymol., 17A, 439-446.
18.Cleland, W. W. (1979) Meth. Enzymol.,
63, 103-138.
19.Glasoe, P. V., and Long, F. A. (1960) J. Phys.
Chem., 64, 188-194.
20.Berezin, A. V., and Klesov, A. A. (1976)
Apprentice Course of Chemical and Enzyme Kinetics [in Russian],
MGU Publishers, Moscow, p. 168.
21.Peterson, E. A., and Sober, H. A. (1954) J.
Amer. Chem. Soc., 76, 169-175.
22.Morino, Y., and Snell, E. E. (1967) J. Biol.
Chem., 242, 5591-5601.
23.Klotz, I. M., and Hunston, D. L. (1971)
Biochemistry, 76, 169-175.
24.Altschul, S. F., Madden, T. L., Schaffer, A. A.,
Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997) Nucleic
Acids Res., 25, 3389-3402.